
Poly ADP Ribose Polymerase Inhibitor Olaparib Targeting Microhomology End Joining in Retinoblastoma Protein Defective Cancer: Analysis of the Retinoblastoma Cell-Killing Effects by Olaparib after Inducing Double-Strand Breaks




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
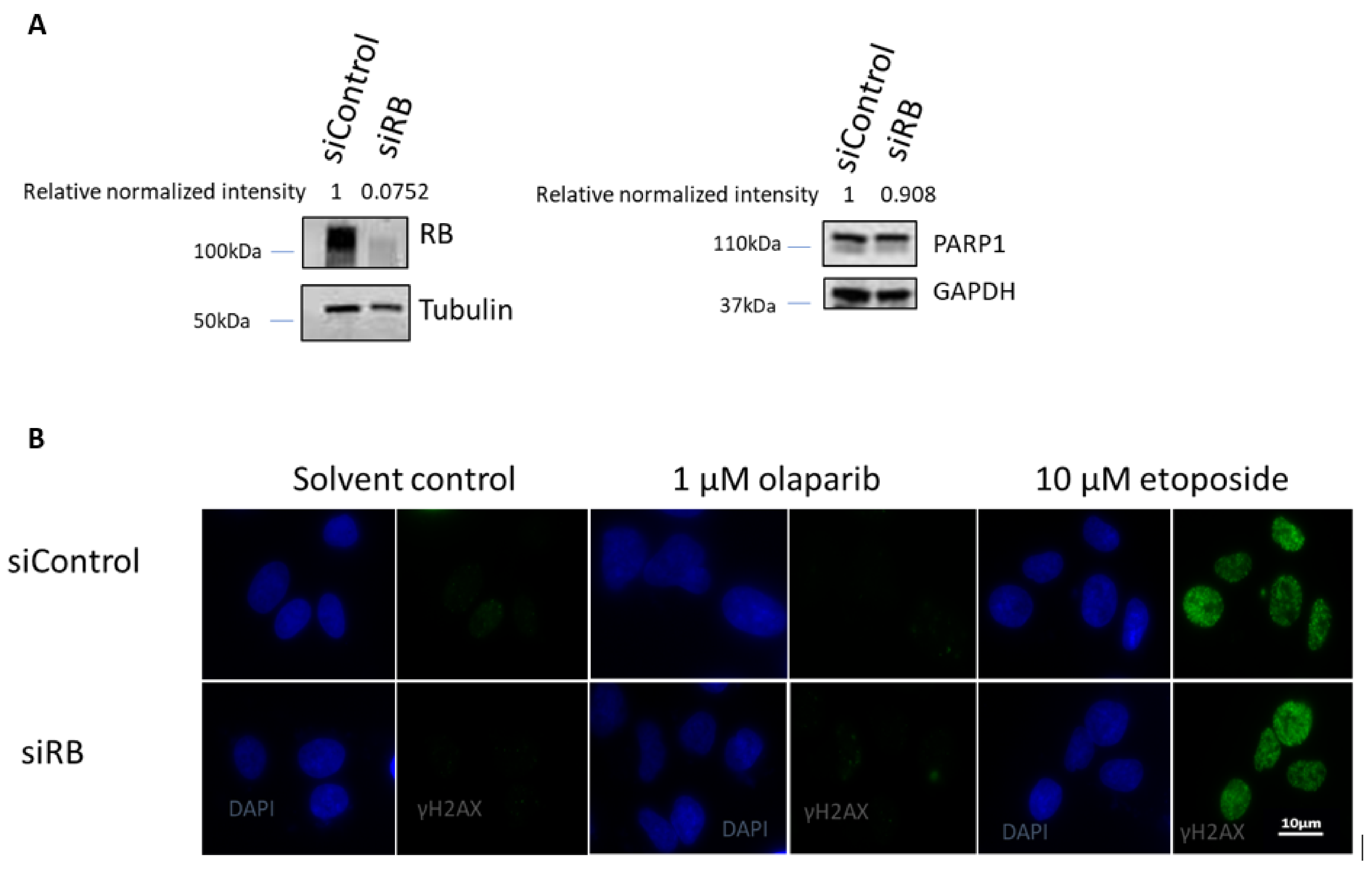
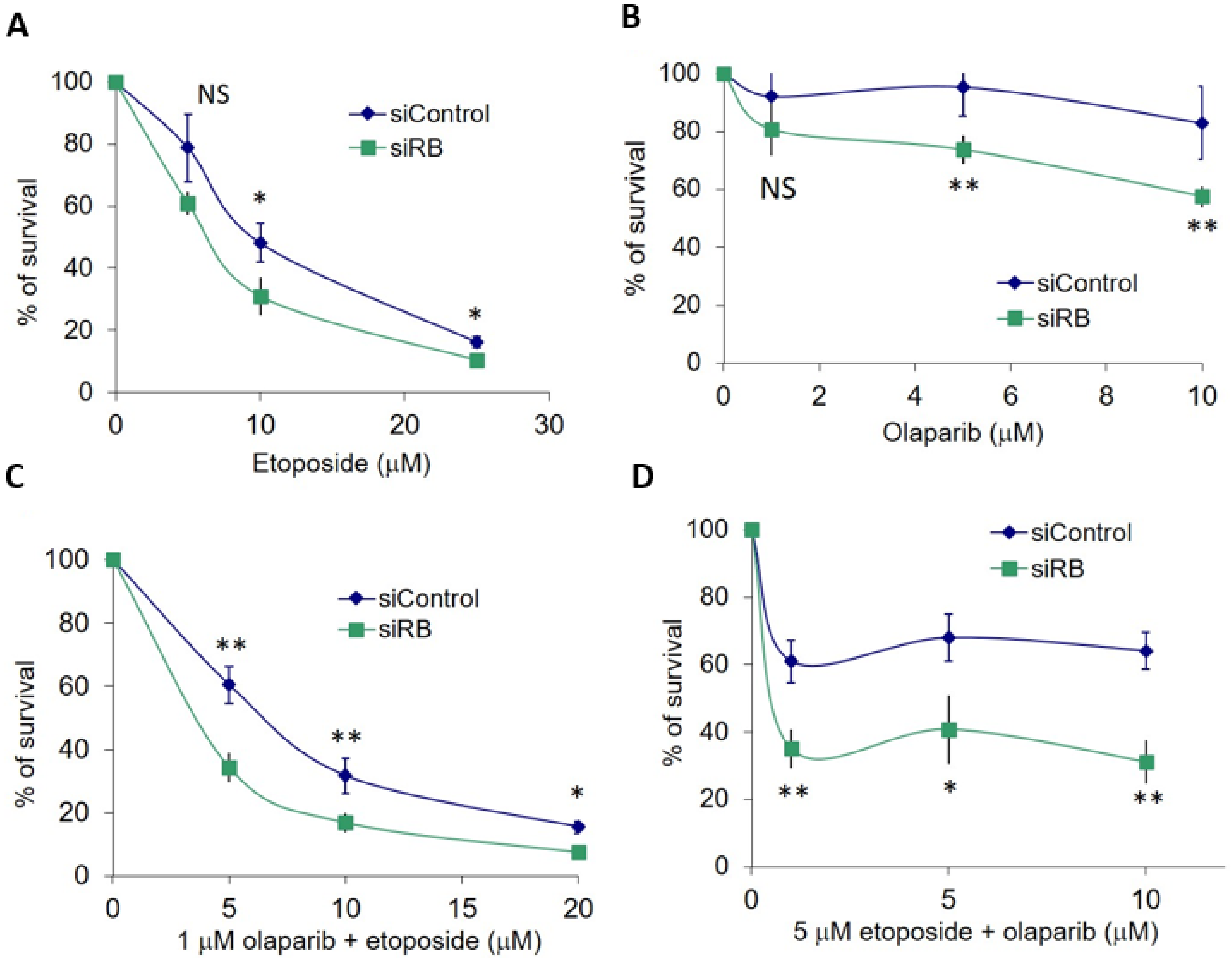
2.1. RB Deficient Cells Depend on MMEJ to Repair DSBs and Are Hypersensitive to Etoposide Treatment
2.2. RB Expression in Primary Retinoblastoma Cells
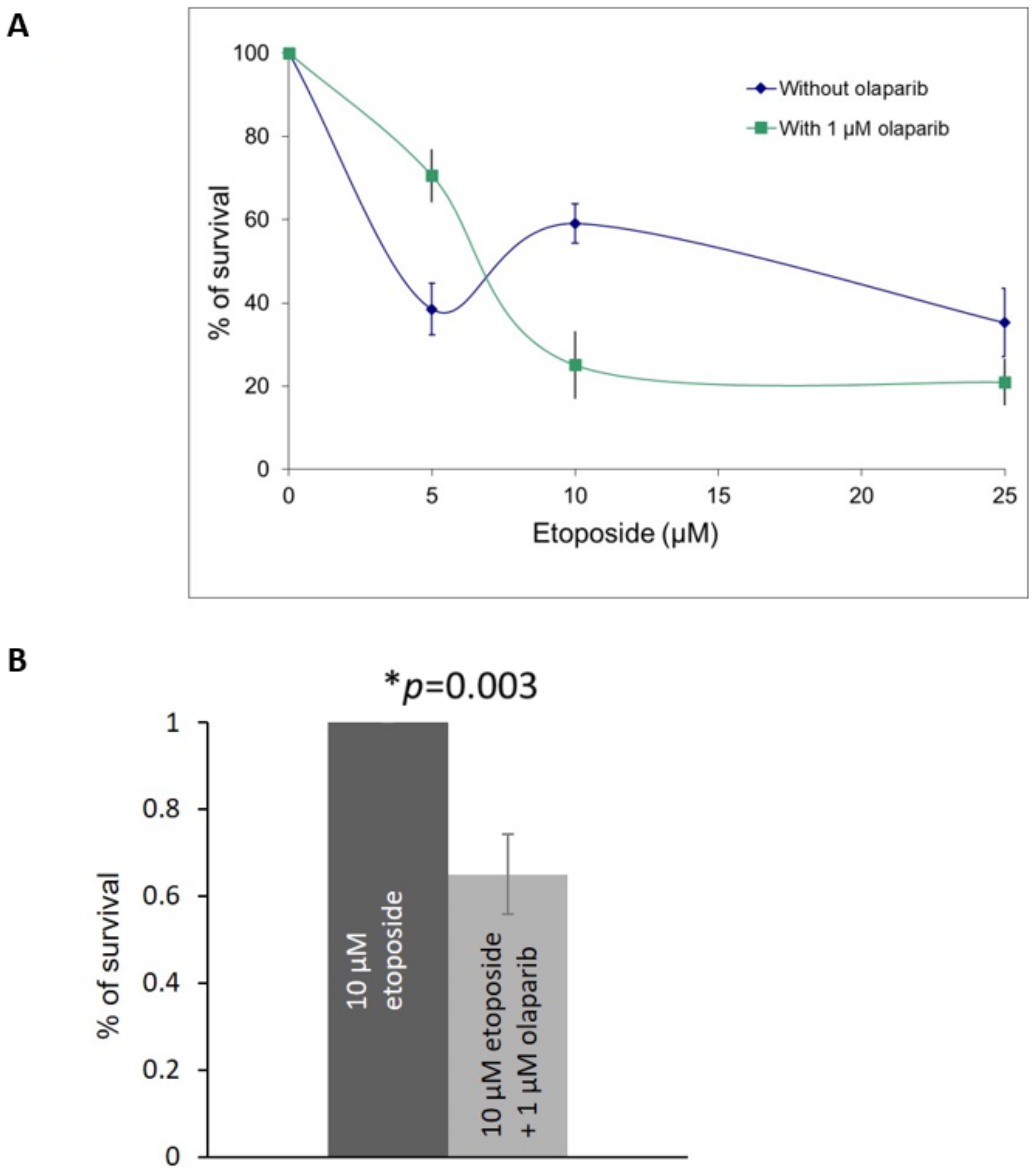
2.3. Olaparib and Etoposide Co-Treatment in Primary Retinoblastoma Cells
3. Discussion
4. Methods
4.1. Cell Culture and siRNA Transfection
4.2. MMEJ Reporter Assay
4.3. MTT Cell Proliferation Assay
4.4. Immunofluorescence
4.5. Western Blotting
4.6. Click-It Plus Edu Flow Cytometry Assay to Detect Cell Proliferation
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Correction Statement
References
- Rodriguez-Galindo, C.; Wilson, M.W.; Haik, B.G.; Lipson, M.J.; Cain, A.; E Merchant, T.; Kaste, S.; Pratt, C.B. Treatment of metastatic retinoblastoma. Ophthalmol. 2003, 110, 1237–1240. [Google Scholar] [CrossRef]
- Lohmann, D. Retinoblastoma. Adv. Exp. Med. Biol. 2010, 685, 220–227. [Google Scholar]
- Xu, X.L.; Fang, Y.; Lee, T.C.; Forrest, D.; Gregory-Evans, C.; Almeida, D.; Liu, A.; Jhanwar, S.C.; Abramson, D.H.; Cobrinik, D. Retinoblastoma Has Properties of a Cone Precursor Tumor and Depends Upon Cone-Specific MDM2 Signaling. Cell 2009, 137, 1018–1031. [Google Scholar] [CrossRef]
- Friend, S.H.; Bernards, R.; Rogelj, S.; Weinberg, R.A.; Rapaport, J.M.; Albert, D.M.; Dryja, T.P. A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nat. Cell Biol. 1986, 323, 643–646. [Google Scholar] [CrossRef] [PubMed]
