
Brainstem and Cortical Spreading Depolarization in a Closed Head Injury Rat Model

 , ,
, ,
Abstract
:1. Introduction
2. Results
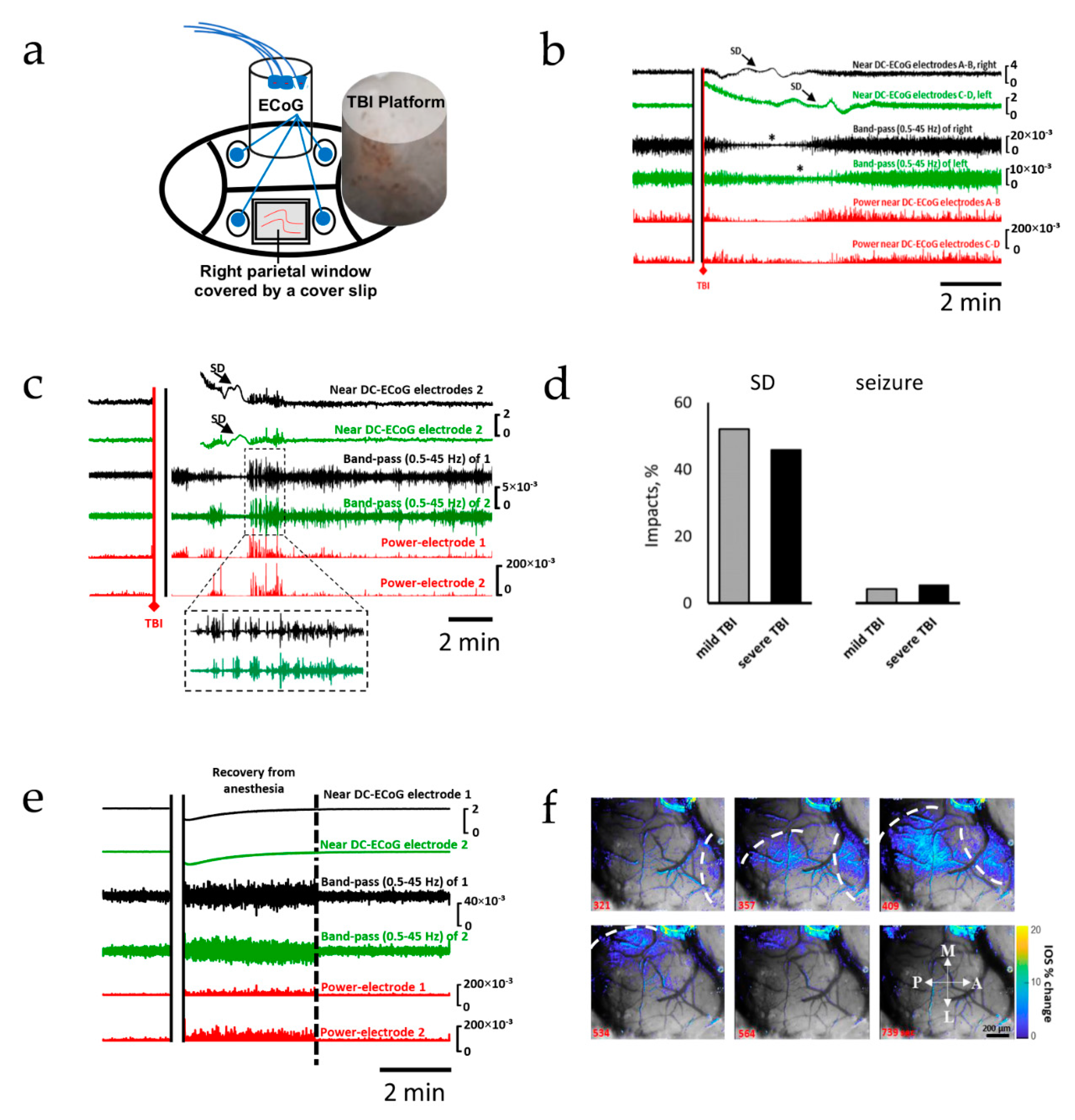
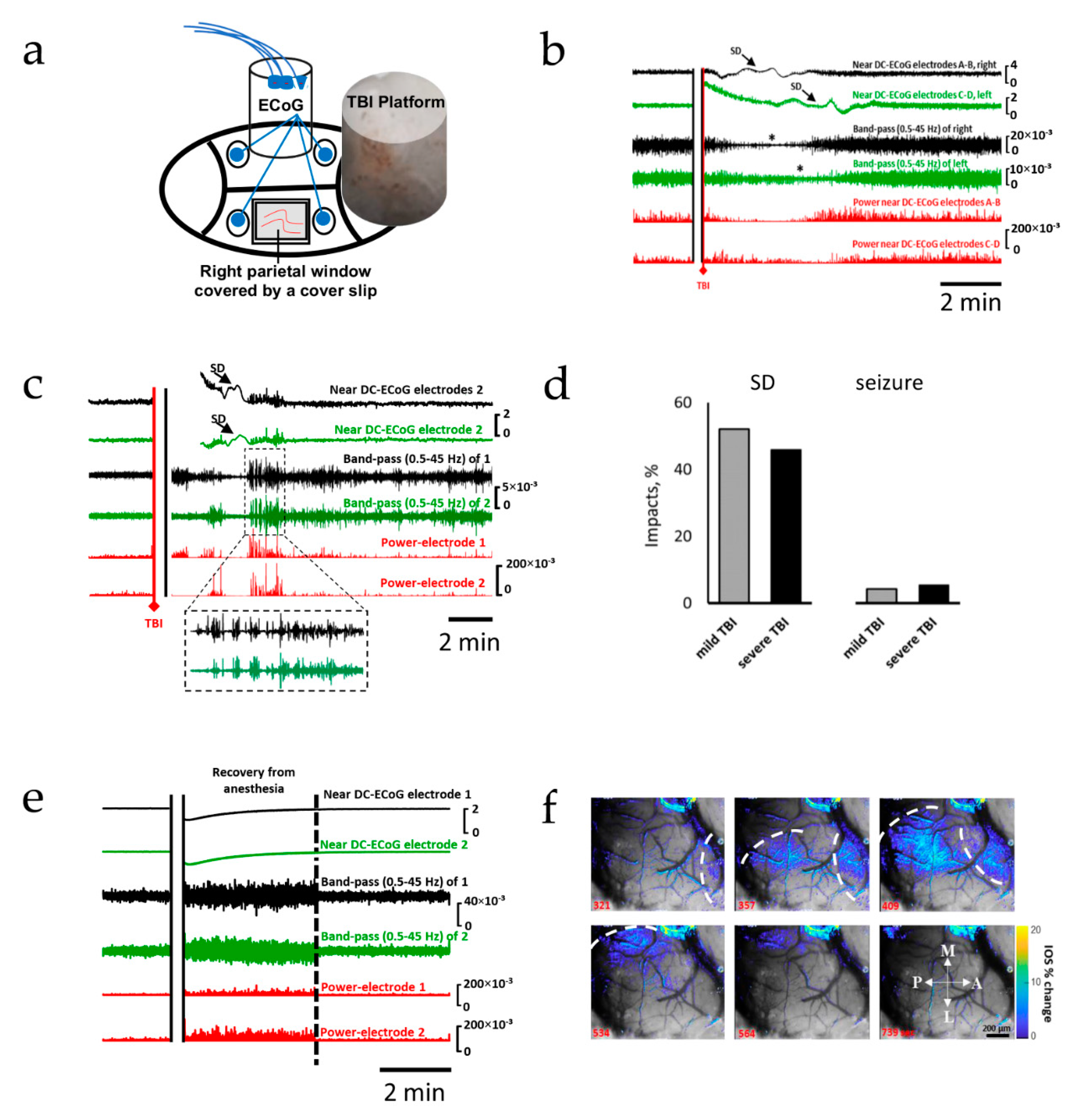
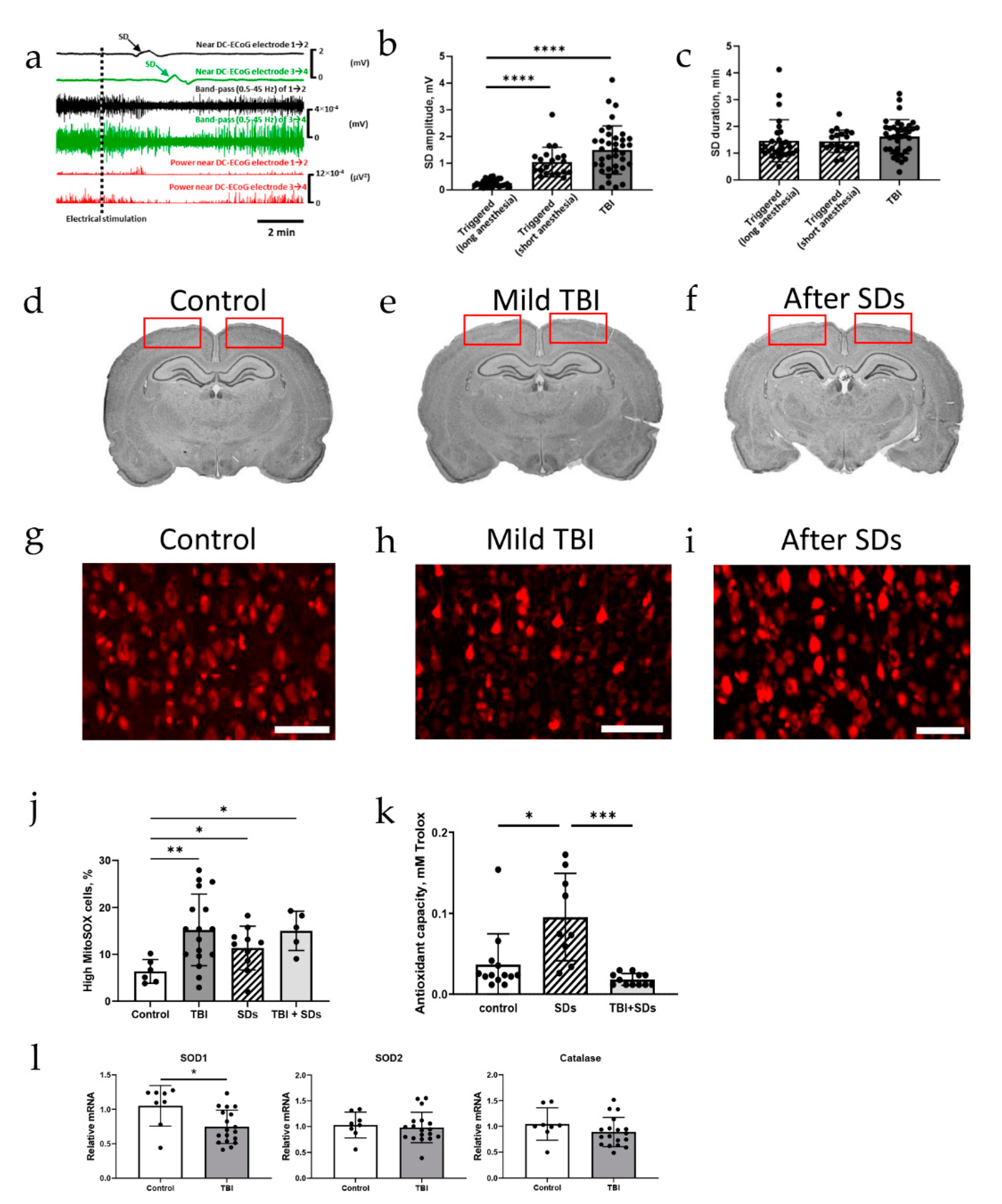
2.1. Spreading Depolarization Is Common Following Mild and Severe TBI
2.2. Oxidative Stress Defense Is Compromised by TBI
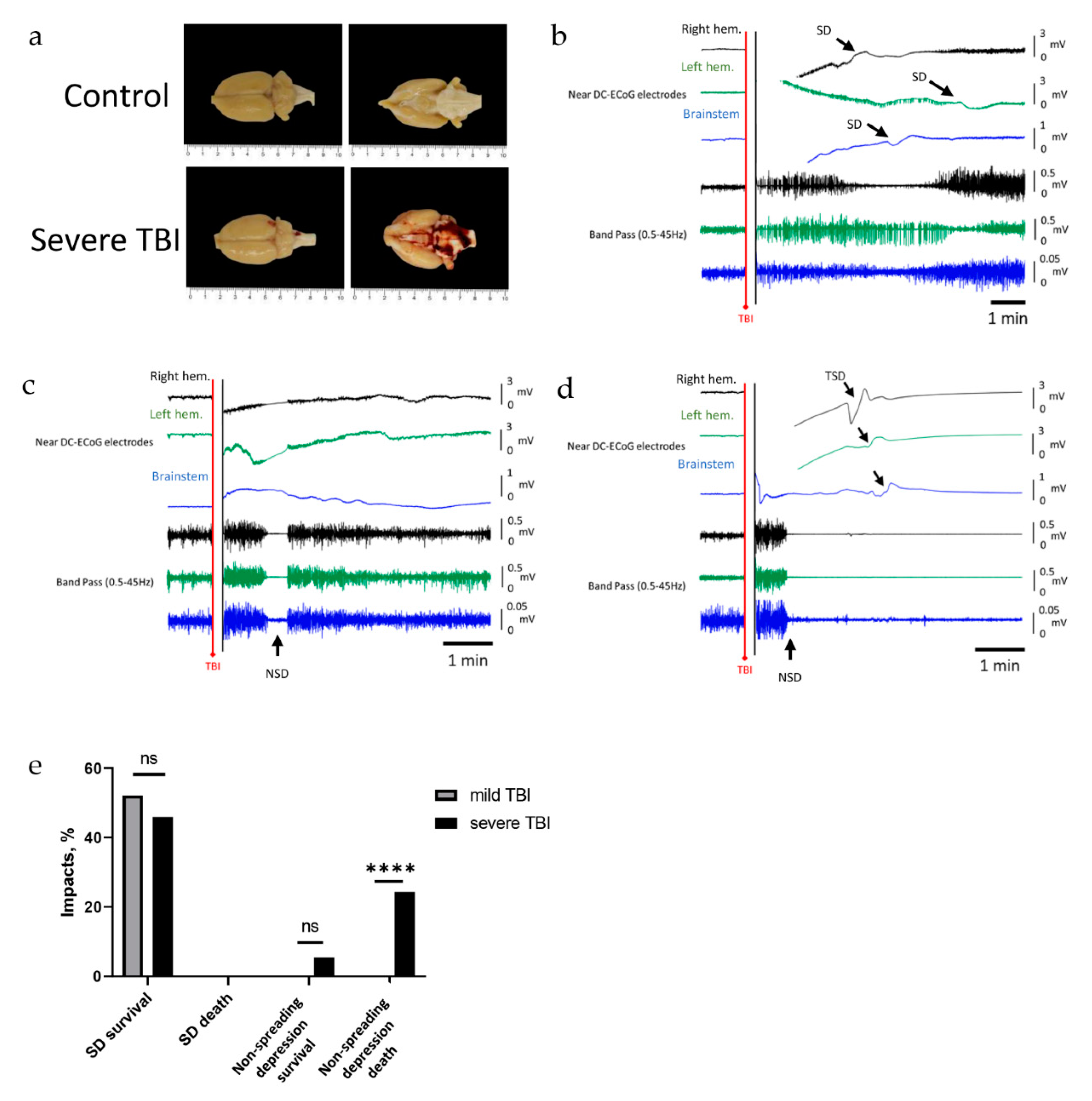
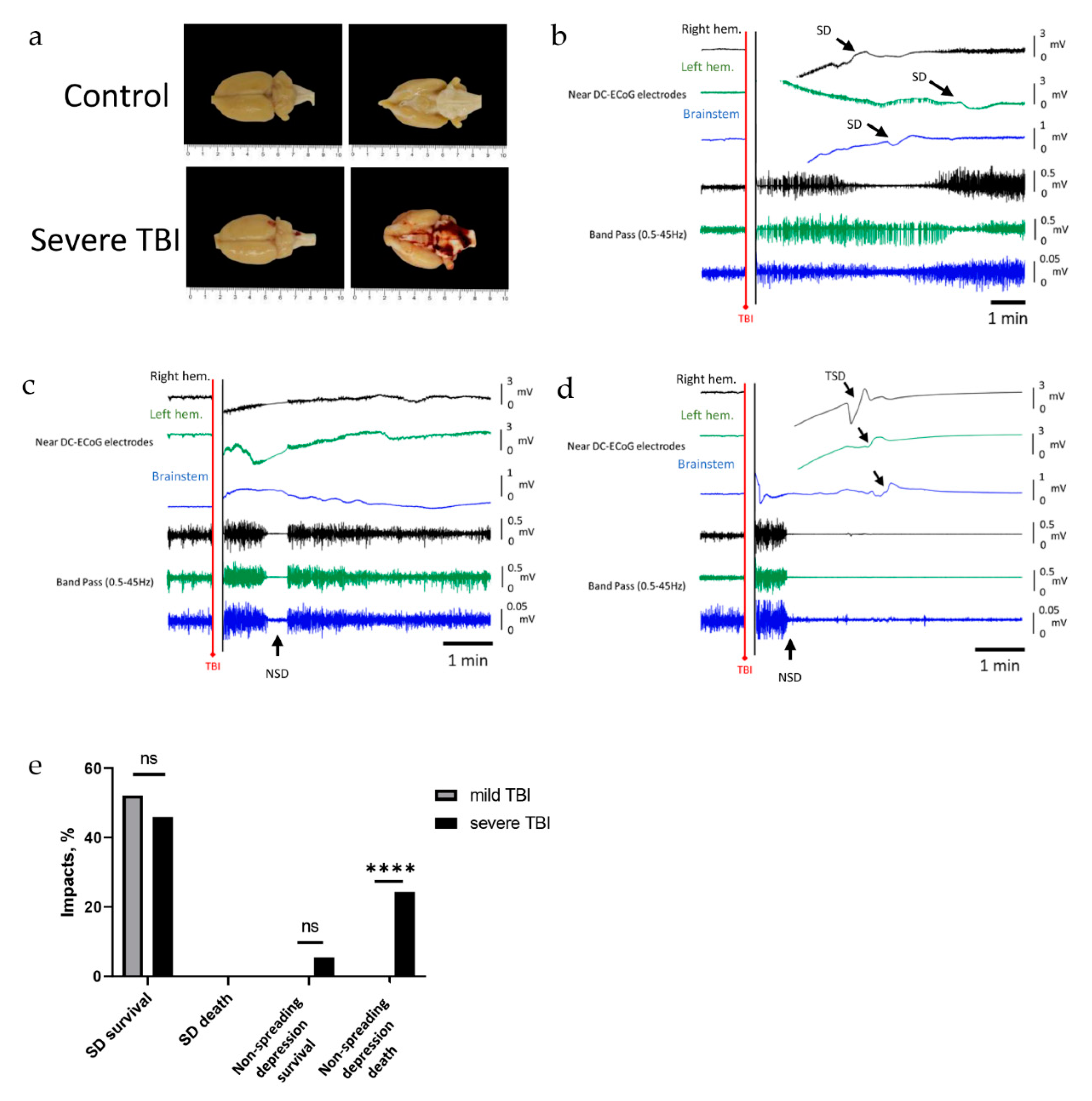
2.3. Severe TBI-Induced Immediate Death Is Associated with the Typical Electrocorticographic Signature of NSD in Both Cortical Hemispheres and Brainstem, Followed by Terminal SD
3. Discussion
4. Materials and Methods
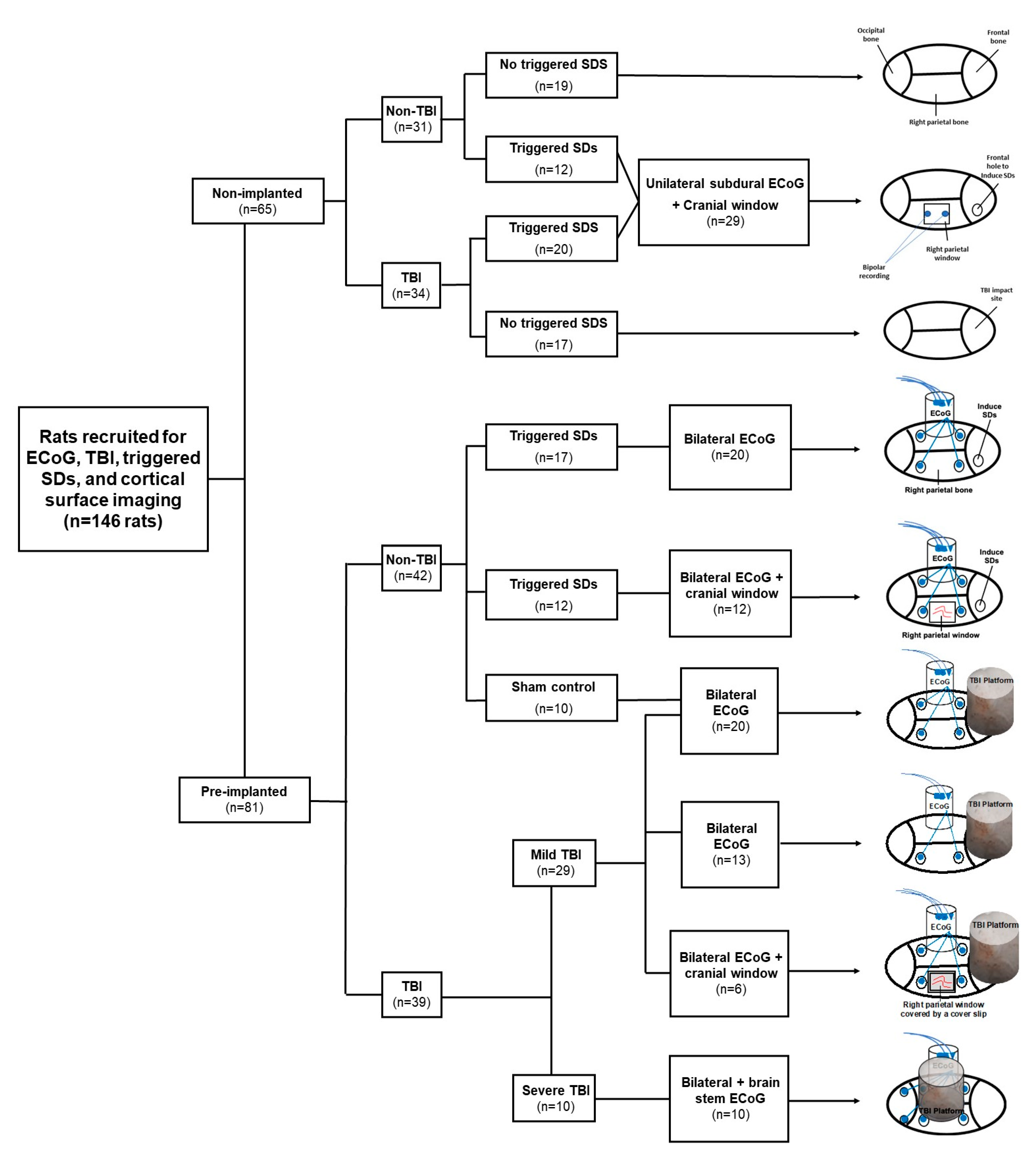
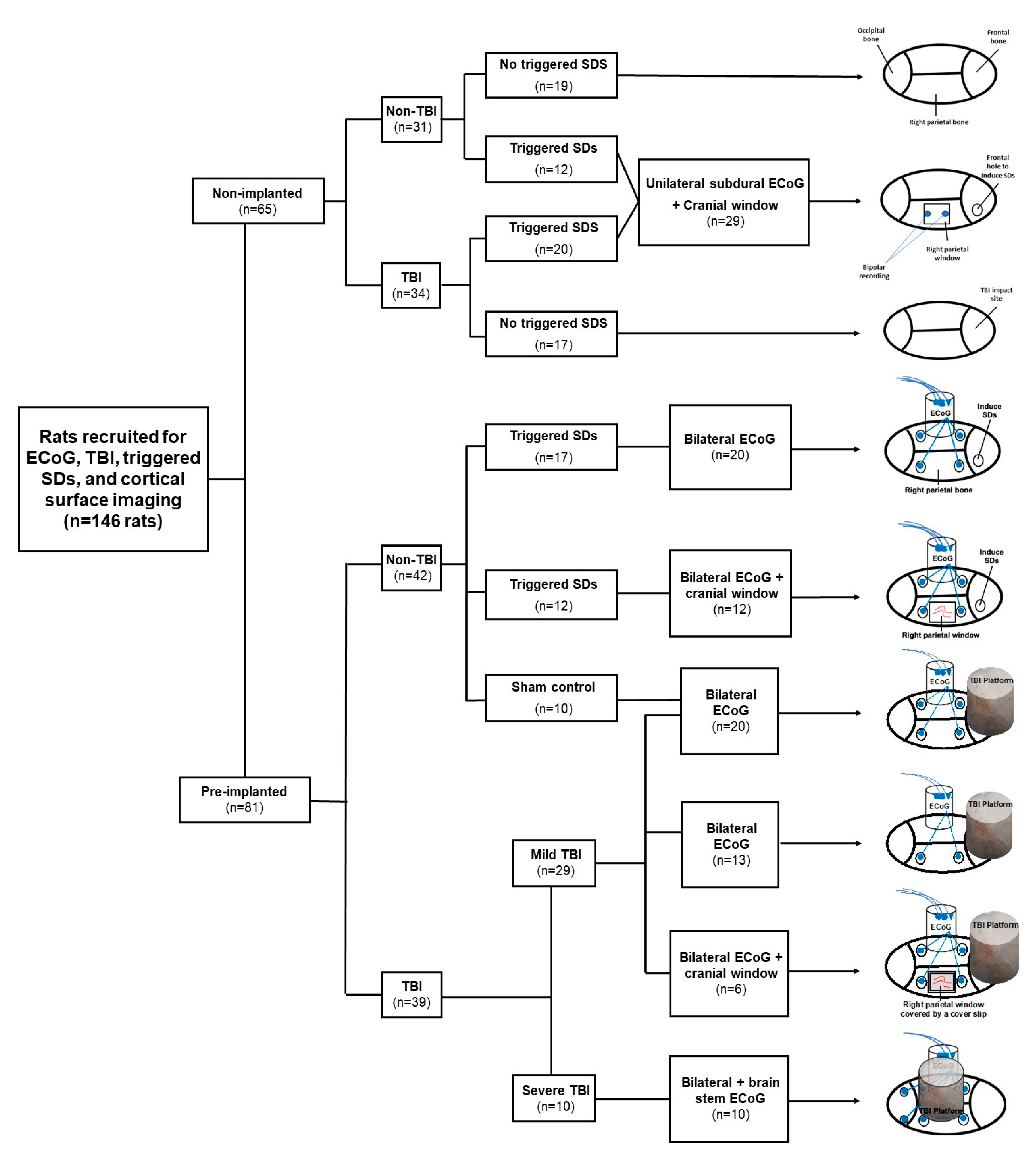
4.1. Animals
4.2. Recording of Cortical Activity
4.3. Traumatic Brain Injury
4.4. Electrocorticographic Recordings
4.5. Electrical Induction of Spreading Depolarizations
4.6. Cortical Surface Imaging
4.7. Histochemistry
4.8. Antioxidant Assay
4.9. Real-Time Quantitative PCR
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Laker, S.R. Epidemiology of Concussion and Mild Traumatic Brain Injury. PM&R 2011, 3, S354–S358. [Google Scholar] [CrossRef]
- Gardner, R.C.; Yaffe, K. Epidemiology of Mild Traumatic Brain Injury and Neurodegenerative Disease. Mol. Cell. Neurosci. 2015, 66, 75–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maas, A.I.R.; Menon, D.K.; Adelson, P.D.; Andelic, N.; Bell, M.J.; Belli, A.; Bragge, P.; Brazinova, A.; Büki, A.; Chesnut, R.M.; et al. Traumatic Brain Injury: Integrated Approaches to Improve Prevention, Clinical Care, and Research. Lancet Neurol. 2017, 16, 987–1048. [Google Scholar] [CrossRef] [Green Version]
- Sharp, D.J.; Jenkins, P.O. Concussion Is Confusing Us All. Pract. Neurol. 2015, 15, 172–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, N.A. The Neurophysiology of Concussion. Prog. Neurobiol. 2002, 67, 281–344. [Google Scholar] [CrossRef]
- Maas, A.I.; Stocchetti, N.; Bullock, R. Moderate and Severe Traumatic Brain Injury in Adults. Lancet Neurol. 2008, 7, 728–741. [Google Scholar] [CrossRef]
- Van Eijck, M.M.; Schoonman, G.G.; van der Naalt, J.; de Vries, J.; Roks, G. Diffuse Axonal Injury after Traumatic Brain Injury Is a Prognostic Factor for Functional Outcome: A Systematic Review and Meta-Analysis. Brain Inj. 2018, 32, 395–402. [Google Scholar] [CrossRef]
- Patel, H.; Bouamra, O.; Woodford, M.; King, A.; Yates, D.; Lecky, F. Trends in Head Injury Outcome from 1989 to 2003 and the Effect of Neurosurgical Care: An Observational Study. Lancet 2005, 366, 1538–1544. [Google Scholar] [CrossRef]
- Schoenhuber, R.; Gentilini, M. Anxiety and Depression after Mild Head Injury: A Case Control Study. J. Neurol. Neurosurg. Psychiatry 1988, 51, 722–724. [Google Scholar] [CrossRef] [Green Version]
- Christensen, J. Traumatic Brain Injury: Risks of Epilepsy and Implications for Medicolegal Assessment. Epilepsia 2012, 53 (Suppl. 4), 43–47. [Google Scholar] [CrossRef]
