
Metabolic Remodeling and Implicated Calcium and Signal Transduction Pathways in the Pathogenesis of Heart Failure


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
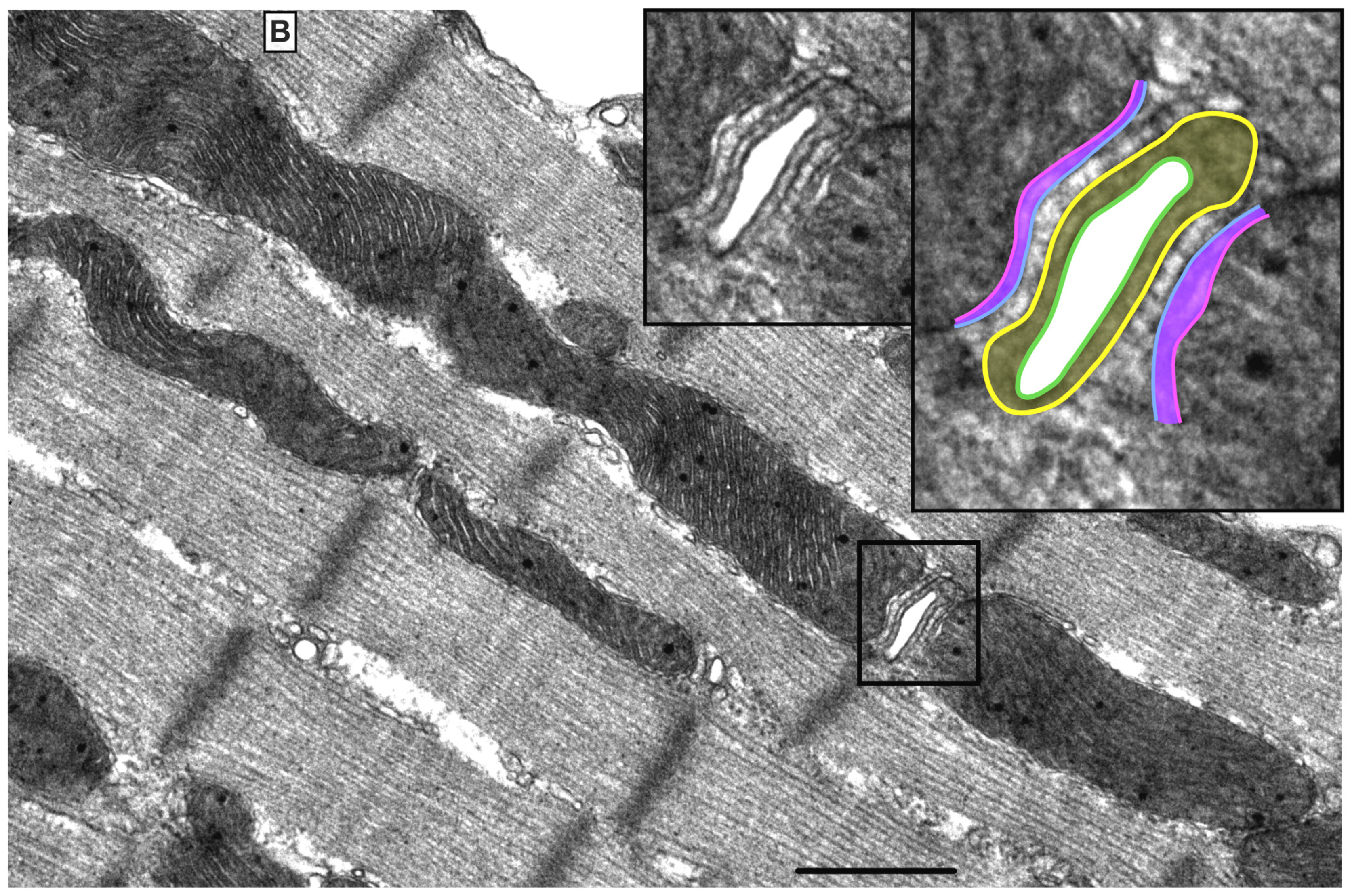
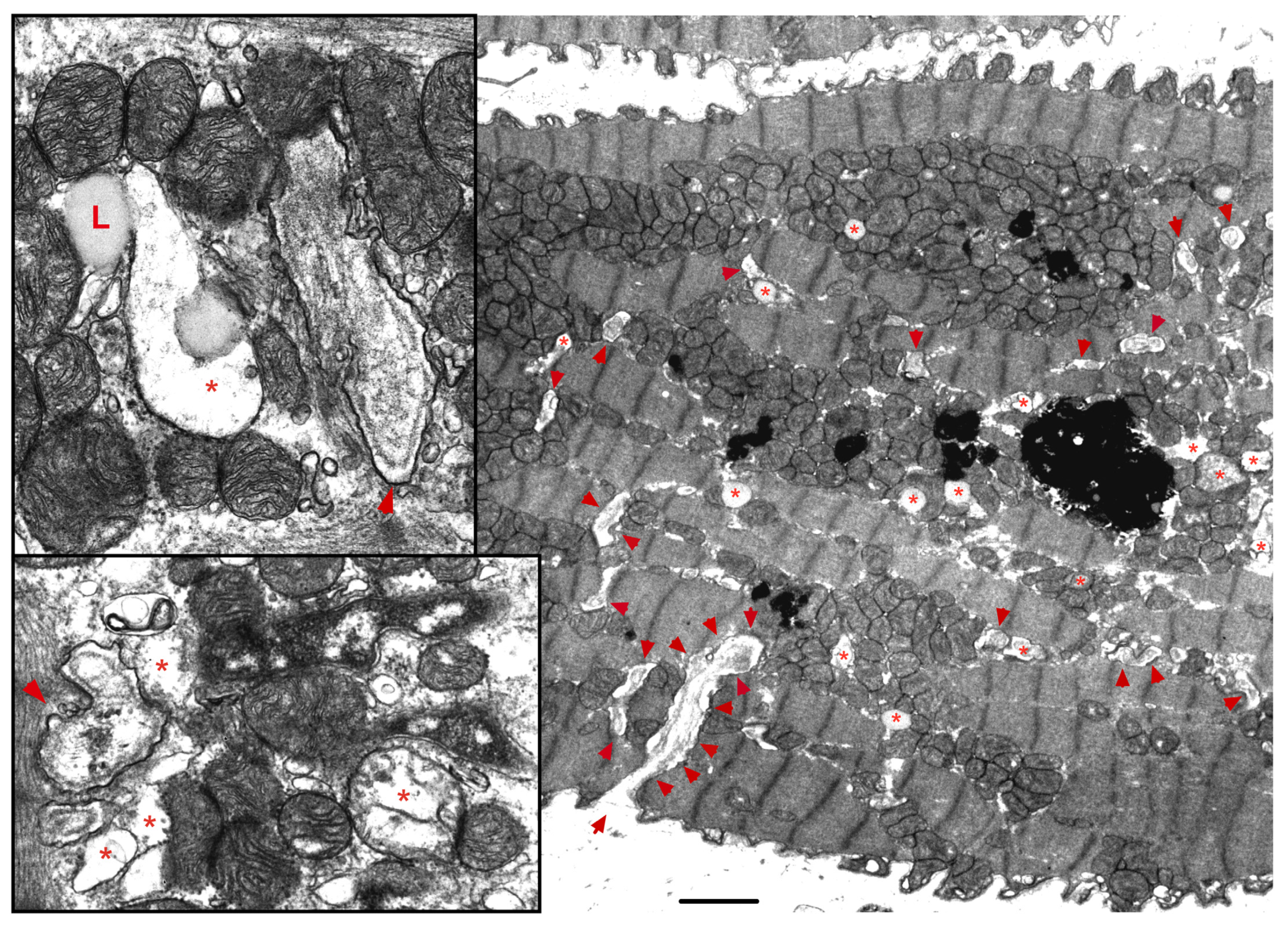
2. Mitochondrial Morphology, Topology and Function
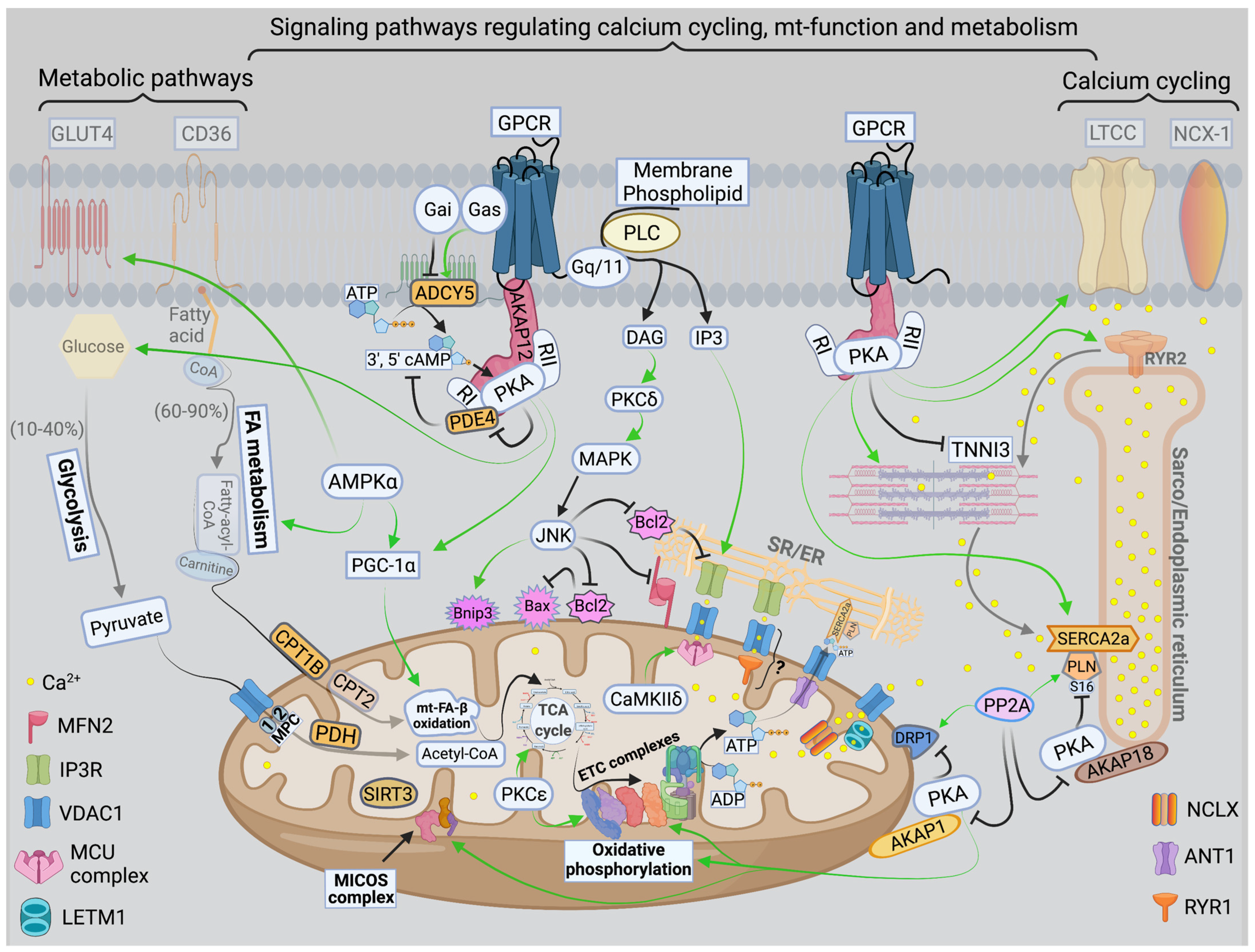
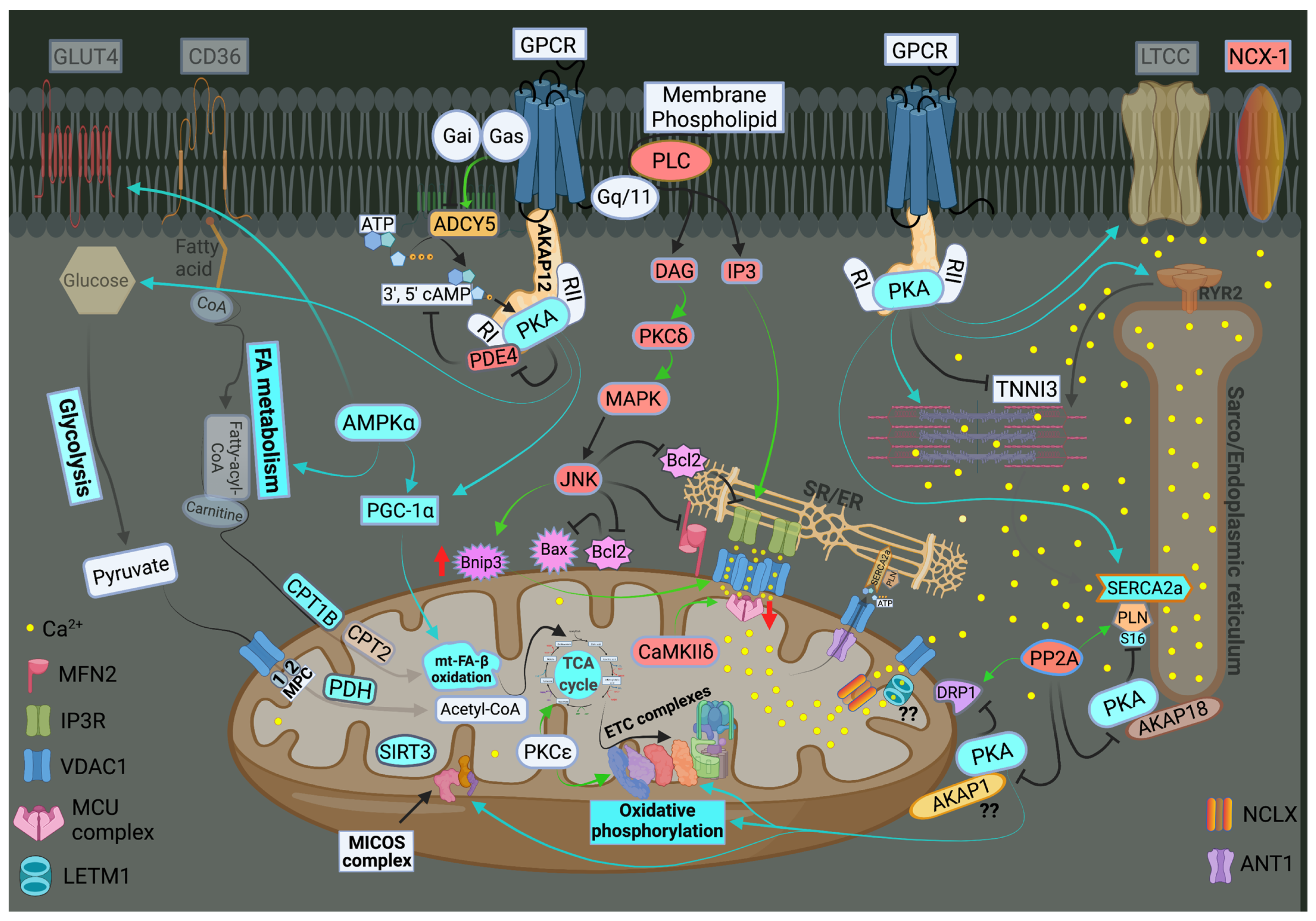
3. Calcium Signaling and Mitochondrial Oxidative Capacity
4. Bcl-2 Family of Proteins and Their Implication in Calcium Homeostasis and Metabolism
5. Signal Transduction Pathways Modulating Mitochondrial Dynamics and Function
5.1. PKA
5.2. AMPK
5.3. Other Kinases
6. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ATP | Adenosine triphosphate |
| AC | Adenylyl cyclases |
| AKAP | A-kinase anchoring protein |
| AKT | RAC-alpha serine/threonine-protein kinase |
| AMPK | Adenosine-monophosphate-activated protein kinase |
| Bcl-2 | B-cell lymphoma-2 |
| BNIP3 | BCL2/adenovirus E1B 19 kDa protein-interacting protein 3 |
| CaMK | Calcium/calmodulin-dependent protein kinase |
| CaMKIIδ | Calcium/calmodulin-dependent protein kinase type II subunit delta |
| cAMP | Cyclic adenosine monophosphate |
| CICR | Calcium induced calcium release |
| COXIV | Cytochrome c oxidase subunit IV |
| DAG | Diacylglycerol |
| DRP1 | Dynamin related protein 1 |
| ER/SR | Endo/Sarcoplasmic reticulum |
| ETC | Electron transport chain |
| FA | Fatty acid |
| FADH2 | Flavin adenine dinucleotide |
| FOXO3a | Forkhead box protein O3a |
| Hsp70 | Heat shock 70 kDa protein 1A |
| IMM | Inner mitochondrial membrane |
| IMS | Inter-membrane space |
| JNK | Jun-N-terminal kinase |
| LETM1 | Leucine zipper EF-hand containing transmembrane protein 1 |
| LTCC | L-type calcium channels |
| MAM | Mitochondria associated membranes |
| MAPK | Mitogen-activated protein kinase |
| MCU | Mitochondrial calcium uniporter |
| MFN2 | Mitofusin 2 |
| MOMP | Mitochondrial outer membrane permeabilization |
| mt | Mitochondrial |
| NADH | Nicotinamide adenine dinucleotide |
| NCLX | Sodium/calcium/lithium exchanger |
| NDUFA9 | NADH dehydrogenase [ubiquinone] 1 alpha subcomplex subunit 9 |
| NDUFS4 | NADH:ubiquinone oxidoreductase subunit S4 |
| NDUFV2 | NADH dehydrogenase [ubiquinone] flavoprotein 2 |
| NFAT | Nuclear factor of activated T-cells |
| OMM | Outer mitochondrial membrane |
| OPA1 | Optic atrophy 1 |
| OXPHOS | Oxidative Phosphorylation |
| PDE | Phosphodiesterase |
