
Stable and Flexible Synaptic Transmission Controlled by the Active Zone Protein Interactions

Abstract
:1. Introduction
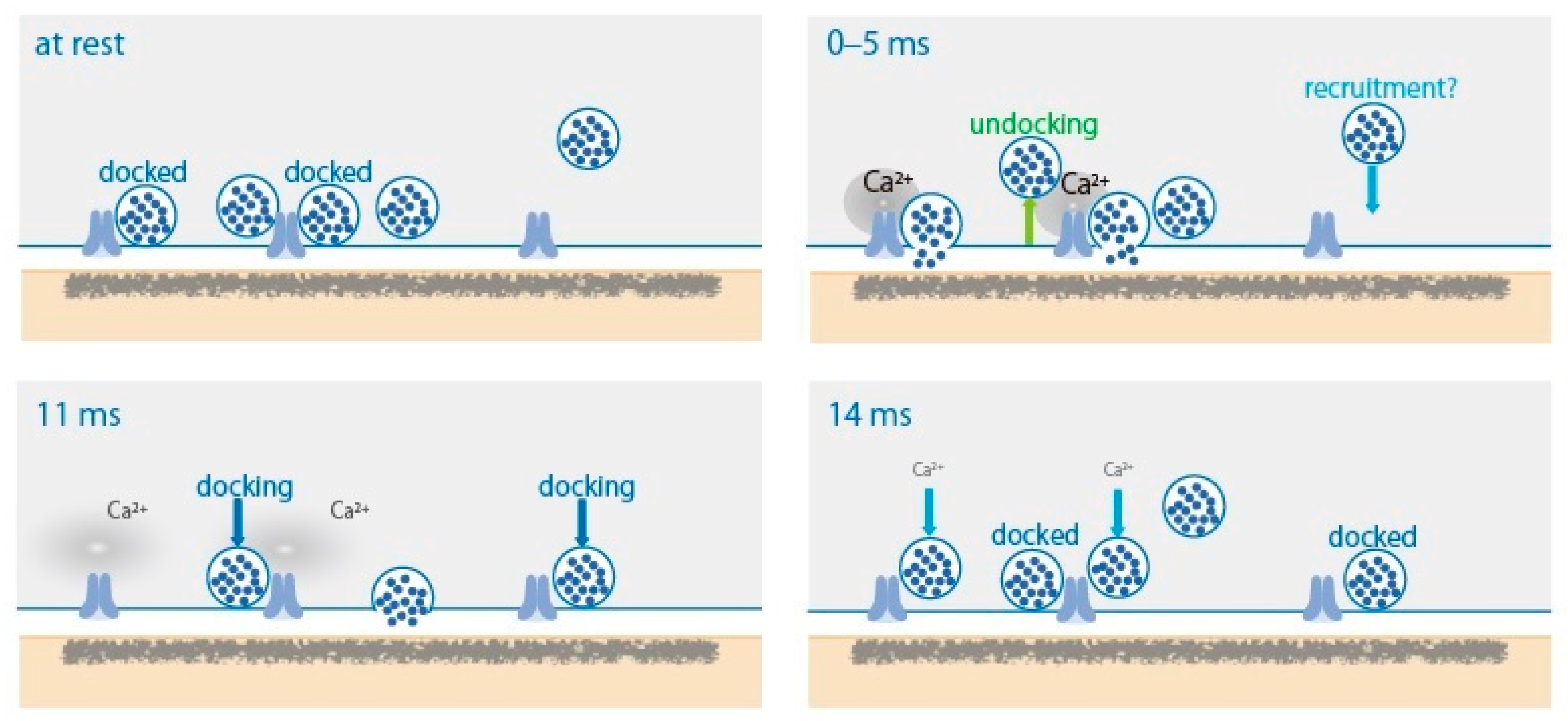
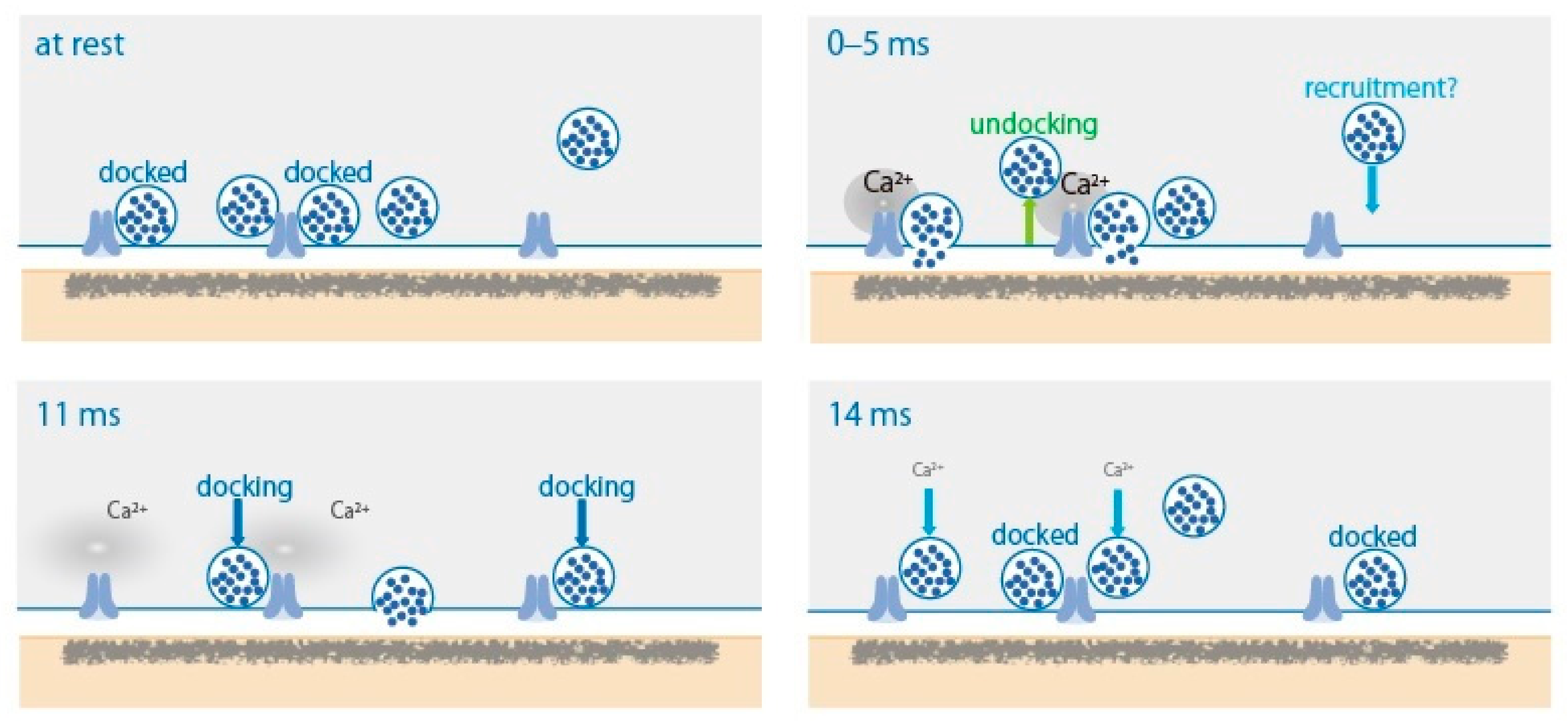
2. SV Dynamics in the AZ after AP
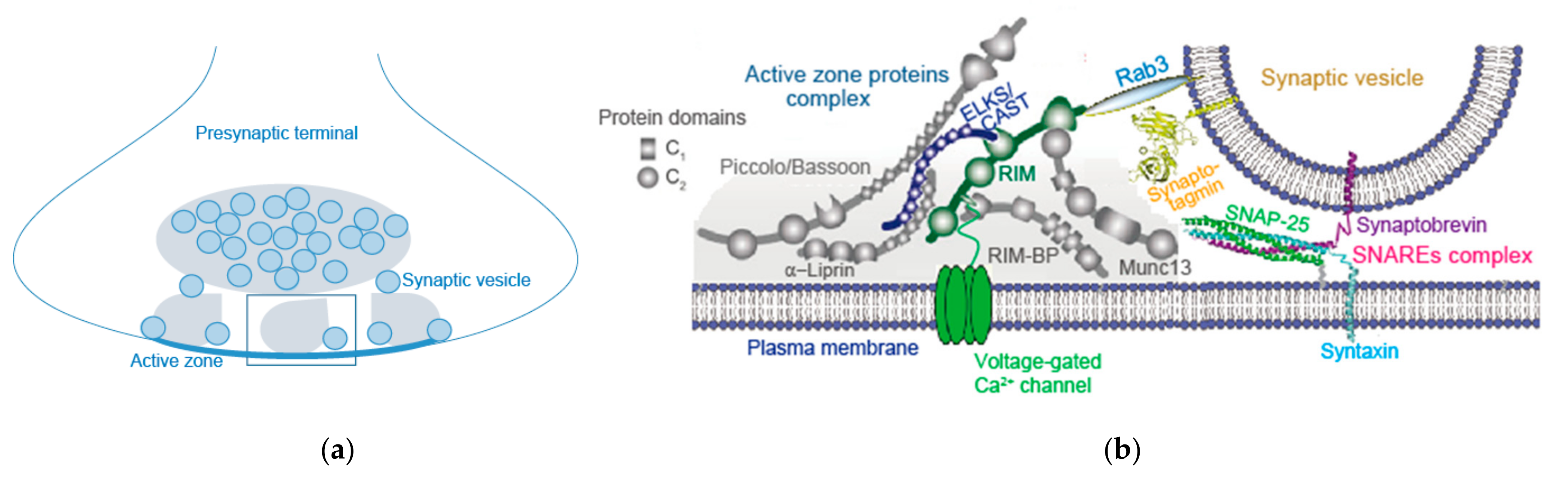
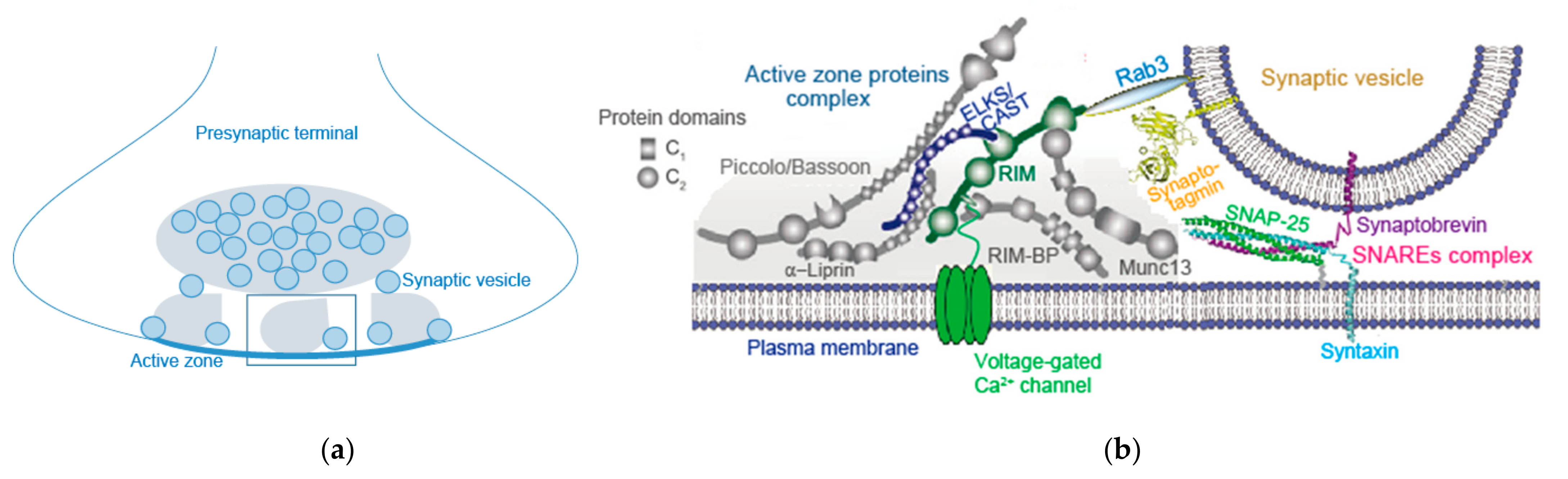
3. AZ Proteins and Their Complex
3.1. Munc13
3.2. RIM, RIM-BP, and Others
3.3. Bruchpilot and CAST/ELKS
3.4. AZ Assembly Models
3.5. AZ Assembly Stability and Degradation
4. Control of SV States and Localization
4.1. SV States
4.2. SV Docking and SV Priming
4.3. SV Fusion
4.4. SV Localization
5. SV Replenishment
5.1. CAST/ELKS
5.2. Bassoon
5.3. RIM-BP
6. Presynaptic Short-Term Plasticity
6.1. RIM
6.2. Munc13 and Calmodulin
6.3. Munc18, Diacylglycerol, and PKC
6.4. Bassoon and Piccolo
6.5. CAST and SAD-Kinase
7. Presynaptic Homeostasis Plasticity
7.1. RIM
7.2. RIM-BP
7.3. Bassoon
8. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Südhof, T.C. The presynaptic active zone. Neuron 2012, 75, 11–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biederer, T.; Kaeser, P.S.; Blanpied, T.A. Transcellular Nanoalignment of Synaptic Function. Neuron 2017, 96, 680–696. [Google Scholar] [CrossRef]
- Wang, S.S.; Held, R.G.; Wong, M.Y.; Liu, C.; Karakhanyan, A.; Kaeser, P.S. Fusion Competent Syenaptic Vesicles Persist upon Active Zone Disruption and Loss of Vesicle Docking. Neuron 2016, 91, 777–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kusick, G.F.; Chin, M.; Raychaudhuri, S.; Lippmann, K.; Adula, K.P.; Hujber, E.J.; Vu, T.; Davis, M.W.; Jorgensen, E.M.; Watanabe, S. Synaptic vesicles transiently dock to refill release sites. Nat. Neurosci. 2020, 23, 1329–1338. [Google Scholar] [CrossRef] [PubMed]
- Emperador-Melero, J.; Kaeser, P.S. Assembly of the presynaptic active zone. Curr. Opin. Neurobiol. 2020, 63, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Tang, A.H.; Chen, H.; Li, T.P.; Metzbower, S.R.; MacGillavry, H.D.; Blanpied, T.A. A trans-synaptic nanocolumn aligns neurotransmitter release to receptors. Nature 2016, 536, 210–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakamoto, H.; Ariyoshi, T.; Kimpara, N.; Sugao, K.; Taiko, I.; Takikawa, K.; Asanuma, D.; Namiki, S.; Hirose, K. Synaptic weight set by Munc13-1 supramolecular assemblies. Nat. Neurosci. 2018, 21, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Zarebidaki, F.; Camacho, M.; Brockmann, M.M.; Trimbuch, T.; Herman, M.A.; Rosenmund, C. Disentangling the Roles of RIM and Munc13 in Synaptic Vesicle Localization and Neurotransmission. J. Neurosci. 2020, 40, 9372–9385. [Google Scholar] [CrossRef]
- Junge, H.J.; Rhee, J.S.; Jahn, O.; Varoqueaux, F.; Spiess, J.; Waxham, M.N.; Rosenmund, C.; Brose, N. Calmodulin and Munc13 form a Ca2+ sensor/effector complex that controls short-term synaptic plasticity. Cell 2004, 118, 389–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mochida, S.; Hida, Y.; Tanifuji, S.; Hagiwara, A.; Hamada, S.; Abe, M.; Ma, H.; Yasumura, M.; Kitajima, I.; Sakimura, K.; et al. SAD-B Phosphorylation of CAST Controls Active Zone Vesicle Recycling for Synaptic Depression. Cell Rep. 2016, 16, 2901–2913. [Google Scholar] [CrossRef] [Green Version]