- Knudson, A.G. Mutation and Cancer: Statistical Study of Retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823. [Google Scholar] [CrossRef] [PubMed]
- Fung, Y.; Murphree, A.; T’Ang, A.; Qian, J.; Hinrichs, S.; Benedict, W. Structural evidence for the authenticity of the human retinoblastoma gene. Science 1987, 236, 1657–1661. [Google Scholar] [CrossRef]
- Berry, J.L.; Polski, A.; Cavenee, W.K.; Dryja, T.P.; Murphree, A.L.; Gallie, B.L. The RB1 Story: Characterization and Cloning of the First Tumor Suppressor Gene. Genes 2019, 10, 879. [Google Scholar] [CrossRef] [PubMed]
- Comings, D.E. A General Theory of Carcinogenesis. Proc. Natl. Acad. Sci. USA 1973, 70, 3324–3328. [Google Scholar] [CrossRef]
- Choy, K.W.; Pang, C.P.; Yu, C.B.; Wong, H.L.; Ng, J.S.; Fan, D.S.; Lo, K.W.; Chai, J.T.; Wang, J.; Fu, W.; et al. Loss of heterozygosity and mutations are the major mechanisms of RB1 gene inactivation in Chinese with sporadic retinoblastoma. Hum. Mutat. 2002, 20, 408. [Google Scholar] [CrossRef]
- Mohney, B.G.; Robertson, D.M.; Schomberg, P.J.; O Hodge, D. Second nonocular tumors in survivors of heritable retinoblastoma and prior radiation therapy. Am. J. Ophthalmol. 1998, 126, 269–277. [Google Scholar] [CrossRef]
- Shields, C.L.; Mashayekhi, A.; Au, A.K.; Czyz, C.; Leahey, A.; Meadows, A.T.; Shields, J.A. The International Classification of Retinoblastoma Predicts Chemoreduction Success. Ophthalmology 2006, 113, 2276–2280. [Google Scholar] [CrossRef] [PubMed]