| PGC-1α | Peroxisome proliferator-activated receptor gamma coactivator 1-alpha |
| PINK1 | PTEN-induced putative kinase 1 |
| PKA | Protein kinase A |
| PKCε | Protein kinase C epsilon |
| PLN | Phospholamban |
| PP1 | Protein phosphatase 1 |
| PP2A | Calcineurin |
| PPARα | Peroxisome proliferator-activated receptor alpha |
| PTM | Post-translational modification |
| ROS | Reactive oxygen species |
| RYR1 | Ryanodine receptor isoform 1 |
| RYR2 | Ryanodine receptors isoform 2 |
| Sab | Mitochondrial JNK binding protein |
| SERCA2a | Sarco/Endoplasmic reticulum ATPase |
| SHF | Systolic heart failure |
| SIRT3 | NAD-dependent protein deacetylase sirtuin-3 |
| SIRT5 | NAD-dependent protein deacylase sirtuin-5 |
| TCA | Tricarboxylic acid |
| TNNI3 | Troponin I, cardiac muscle |
| UCP3 | Uncoupling protein 3 |
| ULK1 | Serine/threonine-protein kinase ULK1 |
| VDAC1 | Voltage dependent anion channel isoform 1 |
References
- Neubauer, S. The failing heart—An engine out of fuel. N. Engl. J. Med. 2007, 356, 1140–1151. [Google Scholar] [CrossRef] [PubMed]
- D’Erchia, A.M.; Atlante, A.; Gadaleta, G.; Pavesi, G.; Chiara, M.; De Virgilio, C.; Manzari, C.; Mastropasqua, F.; Prazzoli, G.M.; Picardi, E.; et al. Tissue-specific mtDNA abundance from exome data and its correlation with mitochondrial transcription, mass and respiratory activity. Mitochondrion 2015, 20, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Tian, R. Mitochondrial dysfunction in pathophysiology of heart failure. J. Clin. Investig. 2018, 128, 3716–3726. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Kepp, O.; Trojel-Hansen, C.; Kroemer, G. Mitochondrial Control of Cellular Life, Stress, and Death. Circ. Res. 2012, 111, 1198–1207. [Google Scholar] [CrossRef] [PubMed]
- Sack, M.N.; Rader, T.A.; Park, S.; Bastin, J.; McCune, S.A.; Kelly, D.P. Fatty acid oxidation enzyme gene expression is down-regulated in the failing heart. Circulation 1996, 94, 2837–2842. [Google Scholar] [CrossRef]
- Doenst, T.; Pytel, G.; Schrepper, A.; Amorim, P.; Färber, G.; Shingu, Y.; Mohr, F.W.; Schwarzer, M. Decreased rates of substrate oxidation ex vivo predict the onset of heart failure and contractile dysfunction in rats with pressure overload. Cardiovasc. Res. 2009, 86, 461–470. [Google Scholar] [CrossRef]
- Stride, N.; Larsen, S.; Hey-Mogensen, M.; Sander, K.; Lund, J.T.; Gustafsson, F.; Køber, L.; Dela, F. Decreased mitochondrial oxidative phosphorylation capacity in the human heart with left ventricular systolic dysfunction. Eur. J. Heart Fail. 2013, 15, 150–157. [Google Scholar] [CrossRef]