- Shlosberg, D.; Benifla, M.; Kaufer, D.; Friedman, A. Blood-Brain Barrier Breakdown as a Therapeutic Target in Traumatic Brain Injury. Nat. Rev. Neurol. 2010, 6, 393–403. [Google Scholar] [CrossRef] [Green Version]
- Stein, T.D.; Alvarez, V.E.; McKee, A.C. Concussion in Chronic Traumatic Encephalopathy. Curr. Pain Headache Rep. 2015, 19, 47. [Google Scholar] [CrossRef] [Green Version]
- Jafari, S.; Etminan, M.; Aminzadeh, F.; Samii, A. Head Injury and Risk of Parkinson Disease: A Systematic Review and Meta-Analysis. Mov. Disord. Off. J. Mov. Disord. Soc. 2013, 28, 1222–1229. [Google Scholar] [CrossRef] [PubMed]
- Mortimer, J.A.; van Duijn, C.M.; Chandra, V.; Fratiglioni, L.; Graves, A.B.; Heyman, A.; Jorm, A.F.; Kokmen, E.; Kondo, K.; Rocca, W.A.; et al. Head Trauma as a Risk Factor for Alzheimer’s Disease: A Collaborative Re-Analysis of Case-Control Studies. EURODEM Risk Factors Research Group. Int. J. Epidemiol. 1991, 20 (Suppl. 2), S28–S35. [Google Scholar] [CrossRef] [Green Version]
- Blyth, B.J.; Bazarian, J.J. Traumatic Alterations in Consciousness: Traumatic Brain Injury. Emerg. Med. Clin. N. Am. 2010, 28, 571–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verellen, R.M.; Cavazos, J.E. Post-Traumatic Epilepsy: An Overview. Ther. Lond. Engl. 2010, 7, 527–531. [Google Scholar] [CrossRef] [Green Version]
- Waziri, A.; Claassen, J.; Stuart, R.M.; Arif, H.; Schmidt, J.M.; Mayer, S.A.; Badjatia, N.; Kull, L.L.; Connolly, E.S.; Emerson, R.G.; et al. Intracortical Electroencephalography in Acute Brain Injury. Ann. Neurol. 2009, 66, 366–377. [Google Scholar] [CrossRef]
- Von Baumgarten, L.; Trabold, R.; Thal, S.; Back, T.; Plesnila, N. Role of Cortical Spreading Depressions for Secondary Brain Damage after Traumatic Brain Injury in Mice. J. Cereb. Blood Flow Metab. 2008, 28, 1353–1360. [Google Scholar] [CrossRef] [Green Version]
- Hartings, J.A.; Andaluz, N.; Bullock, M.R.; Hinzman, J.M.; Mathern, B.; Pahl, C.; Puccio, A.; Shutter, L.A.; Strong, A.J.; Vagal, A.; et al. Prognostic Value of Spreading Depolarizations in Patients With Severe Traumatic Brain Injury. JAMA Neurol. 2020, 77, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Fabricius, M.; Fuhr, S.; Bhatia, R.; Boutelle, M.; Hashemi, P.; Strong, A.J.; Lauritzen, M. Cortical Spreading Depression and Peri-Infarct Depolarization in Acutely Injured Human Cerebral Cortex. Brain J. Neurol. 2006, 129, 778–790. [Google Scholar] [CrossRef] [Green Version]
- Dreier, J.P.; Isele, T.; Reiffurth, C.; Offenhauser, N.; Kirov, S.A.; Dahlem, M.A.; Herreras, O. Is Spreading Depolarization Characterized by an Abrupt, Massive Release of Gibbs Free Energy from the Human Brain Cortex? Neuroscientist 2012, 19, 25–42. [Google Scholar] [CrossRef] [Green Version]
- Dreier, J.P.; Lemale, C.L.; Kola, V.; Friedman, A.; Schoknecht, K. Spreading Depolarization Is Not an Epiphenomenon but the Principal Mechanism of the Cytotoxic Edema in Various Gray Matter Structures of the Brain during Stroke. Cereb. Ischemia 2018, 134, 189–207. [Google Scholar] [CrossRef]