- Temming, P.; Arendt, M.; Viehmann, A.; Eisele, L.; Le Guin, C.H.D.; Schündeln, M.M.; Biewald, E.; Astrahantseff, K.; Wieland, R.; Bornfeld, N.; et al. Incidence of second cancers after radiotherapy and systemic chemotherapy in heritable retinoblastoma survivors: A report from the German reference center. Pediatr. Blood Cancer 2017, 64, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Ishak, C.; A Dick, F. Conditional haploinsufficiency of the retinoblastoma tumor suppressor gene. Mol. Cell. Oncol. 2014, 2, e968069. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Yam, J.C.; Tham, C.C.; Pang, C.P.; Chu, W.K. RB Regulates DNA Double Strand Break Repair Pathway Choice by Mediating CtIP Dependent End Resection. Int. J. Mol. Sci. 2020, 21, 9176. [Google Scholar] [CrossRef]
- Cook, R.; Zoumpoulidou, G.; Luczynski, M.T.; Rieger, S.; Moquet, J.; Spanswick, V.J.; Hartley, J.A.; Rothkamm, K.; Huang, P.H.; Mittnacht, S. Direct Involvement of Retinoblastoma Family Proteins in DNA Repair by Non-homologous End-Joining. Cell Rep. 2015, 10, 2006–2018. [Google Scholar] [CrossRef]
- Velez-Cruz, R.; Manickavinayaham, S.; Biswas, A.K.; Clary, R.W.; Premkumar, T.; Cole, F.; Johnson, D.G. RB localizes to DNA double-strand breaks and promotes DNA end resection and homologous recombination through the recruitment of BRG1. Genes Dev. 2016, 30, 2500–2512. [Google Scholar] [CrossRef]
- Canning, S.; Dryja, T.P. Short, direct repeats at the breakpoints of deletions of the retinoblastoma gene. Proc. Natl. Acad. Sci. USA 1989, 86, 5044–5048. [Google Scholar] [CrossRef]
- Chopra, N.; Tovey, H.; Pearson, A.; Cutts, R.; Toms, C.; Proszek, P.; Hubank, M.; Dowsett, M.; Dodson, A.; Daley, F.; et al. Homologous recombination DNA repair deficiency and PARP inhibition activity in primary triple negative breast cancer. Nat. Commun. 2020, 11, 2662. [Google Scholar] [CrossRef]