- Barger, P.M.; Brandt, J.M.; Leone, T.C.; Weinheimer, C.J.; Kelly, D.P. Deactivation of peroxisome proliferator-activated receptor-alpha during cardiac hypertrophic growth. J. Clin. Investig. 2000, 105, 1723–1730. [Google Scholar] [CrossRef]
- Chaanine, A.H.; Sreekumaran Nair, K.; Bergen, R.H.; Klaus, K.; Guenzel, A.J.; Hajjar, R.J.; Redfield, M.M. Mitochondrial Integrity and Function in the Progression of Early Pressure Overload-Induced Left Ventricular Remodeling. J. Am. Heart Assoc. 2017, 6, e005869. [Google Scholar] [CrossRef]
- Huss, J.M.; Kelly, D.P. Nuclear Receptor Signaling and Cardiac Energetics. Circ. Res. 2004, 95, 568–578. [Google Scholar] [CrossRef]
- Sutanto, H.; Lyon, A.; Lumens, J.; Schotten, U.; Dobrev, D.; Heijman, J. Cardiomyocyte calcium handling in health and disease: Insights from in vitro and in silico studies. Prog. Biophys. Mol. Biol. 2020, 157, 54–75. [Google Scholar] [CrossRef]
- Kurland, C.G.; Andersson, S.G. Origin and evolution of the mitochondrial proteome. Microbiol. Mol. Biol. Rev. MMBR 2000, 64, 786–820. [Google Scholar] [CrossRef]
- D’Souza, A.R.; Minczuk, M. Mitochondrial transcription and translation: Overview. Essays Biochem. 2018, 62, 309–320. [Google Scholar] [CrossRef]
- Taanman, J.-W. The mitochondrial genome: Structure, transcription, translation and replication. Biochim. Biophys. Acta BBA Bioenerg. 1999, 1410, 103–123. [Google Scholar] [CrossRef]
- Wiedemann, N.; Pfanner, N. Mitochondrial Machineries for Protein Import and Assembly. Annu. Rev. Biochem. 2017, 86, 685–714. [Google Scholar] [CrossRef] [PubMed]
- Hoppel, C.L.; Tandler, B.; Fujioka, H.; Riva, A. Dynamic organization of mitochondria in human heart and in myocardial disease. Int. J. Biochem. Cell Biol. 2009, 41, 1949–1956. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, A.V.; Margreiter, R. Heterogeneity of Mitochondria and Mitochondrial Function within Cells as Another Level of Mitochondrial Complexity. Int. J. Mol. Sci. 2009, 10, 1911–1929. [Google Scholar] [CrossRef] [PubMed]
- Boncompagni, S.; Rossi, A.E.; Micaroni, M.; Beznoussenko, G.V.; Polishchuk, R.; Dirksen, R.T.; Protasi, F. Mitochondria Are Linked to Calcium Stores in Striated Muscle by Developmentally Regulated Tethering Structures. Mol. Biol. Cell 2009, 20, 1058–1067. [Google Scholar] [CrossRef] [PubMed]