- Dagostin, A.; Kushmerick, C.; von Gersdorff, H. Allegro giusto: Piccolo, bassoon and clarinet set the tempo of vesicle pool replenishment. J. Physiol. 2018, 596, 1315–1316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wierda, K.D.; Toonen, R.F.; de Wit, H.; Brussaard, A.B.; Verhage, M. Interdependence of PKC-dependent and PKC-independent pathways for presynaptic plasticity. Neuron 2007, 54, 275–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genc, O.; Kochubey, O.; Toonen, R.F.; Verhage, M.; Schneggenburger, R. Munc18-1 is a dynamically regulated PKC target during short-term enhancement of transmitter release. eLife 2014, 3, e01715. [Google Scholar] [CrossRef] [Green Version]
- Hong, H.; Zhao, K.; Huang, S.; Huang, S.; Yao, A.; Jiang, Y.; Sigrist, S.; Zhao, L.; Zhang, Y.Q. Structural Remodeling of Active Zones Is Associated with Synaptic Homeostasis. J. Neurosci. 2020, 40, 2817–2827. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.; Liu, K.S.; Sigrist, S.J.; Davis, G.W. RIM controls homeostatic plasticity through modulation of the readily-releasable vesicle pool. J. Neurosci. 2012, 32, 16574–16585. [Google Scholar] [CrossRef]
- Mendoza Schulz, A.; Jing, Z.; Sánchez Caro, J.M.; Wetzel, F.; Dresbach, T.; Strenzke, N.; Wichmann, C.; Moser, T. Bassoon-disruption slows vesicle replenishment and induces homeostatic plasticity at a CNS synapse. EMBO J. 2014, 33, 512–527. [Google Scholar] [CrossRef] [Green Version]
- Heuser, J.E.; Reese, T.S. Structural changes after transmitter release at the frog neuromuscular junction. J. Cell Biol. 1981, 88, 564–580. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Harada, H.; Kamasawa, N.; Matsui, K.; Rothman, J.S.; Shigemoto, R.; Silver, R.A.; DiGregorio, D.A.; Takahashi, T. Nanoscale distribution of presynaptic Ca2+ channels and its impact on vesicular release during development. Neuron 2015, 85, 145–158. [Google Scholar] [CrossRef] [Green Version]
- Lipstein, N.; Sakaba, T.; Cooper, B.H.; Lin, K.H.; Strenzke, N.; Ashery, U.; Rhee, J.S.; Taschenberger, H.; Neher, E.; Brose, N. Dynamic control of synaptic vesicle replenishment and short-term plasticity by Ca2+-calmodulin-Munc13-1 signaling. Neuron 2013, 79, 82–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borst, J.G.; Helmchen, F.; Sakmann, B. Pre- and postsynaptic whole-cell recordings in the medial nucleus of the trapezoid body of the rat. J. Physiol. 1995, 489 Pt 3, 825–840. [Google Scholar] [CrossRef] [Green Version]
- Neher, E.; Brose, N. Dynamically Primed Synaptic Vesicle States: Key to Understand Synaptic Short-Term Plasticity. Neuron 2018, 100, 1283–1291. [Google Scholar] [CrossRef] [Green Version]
- Pofantis, E.; Neher, E.; Dresbach, T. Regulation of a subset of release-ready vesicles by the presynaptic protein Mover. Proc. Natl. Acad. Sci. USA 2021, 118, e2022551118. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Ganzella, M.; Zhou, J.; Zhu, S.; Jahn, R.; Zhang, M. Vesicle Tethering on the Surface of Phase-Separated Active Zone Condensates. Mol. Cell 2021, 81, 13–24. [Google Scholar] [CrossRef]
- Ikeda, S.R. Voltage-dependent modulation of N-type calcium channels by G-protein beta gamma subunits. Nature 1996, 380, 255–258. [Google Scholar] [CrossRef]