- Delgado, J.L.; Hsieh, C.-M.; Chan, N.-L.; Hiasa, H. Topoisomerases as anticancer targets. Biochem. J. 2018, 475, 373–398. [Google Scholar] [CrossRef]
- Ashworth, A. A Synthetic Lethal Therapeutic Approach: Poly (ADP) Ribose Polymerase Inhibitors for the Treatment of Cancers Deficient in DNA Double-Strand Break Repair. J. Clin. Oncol. 2008, 26, 3785–3790. [Google Scholar] [CrossRef]
- Treszezamsky, A.D.; Kachnic, L.A.; Feng, Z.; Zhang, J.; Tokadjian, C.; Powell, S.N. BRCA1-and BRCA2-Deficient Cells Are Sensitive to Etoposide-Induced DNA Double-Strand Breaks via Topoisomerase II. Cancer Res. 2007, 67, 7078–7081. [Google Scholar] [CrossRef] [PubMed]
- Krokan, H.E.; Bjoras, M. Base excision repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012583. [Google Scholar] [CrossRef] [PubMed]
- McEvoy, J.; Nagahawatte, P.; Finkelstein, D.; Richards-Yutz, J.; Valentine, M.; Ma, J.; Mullighan, C.; Song, G.; Chen, X.; Wilson, M.; et al. RB1 gene inactivation by chromothripsis in human retinoblastoma. Oncotarget 2014, 5, 438–450. [Google Scholar] [CrossRef] [PubMed]
- Colicchia, V.; Petroni, M.; Guarguaglini, G.; Sardina, F.; Sahún-Roncero, M.; Carbonari, M.; Ricci, B.; Heil, C.; Capalbo, C.; Belardinilli, F.; et al. PARP inhibitors enhance replication stress and cause mitotic catastrophe in MYCN-dependent neuroblastoma. Oncogene 2017, 36, 4682–4691. [Google Scholar] [CrossRef]
- Yun, J.; Li, Y.; Xu, C.-T.; Pan, B.-R. Epidemiology and Rb1 gene of retinoblastoma. Int. J. Ophthalmol. 2011, 4, 103–109. [Google Scholar] [CrossRef]
- Kleinerman, R.A.; Tucker, M.A.; Tarone, R.E.; Abramson, D.H.; Seddon, J.M.; Stovall, M.; Li, F.P.; Jr, J.F.F. Risk of New Cancers After Radiotherapy in Long-Term Survivors of Retinoblastoma: An Extended Follow-Up. J. Clin. Oncol. 2005, 23, 2272–2279. [Google Scholar] [CrossRef]