- Bertero, E.; Maack, C. Metabolic remodelling in heart failure. Nat. Rev. Cardiol. 2018, 15, 457–470. [Google Scholar] [CrossRef]
- McCormack, J.G.; Halestrap, A.P.; Denton, R.M. Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol. Rev. 1990, 70, 391–425. [Google Scholar] [CrossRef]
- Görlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A mutual interplay. Redox Biol. 2015, 6, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Fabiato, A. Calcium-induced release of calcium from the cardiac sarcoplasmic reticulum. Am. J. Physiol. 1983, 245, C1–C14. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.S.; Huertas, M.A.; Sobie, E.A.; Jafri, M.S.; Smith, G.D. A Probability Density Approach to Modeling Local Control of Calcium-Induced Calcium Release in Cardiac Myocytes. Biophys. J. 2007, 92, 2311–2328. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Williams, G.S.B.; Huertas, M.A.; Sobie, E.A.; Jafri, M.S.; Smith, G.D. Moment Closure for Local Control Models of Calcium-Induced Calcium Release in Cardiac Myocytes. Biophys. J. 2008, 95, 1689–1703. [Google Scholar] [CrossRef]
- Minamisawa, S.; Hoshijima, M.; Chu, G.; Ward, C.A.; Frank, K.; Gu, Y.; Martone, M.E.; Wang, Y.; Ross, J.; Kranias, E.G.; et al. Chronic Phospholamban–Sarcoplasmic Reticulum Calcium ATPase Interaction Is the Critical Calcium Cycling Defect in Dilated Cardiomyopathy. Cell 1999, 99, 313–322. [Google Scholar] [CrossRef]
- Gustavsson, M.; Verardi, R.; Mullen, D.G.; Mote, K.R.; Traaseth, N.J.; Gopinath, T.; Veglia, G. Allosteric regulation of SERCA by phosphorylation-mediated conformational shift of phospholamban. Proc. Natl. Acad. Sci. USA 2013, 110, 17338–17343. [Google Scholar] [CrossRef]
- Rimessi, A.; Pedriali, G.; Vezzani, B.; Tarocco, A.; Marchi, S.; Wieckowski, M.; Giorgi, C.; Pinton, P. Interorganellar calcium signaling in the regulation of cell metabolism: A cancer perspective. Semin. Cell Dev. Biol. 2019, 98, 167–180. [Google Scholar] [CrossRef]
- Michalak, M.; Parker, J.R.; Opas, M. Ca2+ signaling and calcium binding chaperones of the endoplasmic reticulum. Cell Calcium 2002, 32, 269–278. [Google Scholar] [CrossRef]
- Pan, X.; Liu, J.; Nguyen, T.; Liu, C.; Sun, J.; Teng, Y.; Fergusson, M.M.; Rovira, I.I.; Allen, M.; Springer, D.A.; et al. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat. Cell Biol. 2013, 15, 1464–1472. [Google Scholar] [CrossRef]