- Kirov, S.A.; Fomitcheva, I.V.; Sword, J. Rapid Neuronal Ultrastructure Disruption and Recovery during Spreading Depolarization-Induced Cytotoxic Edema. Cereb. Cortex 2020, 30, 5517–5531. [Google Scholar] [CrossRef]
- Kager, H.; Wadman, W.J.; Somjen, G.G. Conditions for the Triggering of Spreading Depression Studied with Computer Simulations. J. Neurophysiol. 2002, 88, 2700–2712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dreier, J.P.; Fabricius, M.; Ayata, C.; Sakowitz, O.W.; Shuttleworth, C.W.; Dohmen, C.; Graf, R.; Vajkoczy, P.; Helbok, R.; Suzuki, M.; et al. Recording, Analysis, and Interpretation of Spreading Depolarizations in Neurointensive Care: Review and Recommendations of the COSBID Research Group. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2017, 37, 1595–1625. [Google Scholar] [CrossRef]
- Dreier, J.P.; Major, S.; Foreman, B.; Winkler, M.K.L.; Kang, E.-J.; Milakara, D.; Lemale, C.L.; DiNapoli, V.; Hinzman, J.M.; Woitzik, J.; et al. Terminal Spreading Depolarization and Electrical Silence in Death of Human Cerebral Cortex. Ann. Neurol. 2018, 83, 295–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dreier, J.P. The Role of Spreading Depression, Spreading Depolarization and Spreading Ischemia in Neurological Disease. Nat. Med. 2011, 17, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Hartings, J.A.; Strong, A.J.; Fabricius, M.; Manning, A.; Bhatia, R.; Dreier, J.P.; Mazzeo, A.T.; Tortella, F.C.; Bullock, M.R. Co-Operative Study of Brain Injury Depolarizations Spreading Depolarizations and Late Secondary Insults after Traumatic Brain Injury. J. Neurotrauma 2009, 26, 1857–1866. [Google Scholar] [CrossRef]
- Fabricius, M.; Fuhr, S.; Willumsen, L.; Dreier, J.P.; Bhatia, R.; Boutelle, M.G.; Hartings, J.A.; Bullock, R.; Strong, A.J.; Lauritzen, M. Association of Seizures with Cortical Spreading Depression and Peri-Infarct Depolarisations in the Acutely Injured Human Brain. Clin. Neurophysiol. Off. J. Int. Fed. Clin. Neurophysiol. 2008, 119, 1973–1984. [Google Scholar] [CrossRef] [Green Version]
- Dreier, J.P.; Major, S.; Pannek, H.-W.; Woitzik, J.; Scheel, M.; Wiesenthal, D.; Martus, P.; Winkler, M.K.L.; Hartings, J.A.; Fabricius, M.; et al. Spreading Convulsions, Spreading Depolarization and Epileptogenesis in Human Cerebral Cortex. Brain J. Neurol. 2012, 135, 259–275. [Google Scholar] [CrossRef]
- Revankar, G.S.; Winkler, M.K.L.; Major, S.; Schoknecht, K.; Heinemann, U.; Woitzik, J.; Claassen, J.; Hartings, J.A.; Dreier, J.P. Spreading Depolarizations and Seizures in Clinical Subdural Electrocorticographic Recordings. In Seizures in Critical Care: A Guide to Diagnosis and Therapeutics; Varelas, P.N., Claassen, J., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 77–90. ISBN 978-3-319-49557-6. [Google Scholar]