- Wang, X.; Kibschull, M.; Laue, M.M.; Lichte, B.; Petrasch-Parwez, E.; Kilimann, M.W. Aczonin, a 550-kD putative scaffolding protein of presynaptic active zones, shares homology regions with Rim and Bassoon and binds profilin. J. Cell Biol. 1999, 147, 151–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brose, N.; Hofmann, K.; Hata, Y.; Südhof, T.C. Mammalian homologues of Caenorhabditis elegans unc-13 gene define novel family of C2-domain proteins. J. Biol. Chem. 1995, 270, 25273–25280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fenster, S.D.; Chung, W.J.; Zhai, R.; Cases-Langhoff, C.; Voss, B.; Garner, A.M.; Kaempf, U.; Kindler, S.; Gundelfinger, E.D.; Garner, C.C. Piccolo, a presynaptic zinc finger protein structurally related to bassoon. Neuron 2000, 25, 203–214. [Google Scholar] [CrossRef] [Green Version]
- Ohtsuka, T.; Takao-Rikitsu, E.; Inoue, E.; Inoue, M.; Takeuchi, M.; Matsubara, K.; Deguchi-Tawarada, M.; Satoh, K.; Morimoto, K.; Nakanishi, H.; et al. Cast: A novel protein of the cytomatrix at the active zone of synapses that forms a ternary complex with RIM1 and munc13-1. J. Cell Biol. 2002, 158, 577–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takao-Rikitsu, E.; Mochida, S.; Inoue, E.; Deguchi-Tawarada, M.; Inoue, M.; Ohtsuka, T.; Takai, Y. Physical and functional interaction of the active zone proteins, CAST, RIM1, and Bassoon, in neurotransmitter release. J. Cell Biol. 2004, 164, 301–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- tom Dieck, S.; Sanmarti-Vila, L.; Langnaese, K.; Richter, K.; Kindler, S.; Soyke, A.; Wex, H.; Smalla, K.H.; Kampf, U.; Franzer, J.T.; et al. Bassoon, a novel zinc-finger CAG/glutamine-repeat protein selectively localized at the active zone of presynaptic nerve terminals. J. Cell Biol. 1998, 142, 499–509. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Liu, X.; Biederer, T.; Südhof, T.C. A family of RIM-binding proteins regulated by alternative splicing: Implications for the genesis of synaptic active zones. Proc. Natl. Acad. Sci. USA 2002, 99, 14464–14469. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Okamoto, M.; Schmitz, F.; Hofmann, K.; Südhof, T.C. Rim is a putative Rab3 effector in regulating synaptic-vesicle fusion. Nature 1997, 388, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Rebola, N.; Reva, M.; Kirizs, T.; Szoboszlay, M.; Lőrincz, A.; Moneron, G.; Nusser, Z.; DiGregorio, D.A. Distinct Nanoscale Calcium Channel and Synaptic Vesicle Topographies Contribute to the Diversity of Synaptic Function. Neuron 2019, 104, 693–710. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Kaeser, P.S.; Südhof, T.C.; Schneggenburger, R. RIM determines Ca2+ channel density and vesicle docking at the presynaptic active zone. Neuron 2011, 69, 304–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaeser, P.S.; Deng, L.; Wang, Y.; Dulubova, I.; Liu, X.; Rizo, J.; Südhof, T.C. RIM Proteins Tether Ca2+ Channels to Presynaptic Active Zones via a Direct PDZ-Domain Interaction. Cell 2011, 144, 282–295. [Google Scholar] [CrossRef] [Green Version]
- Deng, L.; Kaeser, P.S.; Xu, W.; Südhof, T.C. RIM Proteins Activate Vesicle Priming by Reversing Autoinhibitory Homodimerization of Munc13. Neuron 2011, 69, 317–331. [Google Scholar] [CrossRef] [Green Version]
- Coppola, T.; Magnin-Luthi, S.; Perret-Menoud, V.; Gattesco, S.; Schiavo, G.; Regazzi, R. Direct interaction of the Rab3 effector RIM with Ca2+ channels, SNAP-25, and synaptotagmin. J. Biol. Chem. 2001, 276, 32756–32762. [Google Scholar] [CrossRef] [Green Version]