- Wu, Y.; Rasmussen, T.P.; Koval, O.M.; Joiner, M.L.; Hall, D.D.; Chen, B.; Luczak, E.D.; Wang, Q.; Rokita, A.G.; Wehrens, X.H.; et al. The mitochondrial uniporter controls fight or flight heart rate increases. Nat. Commun. 2015, 6, 6081. [Google Scholar] [CrossRef]
- Luongo, T.; Lambert, J.; Yuan, A.; Zhang, X.; Gross, P.; Song, J.; Shanmughapriya, S.; Gao, E.; Jain, M.; Houser, S.R.; et al. The Mitochondrial Calcium Uniporter Matches Energetic Supply with Cardiac Workload during Stress and Modulates Permeability Transition. Cell Rep. 2015, 12, 23–34. [Google Scholar] [CrossRef]
- Abu-Hamad, S.; Sivan, S.; Shoshan-Barmatz, V. The expression level of the voltage-dependent anion channel controls life and death of the cell. Proc. Natl. Acad. Sci. USA 2006, 103, 5787–5792. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Chandel, N.S.; Li, X.X.; Schumacker, P.T.; Colombini, M.; Thompson, C.B. Outer mitochondrial mem-brane permeability can regulate coupled respiration and cell survival. Proc. Natl. Acad. Sci. USA 2000, 97, 4666–4671. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T.; Menazza, S.; Holmstrom, K.M.; Parks, R.J.; Liu, J.; Sun, J.; Liu, J.; Pan, X.; Murphy, E. The ins and outs of mitochondrial calcium. Circ. Res. 2015, 116, 1810–1819. [Google Scholar] [CrossRef] [PubMed]
- Doonan, P.J.; Chandramoorthy, H.C.; Hoffman, N.E.; Zhang, X.; Cárdenas, C.; Shanmughapriya, S.; Rajan, S.; Vallem, S.; Chen, X.; Foskett, J.K.; et al. LETM1-dependent mitochondrial Ca2+ flux modulates cellular bioenergetics and proliferation. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2014, 28, 4936–4949. [Google Scholar]
- Chaanine, A.H.; Jeong, D.; Liang, L.; Chemaly, E.R.; Fish, K.; Gordon, R.E.; Hajjar, R.J. JNK modulates FOXO3a for the ex-pression of the mitochondrial death and mitophagy marker BNIP3 in pathological hypertrophy and in heart failure. Cell Death Dis. 2012, 3, 265. [Google Scholar] [CrossRef] [PubMed]
- Chaanine, A.H. Morphological Stages of Mitochondrial Vacuolar Degeneration in Phenylephrine-Stressed Cardiac Myocytes and in Animal Models and Human Heart Failure. Medicina 2019, 55, 239. [Google Scholar] [CrossRef]
- Chaanine, A.H.; Joyce, L.D.; Stulak, J.M.; Maltais, S.; Joyce, D.L.; Dearani, J.A.; Klaus, K.; Nair, K.S.; Hajjar, R.J.; Redfield, M.M. Mitochondrial Morphology, Dynamics, and Function in Human Pressure Overload or Ischemic Heart Disease with Preserved or Reduced Ejection Fraction. Circ. Heart Fail. 2019, 12, e005131. [Google Scholar] [CrossRef]