- Woitzik, J.; Hecht, N.; Pinczolits, A.; Sandow, N.; Major, S.; Winkler, M.K.L.; Weber-Carstens, S.; Dohmen, C.; Graf, R.; Strong, A.J.; et al. Propagation of Cortical Spreading Depolarization in the Human Cortex after Malignant Stroke. Neurology 2013, 80, 1095–1102. [Google Scholar] [CrossRef] [PubMed]
- Dohmen, C.; Sakowitz, O.W.; Fabricius, M.; Bosche, B.; Reithmeier, T.; Ernestus, R.-I.; Brinker, G.; Dreier, J.P.; Woitzik, J.; Strong, A.J.; et al. Spreading Depolarizations Occur in Human Ischemic Stroke with High Incidence. Ann. Neurol. 2008, 63, 720–728. [Google Scholar] [CrossRef] [PubMed]
- Hartings, J.A.; Bullock, M.R.; Okonkwo, D.O.; Murray, L.S.; Murray, G.D.; Fabricius, M.; Maas, A.I.; Woitzik, J.; Sakowitz, O.; Mathern, B.; et al. Spreading Depolarisations and Outcome after Traumatic Brain Injury: A Prospective Observational Study. Lancet Neurol. 2011, 10, 1058–1064. [Google Scholar] [CrossRef]
- Hinzman, J.M.; Andaluz, N.; Shutter, L.A.; Okonkwo, D.O.; Pahl, C.; Strong, A.J.; Dreier, J.P.; Hartings, J.A. Inverse Neurovascular Coupling to Cortical Spreading Depolarizations in Severe Brain Trauma. Brain J. Neurol. 2014, 137, 2960–2972. [Google Scholar] [CrossRef] [Green Version]
- Dreier, J.P.; Major, S.; Manning, A.; Woitzik, J.; Drenckhahn, C.; Steinbrink, J.; Tolias, C.; Oliveira-Ferreira, A.I.; Fabricius, M.; Hartings, J.A.; et al. Cortical Spreading Ischaemia Is a Novel Process Involved in Ischaemic Damage in Patients with Aneurysmal Subarachnoid Haemorrhage. Brain J. Neurol. 2009, 132, 1866–1881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winkler, M.K.L.; Chassidim, Y.; Lublinsky, S.; Revankar, G.S.; Major, S.; Kang, E.-J.; Oliveira-Ferreira, A.I.; Woitzik, J.; Sandow, N.; Scheel, M.; et al. Impaired Neurovascular Coupling to Ictal Epileptic Activity and Spreading Depolarization in a Patient with Subarachnoid Hemorrhage: Possible Link to Blood-Brain Barrier Dysfunction. Epilepsia 2012, 53 (Suppl. 6), 22–30. [Google Scholar] [CrossRef] [PubMed]
- Somjen, G.G. Mechanisms of Spreading Depression and Hypoxic Spreading Depression-like Depolarization. Physiol. Rev. 2001, 81, 1065–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira-Ferreira, A.I.; Milakara, D.; Alam, M.; Jorks, D.; Major, S.; Hartings, J.A.; Lückl, J.; Martus, P.; Graf, R.; Dohmen, C.; et al. Experimental and Preliminary Clinical Evidence of an Ischemic Zone with Prolonged Negative DC Shifts Surrounded by a Normally Perfused Tissue Belt with Persistent Electrocorticographic Depression. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2010, 30, 1504–1519. [Google Scholar] [CrossRef]
- Richter, F.; Bauer, R.; Lehmenkühler, A.; Schaible, H.-G. The Relationship between Sudden Severe Hypoxia and Ischemia-Associated Spreading Depolarization in Adult Rat Brainstem In Vivo. Exp. Neurol. 2010, 224, 146–154. [Google Scholar] [CrossRef]
- Aiba, I.; Noebels, J.L. Spreading Depolarization in the Brainstem Mediates Sudden Cardiorespiratory Arrest in Mouse SUDEP Models. Sci. Transl. Med. 2015, 7, 282ra46. [Google Scholar] [CrossRef] [Green Version]