- Hibino, H.; Pironkova, R.; Onwumere, O.; Vologodskaia, M.; Hudspeth, A.J.; Lesage, F. RIM binding proteins (RBPs) couple Rab3-interacting molecules (RIMs) to voltage-gated Ca2+ channels. Neuron 2002, 34, 411–423. [Google Scholar] [CrossRef] [Green Version]
- Butola, T.; Alvanos, T.; Hintze, A.; Koppensteiner, P.; Kleindienst, D.; Shigemoto, R.; Wichmann, C.; Moser, T. RIM-Binding Protein 2 Organizes Ca2+ Channel Topography and Regulates Release Probability and Vesicle Replenishment at a Fast Central Synapse. J. Neurosci. 2021, 41, 7742–7767. [Google Scholar] [CrossRef]
- Kittel, R.J.; Wichmann, C.; Rasse, T.M.; Fouquet, W.; Schmidt, M.; Schmid, A.; Wagh, D.A.; Pawlu, C.; Kellner, R.R.; Willig, K.I.; et al. Bruchpilot promotes active zone assembly, Ca2+ channel clustering, and vesicle release. Science 2006, 312, 1051–1054. [Google Scholar] [CrossRef] [Green Version]
- Wagh, D.A.; Rasse, T.M.; Asan, E.; Hofbauer, A.; Schwenkert, I.; Durrbeck, H.; Buchner, S.; Dabauvalle, M.C.; Schmidt, M.; Qin, G.; et al. Bruchpilot, a protein with homology to ELKS/CAST, is required for structural integrity and function of synaptic active zones in Drosophila. Neuron 2006, 49, 833–844. [Google Scholar] [CrossRef] [Green Version]
- Kiyonaka, S.; Nakajima, H.; Takada, Y.; Hida, Y.; Yoshioka, T.; Hagiwara, A.; Kitajima, I.; Mori, Y.; Ohtsuka, T. Physical and functional interaction of the active zone protein CAST/ERC2 and the β-subunit of the voltage-dependent Ca2+ channel. J. Biochem. 2012, 152, 149–159. [Google Scholar] [CrossRef]
- Dong, W.; Radulovic, T.; Goral, R.O.; Thomas, C.; Suarez Montesinos, M.; Guerrero-Given, D.; Hagiwara, A.; Putzke, T.; Hida, Y.; Abe, M.; et al. CAST/ELKS Proteins Control Voltage-Gated Ca2+ Channel Density and Synaptic Release Probability at a Mammalian Central Synapse. Cell Rep. 2018, 24, 284–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- tom Dieck, S.; Specht, D.; Strenzke, N.; Hida, Y.; Krishnamoorthy, V.; Schmidt, K.-F.; Inoue, E.; Ishizaki, H.; Tanaka-Okamoto, M.; Miyoshi, J.; et al. Deletion of the Presynaptic Scaffold CAST Reduces Active Zone Size in Rod Photoreceptors and Impairs Visual Processing. J. Neurosci. 2012, 32, 12192–12203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radulovic, T.; Siebert, M.; Mertel, S.; Knoche, E.; Wegener, S.; Wichmann, C.; Matkovic, T.; Muhammad, K.; Depner, H.; Mettke, C.; et al. Presynaptic development is controlled by the core active zone proteins CAST/ELKS. J. Physiol. 2020, 598, 2431–2452. [Google Scholar] [CrossRef]
- Hallermann, S.; Fejtova, A.; Schmidt, H.; Weyhersmüller, A.; Silver, R.A.; Gundelfinger, E.D.; Eilers, J. Bassoon Speeds Vesicle Reloading at a Central Excitatory Synapse. Neuron 2010, 68, 710–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, K.S.; Siebert, M.; Mertel, S.; Knoche, E.; Wegener, S.; Wichmann, C.; Matkovic, T.; Muhammad, K.; Depner, H.; Mettke, C.; et al. RIM-binding protein, a central part of the active zone, is essential for neurotransmitter release. Science 2011, 334, 1565–1569. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Cai, Q.; Shen, Z.; Chen, X.; Zeng, M.; Du, S.; Zhang, M. RIM and RIM-BP Form Presynaptic Active-Zone-like Condensates via Phase Separation. Mol. Cell 2019, 73, 971–984e5. [Google Scholar] [CrossRef] [Green Version]
- Milovanovic, D.; Wu, Y.; Bian, X.; De Camilli, P. A liquid phase of synapsin and lipid vesicles. Science 2018, 361, 604–607. [Google Scholar] [CrossRef] [Green Version]