- Louch, W.E.; Sejersted, O.M.; Swift, F. There goes the neighborhood: Pathological alterations in T-tubule morphology and consequences for cardiomyocyte Ca2+ handling. J. Biomed. Biotechnol. 2010, 2010, 503906. [Google Scholar] [CrossRef]
- Leboucher, G.P.; Tsai, Y.C.; Yang, M.; Shaw, K.C.; Zhou, M.; Veenstra, T.D.; Glickman, M.H.; Weissman, A.M. Stress-Induced Phosphorylation and Proteasomal Degradation of Mitofusin 2 Facilitates Mitochondrial Fragmentation and Apoptosis. Mol. Cell 2012, 47, 547–557. [Google Scholar] [CrossRef]
- Chaanine, A.H.; Kohlbrenner, E.; Gamb, S.I.; Guenzel, A.J.; Klaus, K.; Fayyaz, A.U.; Nair, K.S.; Hajjar, R.J.; Redfield, M.M. FOXO3a regulates BNIP3 and modulates mitochondrial calcium, dynamics, and function in cardiac stress. Am. J. Physiol. Circ. Physiol. 2016, 311, H1540–H1559. [Google Scholar] [CrossRef] [PubMed]
- Cereghetti, G.M.; Stangherlin, A.; Martins de Brito, O.; Chang, C.R.; Blackstone, C.; Bernardi, P.; Scorrano, L. Dephosphory-lation by calcineurin regulates translocation of Drp1 to mitochondria. Proc. Natl. Acad. Sci. USA 2008, 105, 15803–15808. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-R.; Blackstone, C. Cyclic AMP-dependent Protein Kinase Phosphorylation of Drp1 Regulates Its GTPase Activity and Mitochondrial Morphology. J. Biol. Chem. 2007, 282, 21583–21587. [Google Scholar] [CrossRef]
- Bers, D.M.; Despa, S.; Bossuyt, J. Regulation of Ca2+ and Na+ in Normal and Failing Cardiac Myocytes. Ann. N. Y. Acad. Sci. 2006, 1080, 165–177. [Google Scholar] [CrossRef]
- Hajjar, R.J. Potential of gene therapy as a treatment for heart failure. J. Clin. Investig. 2013, 123, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Marks, A.R. Calcium cycling proteins and heart failure: Mechanisms and therapeutics. J. Clin. Investig. 2013, 123, 46–52. [Google Scholar] [CrossRef]
- Santulli, G.; Xie, W.; Reiken, S.R.; Marks, A.R. Mitochondrial calcium overload is a key determinant in heart failure. Proc. Natl. Acad. Sci. USA 2015, 112, 11389–11394. [Google Scholar] [CrossRef]
- Chaanine, A.H.; Gordon, R.E.; Kohlbrenner, E.; Benard, L.; Jeong, D.; Hajjar, R.J. Potential role of BNIP3 in cardiac remodeling, myocardial stiffness, and endoplasmic reticulum: Mitochondrial calcium homeostasis in diastolic and systolic heart failure. Circ. Heart Fail. 2013, 6, 572–583. [Google Scholar] [CrossRef]