- Bouley, J.; Chung, D.Y.; Ayata, C.; Brown, R.H., Jr.; Henninger, N. Cortical Spreading Depression Denotes Concussion Injury. J. Neurotrauma 2019, 36, 1008–1017. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, J.M.; Hines-Lanham, A.; Stratton, C.; Mehos, C.J.; McCurdy, K.E.; Pinkowski, N.J.; Zhang, H.; Shuttleworth, C.W.; Morton, R.A. Spreading Depolarizations Occur in Mild Traumatic Brain Injuries and Are Associated with Postinjury Behavior. eNeuro 2019, 6, ENEURO.0070-19.2019. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, H.; Manaka, S.; Sano, K. Changes in Extracellular Potassium Concentration in Cortex and Brain Stem during the Acute Phase of Experimental Closed Head Injury. J. Neurosurg. 1981, 55, 708–717. [Google Scholar] [CrossRef]
- Dreier, J.P.; Woitzik, J.; Fabricius, M.; Bhatia, R.; Major, S.; Drenckhahn, C.; Lehmann, T.-N.; Sarrafzadeh, A.; Willumsen, L.; Hartings, J.A.; et al. Delayed Ischaemic Neurological Deficits after Subarachnoid Haemorrhage Are Associated with Clusters of Spreading Depolarizations. Brain J. Neurol. 2006, 129, 3224–3237. [Google Scholar] [CrossRef]
- Balança, B.; Meiller, A.; Bezin, L.; Dreier, J.P.; Marinesco, S.; Lieutaud, T. Altered Hypermetabolic Response to Cortical Spreading Depolarizations after Traumatic Brain Injury in Rats. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2017, 37, 1670–1686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petzold, G.C.; Haack, S.; und Halbach, O.v.B.; Priller, J.; Lehmann, T.-N.; Heinemann, U.; Dirnagl, U.; Dreier, J.P. Nitric Oxide Modulates Spreading Depolarization Threshold in the Human and Rodent Cortex. Stroke 2008, 39, 1292–1299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira-Ferreira, A.I.; Major, S.; Przesdzing, I.; Kang, E.-J.; Dreier, J.P. Spreading Depolarizations in the Rat Endothelin-1 Model of Focal Cerebellar Ischemia. J. Cereb. Blood Flow Metab. 2020, 40, 1274–1289. [Google Scholar] [CrossRef]
- Zheng, Z.; Cao, Z.; Luo, J.; Lv, J. Characterization of Intrinsic Optical Signal during Spreading Depolarization. Neuropsychiatry 2018, 8, 302–309. [Google Scholar] [CrossRef] [Green Version]
- Maslarova, A.; Alam, M.; Reiffurth, C.; Lapilover, E.; Gorji, A.; Dreier, J.P. Chronically Epileptic Human and Rat Neocortex Display a Similar Resistance Against Spreading Depolarization In Vitro. Stroke 2011, 42, 2917–2922. [Google Scholar] [CrossRef] [Green Version]
- Mané, M.; Müller, M. Temporo-Spectral Imaging of Intrinsic Optical Signals during Hypoxia-Induced Spreading Depression-like Depolarization. PLoS ONE 2012, 7, e43981. [Google Scholar] [CrossRef] [Green Version]
- Lauritzen, M.; Hansen, A.J. The Effect of Glutamate Receptor Blockade on Anoxic Depolarization and Cortical Spreading Depression. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 1992, 12, 223–229. [Google Scholar] [CrossRef] [Green Version]