- Zeng, M.; Shang, Y.; Araki, Y.; Guo, T.; Huganir, R.L.; Zhang, M. Phase Transition in Postsynaptic Densities Underlies Formation of Synaptic Complexes and Synaptic Plasticity. Cell 2016, 166, 1163–1175. [Google Scholar] [CrossRef] [Green Version]
- Waites, C.L.; Leal-Ortiz, S.A.; Okerlund, N.; Dalke, H.; Fejtova, A.; Altrock, W.D.; Gundelfinger, E.D.; Garner, C.C. Bassoon and Piccolo maintain synapse integrity by regulating protein ubiquitination and degradation. EMBO J. 2013, 32, 954–969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parthier, D.; Kuner, T.; Körber, C. The presynaptic scaffolding protein Piccolo organizes the readily releasable pool at the calyx of Held. J. Physiol. 2018, 596, 1485–1499. [Google Scholar] [CrossRef]
- Kaeser, P.S.; Deng, L.; Chávez, A.E.; Liu, X.; Castillo, P.E.; Südhof, T.C. ELKS2alpha/CAST deletion selectively increases neurotransmitter release at inhibitory synapses. Neuron 2009, 64, 227–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acuna, C.; Liu, X.; Südhof, T.C. How to Make an Active Zone: Unexpected Universal Functional Redundancy between RIMs and RIM-BPs. Neuron 2016, 91, 792–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brockmann, M.M.; Zarebidaki, F.; Camacho, M.; Grauel, M.K.; Trimbuch, T.; Südhof, T.C.; Rosenmund, C. A Trio of Active Zone Proteins Comprised of RIM-BPs, RIMs, and Munc13s Governs Neurotransmitter Release. Cell Rep. 2020, 32, 107960. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Busnadiego, R.; Asano, S.; Oprisoreanu, A.M.; Sakata, E.; Doengi, M.; Kochovski, Z.; Zürner, M.; Stein, V.; Schoch, S.; Baumeister, W.; et al. Cryo-electron tomography reveals a critical role of RIM1α in synaptic vesicle tethering. J. Cell Biol. 2013, 201, 725–740. [Google Scholar] [CrossRef] [Green Version]
- Müller, M.; Genç, Ö.; Davis, G.W. RIM-binding protein links synaptic homeostasis to the stabilization and replenishment of high release probability vesicles. Neuron 2015, 85, 1056–1069. [Google Scholar] [CrossRef] [Green Version]
- Schneggenburger, R.; Meyer, A.C.; Neher, E. Released fraction and total size of a pool of immediately available transmitter quanta at a calyx synapse. Neuron 1999, 23, 399–409. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Bickford, L.S.; Held, R.G.; Nyitrai, H.; Südhof, T.C.; Kaeser, P.S. The active zone protein family ELKS supports Ca2+ influx at nerve terminals of inhibitory hippocampal neurons. J. Neurosci. 2014, 34, 12289–12303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Südhof, T.C. Neurotransmitter release: The last millisecond in the life of a synaptic vesicle. Neuron 2013, 80, 675–690. [Google Scholar] [CrossRef] [Green Version]
- Augustin, I.; Rosenmund, C.; Südhof, T.C.; Brose, N. Munc13-1 is essential for fusion competence of glutamatergic synaptic vesicles. Nature 1999, 400, 457–461. [Google Scholar] [CrossRef]
- Lai, Y.; Choi, U.B.; Leitz, J.; Rhee, H.J.; Lee, C.; Altas, B.; Zhao, M.; Pfuetzner, R.A.; Wang, A.L.; Brose, N.; et al. Molecular Mechanisms of Synaptic Vesicle Priming by Munc13 and Munc18. Neuron 2017, 95, 591–607. [Google Scholar] [CrossRef]
- Butola, T.; Wichmann, C.; Moser, T. Piccolo Promotes Vesicle Replenishment at a Fast Central Auditory Synapse. Front. Synaptic. Neurosci. 2017, 9, 14. [Google Scholar] [CrossRef] [Green Version]