- Suarez, J.; Cividini, F.; Scott, B.; Lehmann, K.; Diaz-Juarez, J.; Diemer, T.; Dai, A.; Jain, M.; Dillmann, W.H. Restoring mitochondrial calcium uniporter expression in diabetic mouse heart improves mitochondrial calcium handling and cardiac function. J. Biol. Chem. 2018, 293, 8182–8195. [Google Scholar] [CrossRef]
- Lu, X.; Ginsburg, K.S.; Kettlewell, S.; Bossuyt, J.; Smith, G.L.; Bers, D.M. Measuring Local Gradients of Intramitochondrial [Ca2+] in Cardiac Myocytes During Sarcoplasmic Reticulum Ca2+ Release. Circ. Res. 2013, 112, 424–431. [Google Scholar] [CrossRef]
- Maack, C.; Cortassa, S.; Aon, M.A.; Ganesan, A.N.; Liu, T.; O’Rourke, B. Elevated Cytosolic Na+ Decreases Mitochondrial Ca2+ Uptake During Excitation-Contraction Coupling and Impairs Energetic Adaptation in Cardiac Myocytes. Circ. Res. 2006, 99, 172–182. [Google Scholar] [CrossRef]
- Rapizzi, E.; Pinton, P.; Szabadkai, G.; Wieckowski, M.; Vandecasteele, G.; Baird, G.; Tuft, R.A.; Fogarty, K.E.; Rizzuto, R. Recombinant expression of the voltage-dependent anion channel enhances the transfer of Ca2+ microdomains to mitochondria. J. Cell Biol. 2002, 159, 613–624. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; De Pinto, V.; Zweckstetter, M.; Raviv, Z.; Keinan, N.; Arbel, N. VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol. Asp. Med. 2010, 31, 227–285. [Google Scholar] [CrossRef] [PubMed]
- Keinan, N.; Tyomkin, D.; Shoshan-Barmatz, V. Oligomerization of the Mitochondrial Protein Voltage-Dependent Anion Channel Is Coupled to the Induction of Apoptosis. Mol. Cell. Biol. 2010, 30, 5698–5709. [Google Scholar] [CrossRef] [PubMed]
- Joiner, M.-L.A.; Koval, O.; Li, J.; He, B.J.; Allamargot, C.; Gao, Z.; Luczak, E.D.; Hall, D.; Fink, B.D.; Chen, B.; et al. CaMKII determines mitochondrial stress responses in heart. Nature 2012, 491, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, E.K.; Koval, O.M.; Noble, P.; Broadhurst, K.; Allamargot, C.; Wu, M.; Strack, S.; Thiel, W.H.; Grumbach, I.M. CaMKII (Ca(2+)/Calmodulin-Dependent Kinase II) in Mitochondria of Smooth Muscle Cells Controls Mitochondrial Mobility, Migration, and Neointima Formation. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1333–1345. [Google Scholar] [CrossRef] [PubMed]
- Luongo, T.; Lambert, J.; Gross, P.; Nwokedi, M.; Lombardi, A.A.; Shanmughapriya, S.; Carpenter, A.C.; Kolmetzky, D.; Gao, E.; van Berlo, J.; et al. The mitochondrial Na+/Ca2+ exchanger is essential for Ca2+ homeostasis and viability. Nature 2017, 545, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Yang, N.; Sidor, A.; O’Rourke, B. MCU Overexpression Rescues Inotropy and Reverses Heart Failure by Reducing SR Ca2+ Leak. Circ. Res. 2021, 128, 1191–1204. [Google Scholar] [CrossRef]