- Reiffurth, C.; Alam, M.; Zahedi-Khorasani, M.; Major, S.; Dreier, J.P. Na+/K+-ATPase α Isoform Deficiency Results in Distinct Spreading Depolarization Phenotypes. J. Cereb. Blood Flow Metab. 2019, 40, 622–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khatri, N.; Thakur, M.; Pareek, V.; Kumar, S.; Sharma, S.; Datusalia, A.K. Oxidative Stress: Major Threat in Traumatic Brain Injury. CNS Neurol. Disord. Drug Targets 2018, 17, 689–695. [Google Scholar] [CrossRef]
- Rodriguez-Rodriguez, A.; Jose Egea-Guerrero, J.; Murillo-Cabezas, F.; Carrillo-Vico, A. Oxidative Stress in Traumatic Brain Injury. Curr. Med. Chem. 2014, 21, 1201–1211. [Google Scholar] [CrossRef] [PubMed]
- Geddes, D.M.; LaPlaca, M.C.; Cargill, R.S., 2nd. Susceptibility of Hippocampal Neurons to Mechanically Induced Injury. Exp. Neurol. 2003, 184, 420–427. [Google Scholar] [CrossRef]
- McDaid, J.; Briggs, C.A.; Barrington, N.M.; Peterson, D.A.; Kozlowski, D.A.; Stutzmann, G.E. Sustained Hippocampal Synaptic Pathophysiology Following Single and Repeated Closed-Head Concussive Impacts. Front. Cell. Neurosci. 2021, 15, 652721. [Google Scholar] [CrossRef]
- Takagaki, M.; Feuerstein, D.; Kumagai, T.; Gramer, M.; Yoshimine, T.; Graf, R. Isoflurane Suppresses Cortical Spreading Depolarizations Compared to Propofol-Implications for Sedation of Neurocritical Care Patients. Exp. Neurol. 2014, 252, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Hartings, J.A.; York, J.; Carroll, C.P.; Hinzman, J.M.; Mahoney, E.; Krueger, B.; Winkler, M.K.L.; Major, S.; Horst, V.; Jahnke, P.; et al. Subarachnoid Blood Acutely Induces Spreading Depolarizations and Early Cortical Infarction. Brain J. Neurol. 2017, 140, 2673–2690. [Google Scholar] [CrossRef]
- Hartings, J.A.; Watanabe, T.; Bullock, M.R.; Okonkwo, D.O.; Fabricius, M.; Woitzik, J.; Dreier, J.P.; Puccio, A.; Shutter, L.A.; Pahl, C.; et al. Spreading Depolarizations Have Prolonged Direct Current Shifts and Are Associated with Poor Outcome in Brain Trauma. Brain 2011, 134, 1529–1540. [Google Scholar] [CrossRef] [Green Version]
- Lückl, J.; Lemale, C.L.; Kola, V.; Horst, V.; Khojasteh, U.; Oliveira-Ferreira, A.I.; Major, S.; Winkler, M.K.L.; Kang, E.-J.; Schoknecht, K.; et al. The Negative Ultraslow Potential, Electrophysiological Correlate of Infarction in the Human Cortex. Brain J. Neurol. 2018, 141, 1734–1752. [Google Scholar] [CrossRef] [Green Version]
- Helbok, R.; Schiefecker, A.J.; Friberg, C.; Beer, R.; Kofler, M.; Rhomberg, P.; Unterberger, I.; Gizewski, E.; Hauerberg, J.; Möller, K.; et al. Spreading Depolarizations in Patients with Spontaneous Intracerebral Hemorrhage: Association with Perihematomal Edema Progression. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2017, 37, 1871–1882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohammad, L.M.; Abbas, M.; Shuttleworth, C.W.; Ahmadian, R.; Bhat, A.; Hill, D.A.; Carlson, A.P. Spreading Depolarization May Represent a Novel Mechanism for Delayed Fluctuating Neurological Deficit after Chronic Subdural Hematoma Evacuation. J. Neurosurg. 2020, 134, 1294–1302. [Google Scholar] [CrossRef]