- de Jong, A.P.H.; Roggero, C.M.; Ho, M.-R.; Wong, M.Y.; Brautigam, C.A.; Rizo, J.; Kaeser, P.S. RIM C2B Domains Target Presynaptic Active Zone Functions to PIP2-Containing Membranes. Neuron 2018, 98, 335–349. [Google Scholar] [CrossRef] [Green Version]
- He, E.; Wierda, K.; van Westen, R.; Broeke, J.H.; Toonen, R.F.; Cornelisse, L.N.; Verhage, M. Munc13-1 and Munc18-1 together prevent NSF-dependent de-priming of synaptic vesicles. Nat. Commun. 2017, 8, 15915. [Google Scholar] [CrossRef]
- Liu, X.; Seven, A.B.; Camacho, M.; Esser, V.; Xu, J.; Trimbuch, T.; Quade, B.; Su, L.; Ma, C.; Rosenmund, C.; et al. Functional synergy between the Munc13 C-terminal C1 and C2 domains. eLife 2016, 5, e13696. [Google Scholar] [CrossRef] [PubMed]
- Davydova, D.; Marini, C.; King, C.; Klueva, J.; Bischof, F.; Romorini, S.; Montenegro-Venegas, C.; Heine, M.; Schneider, R.; Schröder, M.S.; et al. Bassoon specifically controls presynaptic P/Q-type Ca(2+) channels via RIM-binding protein. Neuron 2014, 82, 181–194. [Google Scholar] [CrossRef] [Green Version]
- Sakaba, T.; Neher, E. Calmodulin mediates rapid recruitment of fast-releasing synaptic vesicles at a calyx-type synapse. Neuron 2001, 32, 1119–1131. [Google Scholar] [CrossRef] [Green Version]
- Dulubova, I.; Lou, X.; Lu, J.; Huryeva, I.; Alam, A.; Schneggenburger, R.; Südhof, T.C.; Rizo, J. A Munc13/RIM/Rab3 tripartite complex: From priming to plasticity? EMBO J. 2005, 24, 2839–2850. [Google Scholar] [CrossRef]
- Held, R.G.; Liu, C.; Kaeser, P.S. ELKS controls the pool of readily releasable vesicles at excitatory synapses through its N-terminal coiled-coil domains. eLife 2016, 5, e14862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, O.-H.; Lu, J.; Rhee, J.-S.; Tomchick, D.R.; Pang, Z.P.; Wojcik, S.M.; Camacho-Perez, M.; Brose, N.; Machius, M.; Rizo, J.; et al. Munc13 C2B domain is an activity-dependent Ca2+ regulator of synaptic exocytosis. Nat. Struct. Mol. Biol. 2010, 17, 280–288. [Google Scholar] [CrossRef]
- Shen, J.; Tareste, D.C.; Paumet, F.; Rothman, J.E.; Melia, T.J. Selective activation of cognate SNAREpins by Sec1/Munc18 proteins. Cell 2007, 128, 183–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhage, M.; Maia, A.S.; Plomp, J.J.; Brussaard, A.B.; Heeroma, J.H.; Vermeer, H.; Toonen, R.F.; Hammer, R.E.; van den Berg, T.K.; Missler, M.; et al. Synaptic Assembly of the Brain in the Absence of Neurotransmitter Secretion. Science 2000, 287, 864–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Camacho, M.; Xu, Y.; Esser, V.; Liu, X.; Trimbuch, T.; Pan, Y.Z.; Ma, C.; Tomchick, D.R.; Rosenmund, C.; et al. Mechanistic insights into neurotransmitter release and presynaptic plasticity from the crystal structure of Munc13-1 C(1)C(2)BMUN. eLife 2017, 6, e22567. [Google Scholar] [CrossRef]
- Yang, X.; Wang, S.; Sheng, Y.; Zhang, M.; Zou, W.; Wu, L.; Kang, S.L.; Rizo, J.; Zhang, R.; Xu, T.; et al. Syntaxin opening by the MUN domain underlies the function of Munc13 in synaptic-vesicle priming. Nat. Struct. Mol. Biol. 2015, 22, 547–554. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Li, Y.; Gong, J.; Ye, S.; Yang, X.; Zhang, R.; Ma, C. Munc18 and Munc13 serve as a functional template to orchestrate neuronal SNARE complex assembly. Nat. Commun. 2019, 10, 69. [Google Scholar] [CrossRef] [Green Version]