- Bennardo, N.; Cheng, A.; Huang, N.; Stark, J.M. Alternative-NHEJ Is a Mechanistically Distinct Pathway of Mammalian Chromosome Break Repair. PLoS Genet. 2008, 4, e1000110. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, Y.; Yam, J.C.; Chu, W.K. Poly ADP Ribose Polymerase Inhibitor Olaparib Targeting Microhomology End Joining in Retinoblastoma Protein Defective Cancer: Analysis of the Retinoblastoma Cell-Killing Effects by Olaparib after Inducing Double-Strand Breaks. Int. J. Mol. Sci. 2021, 22, 10687. https://doi.org/10.3390/ijms221910687
Jiang Y, Yam JC, Chu WK. Poly ADP Ribose Polymerase Inhibitor Olaparib Targeting Microhomology End Joining in Retinoblastoma Protein Defective Cancer: Analysis of the Retinoblastoma Cell-Killing Effects by Olaparib after Inducing Double-Strand Breaks. International Journal of Molecular Sciences. 2021; 22(19):10687. https://doi.org/10.3390/ijms221910687
Chicago/Turabian StyleJiang, Yuning, Jason C. Yam, and Wai Kit Chu. 2021. "Poly ADP Ribose Polymerase Inhibitor Olaparib Targeting Microhomology End Joining in Retinoblastoma Protein Defective Cancer: Analysis of the Retinoblastoma Cell-Killing Effects by Olaparib after Inducing Double-Strand Breaks" International Journal of Molecular Sciences 22, no. 19: 10687. https://doi.org/10.3390/ijms221910687
APA StyleJiang, Y., Yam, J. C., & Chu, W. K. (2021). Poly ADP Ribose Polymerase Inhibitor Olaparib Targeting Microhomology End Joining in Retinoblastoma Protein Defective Cancer: Analysis of the Retinoblastoma Cell-Killing Effects by Olaparib after Inducing Double-Strand Breaks. International Journal of Molecular Sciences, 22(19), 10687. https://doi.org/10.3390/ijms221910687