- Tweedie, D.; Fukui, K.; Li, Y.; Yu, Q.; Barak, S.; Tamargo, I.A.; Rubovitch, V.; Holloway, H.W.; Lehrmann, E.; Wood, W.H., III; et al. Cognitive Impairments Induced by Concussive Mild Traumatic Brain Injury in Mouse Are Ameliorated by Treatment with Phenserine via Multiple Non-Cholinergic and Cholinergic Mechanisms. PLoS ONE 2016, 11, e0156493. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, G.M.; Ma, A.M.; Ko, A.; Harada, M.Y.; Wyss, L.; Haro, P.S.; Vit, J.-P.; Shelest, O.; Rhee, P.; Svendsen, C.N.; et al. A Model of Recurrent Concussion That Leads to Long-Term Motor Deficits, CTE-like Tauopathy and Exacerbation of an ALS Phenotype. J. Trauma Acute Care Surg. 2016, 81, 1070–1079. [Google Scholar] [CrossRef]
- Jia, J.; Chen, F.; Wu, Y. Recombinant PEP-1-SOD1 Improves Functional Recovery after Neural Stem Cell Transplantation in Rats with Traumatic Brain Injury. Exp. Ther. Med. 2018, 15, 2929–2935. [Google Scholar] [CrossRef]
- Jansen, N.A.; Schenke, M.; Voskuyl, R.A.; Thijs, R.D.; van den Maagdenberg, A.M.J.M.; Tolner, E.A. Apnea Associated with Brainstem Seizures in Cacna1a (S218L) Mice Is Caused by Medullary Spreading Depolarization. J. Neurosci. Off. J. Soc. Neurosci. 2019, 39, 9633–9644. [Google Scholar] [CrossRef]
- Loonen, I.C.M.; Jansen, N.A.; Cain, S.M.; Schenke, M.; Voskuyl, R.A.; Yung, A.C.; Bohnet, B.; Kozlowski, P.; Thijs, R.D.; Ferrari, M.D.; et al. Brainstem Spreading Depolarization and Cortical Dynamics during Fatal Seizures in Cacna1a S218L Mice. Brain J. Neurol. 2019, 142, 412–425. [Google Scholar] [CrossRef] [Green Version]
- Mayevsky, A. Responses of NADH to Physiological and Pathophysiological Conditions. In Mitochondrial Function In Vivo Evaluated by NADH Fluorescence; Mayevsky, A., Ed.; Springer International Publishing: Cham, Switzerland, 2015; pp. 111–204. ISBN 978-3-319-16682-7. [Google Scholar]
- Dreier, J.P.; Major, S.; Lemale, C.L.; Kola, V.; Reiffurth, C.; Schoknecht, K.; Hecht, N.; Hartings, J.A.; Woitzik, J. Correlates of Spreading Depolarization, Spreading Depression, and Negative Ultraslow Potential in Epidural versus Subdural Electrocorticography. Front. Neurosci. 2019, 13, 373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, R.; Graf, R.; Hübel, K.; Fujita, T.; Rosner, G.; Heiss, W.-D. Reduction of Infarct Volume by Halothane: Effect on Cerebral Blood Flow or Perifocal Spreading Depression-Like Depolarizations. J. Cereb. Blood Flow Metab. 1997, 17, 857–864. [Google Scholar] [CrossRef] [PubMed]
- Prager, O.; Chassidim, Y.; Klein, C.; Levi, H.; Shelef, I.; Friedman, A. Dynamic in Vivo Imaging of Cerebral Blood Flow and Blood-Brain Barrier Permeability. NeuroImage 2010, 49, 337–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mychasiuk, R.; Farran, A.; Esser, M.J. Assessment of an Experimental Rodent Model of Pediatric Mild Traumatic Brain Injury. J. Neurotrauma 2014, 31, 749–757. [Google Scholar] [CrossRef] [PubMed]