- Shu, T.; Jin, H.; Rothman, J.E.; Zhang, Y. Munc13-1 MUN domain and Munc18-1 cooperatively chaperone SNARE assembly through a tetrameric complex. Proc. Natl. Acad. Sci. USA 2020, 117, 1036–1041. [Google Scholar] [CrossRef] [PubMed]
- Zucker, R.S.; Regehr, W.G. Short-term synaptic plasticity. Annu. Rev. Physiol. 2002, 64, 355–405. [Google Scholar] [CrossRef] [Green Version]
- Kaeser, P.S.; Südhof, T.C. RIM function in short- and long-term synaptic plasticity. Biochem. Soc. Trans. 2005, 33 Pt 6, 1345–1349. [Google Scholar] [CrossRef]
- Kintscher, M.; Wozny, C.; Johenning, F.W.; Schmitz, D.; Breustedt, J. Role of RIM1α in short- and long-term synaptic plasticity at cerebellar parallel fibres. Nat. Commun. 2013, 4, 2392. [Google Scholar] [CrossRef]
- Nakajima, Y.; Mochida, S.; Okawa, K.; Nakanishi, S. Ca2+-dependent release of Munc18-1 from presynaptic mGluRs in short-term facilitation. Proc. Natl. Acad. Sci. USA 2009, 106, 18385–18389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhee, J.S.; Betz, A.; Pyott, S.; Reim, K.; Varoqueaux, F.; Augustin, I.; Hesse, D.; Südhof, T.C.; Takahashi, M.; Rosenmund, C.; et al. Beta phorbol ester- and diacylglycerol-induced augmentation of transmitter release is mediated by Munc13s and not by PKCs. Cell 2002, 108, 121–133. [Google Scholar] [CrossRef] [Green Version]
- Vandael, D.; Borges-Merjane, C.; Zhang, X.; Jonas, P. Short-Term Plasticity at Hippocampal Mossy Fiber Synapses Is Induced by Natural Activity Patterns and Associated with Vesicle Pool Engram Formation. Neuron 2020, 107, 509–521. [Google Scholar] [CrossRef]
- Wang, C.C.; Weyrer, C.; Fioravante, D.; Kaeser, P.S.; Regehr, W.G. Presynaptic Short-Term Plasticity Persists in the Absence of PKC Phosphorylation of Munc18-1. J. Neurosci. 2021, 41, 7329–7339. [Google Scholar] [PubMed]
- Lu, W.B.; Ma, H.; Sheng, Z.H.; Mochida, S. Dynamin and Activity Regulate Synaptic Vesicle Recycling in Sympathetic Neurons. J. Biol. Chem. 2009, 284, 1930–1937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, M.; Tanifuji, S.; Mochida, S. Kinetic organization of Ca2+ signals that regulate synaptic release efficacy in sympathetic neurons. Mol. Pharm. 2014, 86, 297–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, E.; Mochida, S.; Takagi, H.; Higa, S.; Deguchi-Tawarada, M.; Takao-Rikitsu, E.; Inoue, M.; Yao, I.; Takeuchi, K.; Kitajima, I.; et al. SAD: A presynaptic kinase associated with synaptic vesicles and the active zone cytomatrix that regulates neurotransmitter release. Neuron 2006, 50, 261–275. [Google Scholar] [CrossRef] [Green Version]
- Pozo, K.; Goda, Y. Unraveling mechanisms of homeostatic synaptic plasticity. Neuron 2010, 66, 337–351. [Google Scholar] [CrossRef] [Green Version]
- Davis, G.W.; Müller, M. Homeostatic control of presynaptic neurotransmitter release. Annu. Rev. Physiol. 2015, 77, 251–270. [Google Scholar] [CrossRef] [PubMed]
- Böhme, M.A.; McCarthy, A.W.; Grasskamp, A.T.; Beuschel, C.B.; Goel, P.; Jusyte, M.; Laber, D.; Huang, S.; Rey, U.; Petzoldt, A.G.; et al. Rapid active zone remodeling consolidates presynaptic potentiation. Nat. Commun. 2019, 10, 1085. [Google Scholar] [CrossRef] [Green Version]
- Cull-Candy, S.G.; Miledi, R.; Trautmann, A.; Uchitel, O.D. On the release of transmitter at normal, myasthenia gravis and myasthenic syndrome affected human end-plates. J. Physiol. 1980, 299, 621–638. [Google Scholar] [CrossRef] [PubMed]
- Goel, P.; Dickman, D. Synaptic homeostats: Latent plasticity revealed at the Drosophila neuromuscular junction. Cell Mol. Life Sci. 2021, 78, 3159–3179. [Google Scholar] [CrossRef] [PubMed]
- Younger, M.A.; Müller, M.; Tong, A.; Pym, E.C.; Davis, G.W. A presynaptic ENaC channel drives homeostatic plasticity. Neuron 2013, 79, 1183–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mochida, S. Millisecond Ca2+ dynamics activate multiple protein cascades for synaptic vesicle control. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2017, 93, 802–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mochida, S. Neurotransmitter Release Site Replenishment and Presynaptic Plasticity. Int. J. Mol. Sci. 2020, 22, 327. [Google Scholar] [CrossRef] [PubMed]
- Xuan, Z.; Manning, L.; Nelson, J.; Richmond, J.E.; Colón-Ramos, D.A.; Shen, K.; Kurshan, P.T. Clarinet (CLA-1), a novel active zone protein required for synaptic vesicle clustering and release. eLife 2017, 6, e29276. [Google Scholar] [CrossRef] [PubMed]
- Katz, B.; Miledi, R. The role of calcium in neuromuscular facilitation. J. Physiol. 1968, 195, 481–492. [Google Scholar] [CrossRef]
| 1 | Fusion machinery interaction |
| 2 | SV replenishment |
| 3 | Presynaptic short-term plasticity |
| 4 | Presynaptic homeostatic plasticity |

{kind=link}
{kind=link}
| Function | Protein | References | |
|---|---|---|---|
| AZ assembly | CaV channel recruitment | RIM, RIM-BP, CAST/ELKS | [35,37,38,39,42,43] |
| liquid droplet formation | RIM, RIM-BP, ELKS | [23,48] | |
| stabilization and degradation | Bassoon, Piccolo | [51,52] | |
| Fusion machinery interaction | fusion machinery regulation | Munc13, Munc18 | [55,56,57,58,59,60] |
| SV states | tethering | Bassoon, Piccolo | [46,52,61] |
| docking | RIM, CAST/ELKS | [3,10,62] | |
| priming | ELKS, RIM, RIM-BP, Munc13 | [36,55,62,63,64,65] | |
| super priming | Mover | [22] | |
| fusion | Munc13, Munc18 | [55,56,57,58,59,60] | |
| SV replenishment | facilitation | Bassoon, Piccolo | [46,52,61] |
| inhibition | CAST phosphorylation | [10] | |
| Presynaptic short-term plasticity | facilitation | RIM, RIM-BP | [39,66,67] |
| facilitation | Munc13, Munc18 | [9,12,19,68] | |
| post-tetanic potentiation (PTP) | Munc18 | [12,13] | |
| control of depression | Bassoon, Piccolo | [46,52] | |
| depression | CAST phosphorylation | [10] | |
| Presynaptic homeostatic plasticity | RRP enlargement | RIM | [15] |
| promotion of SV priming | RIM-BP | [69] | |
| promotion of SV replenishment | Bassoon | [16] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mochida, S. Stable and Flexible Synaptic Transmission Controlled by the Active Zone Protein Interactions. Int. J. Mol. Sci. 2021, 22, 11775. https://doi.org/10.3390/ijms222111775
Mochida S. Stable and Flexible Synaptic Transmission Controlled by the Active Zone Protein Interactions. International Journal of Molecular Sciences. 2021; 22(21):11775. https://doi.org/10.3390/ijms222111775
Chicago/Turabian StyleMochida, Sumiko. 2021. "Stable and Flexible Synaptic Transmission Controlled by the Active Zone Protein Interactions" International Journal of Molecular Sciences 22, no. 21: 11775. https://doi.org/10.3390/ijms222111775
APA StyleMochida, S. (2021). Stable and Flexible Synaptic Transmission Controlled by the Active Zone Protein Interactions. International Journal of Molecular Sciences, 22(21), 11775. https://doi.org/10.3390/ijms222111775