- Bogdanov, V.B.; Middleton, N.A.; Theriot, J.J.; Parker, P.D.; Abdullah, O.M.; Ju, Y.S.; Hartings, J.A.; Brennan, K.C. Susceptibility of Primary Sensory Cortex to Spreading Depolarizations. J. Neurosci. Off. J. Soc. Neurosci. 2016, 36, 4733–4743. [Google Scholar] [CrossRef] [Green Version]
- Valls-Lacalle, L.; Barba, I.; Miró-Casas, E.; Alburquerque-Béjar, J.J.; Ruiz-Meana, M.; Fuertes-Agudo, M.; Rodríguez-Sinovas, A.; García-Dorado, D. Succinate Dehydrogenase Inhibition with Malonate during Reperfusion Reduces Infarct Size by Preventing Mitochondrial Permeability Transition. Cardiovasc. Res. 2016, 109, 374–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, N.J.; Rice-Evans, C.; Davies, M.J.; Gopinathan, V.; Milner, A. A Novel Method for Measuring Antioxidant Capacity and Its Application to Monitoring the Antioxidant Status in Premature Neonates. Clin. Sci. 1993, 84, 407–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tagge, C.A.; Fisher, A.M.; Minaeva, O.V.; Gaudreau-Balderrama, A.; Moncaster, J.A.; Zhang, X.-L.; Wojnarowicz, M.W.; Casey, N.; Lu, H.; Kokiko-Cochran, O.N.; et al. Concussion, Microvascular Injury, and Early Tauopathy in Young Athletes after Impact Head Injury and an Impact Concussion Mouse Model. Brain J. Neurol. 2018, 141, 422–458. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primers |
|---|---|
| SOD 1 | AAGTGCTGTTGAGTCCAGGT CCATTTCCTCCAGGGTGACT |
| SOD2 | ACTGGTTTCTCAGCTCCTC TCAGGAGCCACAAGTGAGAG |
| Catalase | ATGGCTCCAAGCGATGTTTC AAGGGTGCTGAATGCCTACT |
| Step | Temperature (°C; Ramp Rate: 1.6 °C/s) | Time (min) |
|---|---|---|
| 1 | 50 | 2:00 |
| 2 | 95 | 10:00 |
| 3 | 95 | 0:15 |
| 4 | 60 | 1:00 |
| 5 | 95 | 0:15 |
| 6 | 60 | 1:00 |
| 7 | 95 | 0:15 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aboghazleh, R.; Parker, E.; Yang, L.T.; Kaufer, D.; Dreier, J.P.; Friedman, A.; van Hameren, G. Brainstem and Cortical Spreading Depolarization in a Closed Head Injury Rat Model. Int. J. Mol. Sci. 2021, 22, 11642. https://doi.org/10.3390/ijms222111642
Aboghazleh R, Parker E, Yang LT, Kaufer D, Dreier JP, Friedman A, van Hameren G. Brainstem and Cortical Spreading Depolarization in a Closed Head Injury Rat Model. International Journal of Molecular Sciences. 2021; 22(21):11642. https://doi.org/10.3390/ijms222111642
Chicago/Turabian StyleAboghazleh, Refat, Ellen Parker, Lynn T. Yang, Daniela Kaufer, Jens P. Dreier, Alon Friedman, and Gerben van Hameren. 2021. "Brainstem and Cortical Spreading Depolarization in a Closed Head Injury Rat Model" International Journal of Molecular Sciences 22, no. 21: 11642. https://doi.org/10.3390/ijms222111642
APA StyleAboghazleh, R., Parker, E., Yang, L. T., Kaufer, D., Dreier, J. P., Friedman, A., & van Hameren, G. (2021). Brainstem and Cortical Spreading Depolarization in a Closed Head Injury Rat Model. International Journal of Molecular Sciences, 22(21), 11642. https://doi.org/10.3390/ijms222111642