- Rosca, M.G.; Tandler, B.; Hoppel, C.L. Mitochondria in cardiac hypertrophy and heart failure. J. Mol. Cell. Cardiol. 2012, 55, 31–41. [Google Scholar] [CrossRef]
- Horton, J.L.; Martin, O.J.; Lai, L.; Riley, N.M.; Richards, A.L.; Vega, R.B.; Leone, T.C.; Pagliarini, D.J.; Muoio, D.M.; Bedi, K.C.; et al. Mitochondrial protein hyperacetylation in the failing heart. JCI Insight 2016, 2, e84897. [Google Scholar] [CrossRef]
- Lee, C.F.; Chavez, J.D.; Garcia-Menendez, L.; Choi, Y.S.; Roe, N.D.; Chiao, Y.A.; Edgar, J.S.; Goo, Y.A.; Goodlett, D.R.; Bruce, J.E.; et al. Normalization of NAD + Redox Balance as a Therapy for Heart Failure. Circulation 2016, 134, 883–894. [Google Scholar] [CrossRef]
- Chen, T.; Liu, J.; Li, N.; Wang, S.; Liu, H.; Li, J.; Zhang, Y.; Bu, P. Correction: Mouse SIRT3 Attenuates Hypertrophy-Related Lipid Accumulation in the Heart through the Deacetylation of LCAD. PLoS ONE 2016, 11, e0155173. [Google Scholar] [CrossRef] [PubMed]
- Koentges, C.; Pfeil, K.; Schnick, T.; Wiese, S.; Dahlbock, R.; Cimolai, M.C.; Meyer-Steenbuck, M.; Cenkerova, K.; Hoffmann, M.M.; Jaeger, C.; et al. SIRT3 deficiency impairs mitochondrial and contractile function in the heart. Basic Res. Cardiol. 2015, 110, 36. [Google Scholar] [CrossRef] [PubMed]
- Sundaresan, N.R.; Gupta, M.; Kim, G.; Rajamohan, S.B.; Isbatan, A.; Gupta, M.P. Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J. Clin. Investig. 2009, 119, 2758–2771. [Google Scholar] [CrossRef] [PubMed]
- Pillai, V.B.; Bindu, S.; Sharp, W.W.; Fang, Y.H.; Kim, G.; Gupta, M.; Samant, S.; Gupta, M.P. Sirt3 protects mitochondrial DNA damage and blocks the development of doxorubicin-induced cardiomyopathy in mice. Am. J. Physiol. Circ. Physiol. 2016, 310, H962–H972. [Google Scholar] [CrossRef]
- Pillai, V.B.; Kanwal, A.; Fang, Y.H.; Sharp, W.W.; Samant, S.; Arbiser, J.; Gupta, M.P. Honokiol, an activator of Sirtuin-3 (SIRT3) preserves mitochondria and protects the heart from doxorubicin-induced cardiomyopathy in mice. Oncotarget 2017, 8, 34082–34098. [Google Scholar] [CrossRef] [PubMed]
- Koentges, C.; Bode, C.; Bugger, H. SIRT3 in Cardiac Physiology and Disease. Front. Cardiovasc. Med. 2016, 3, 38. [Google Scholar] [CrossRef]
- Shi, L.; Zhang, T.; Zhou, Y.; Zeng, X.; Ran, L.; Zhang, Q.; Zhu, J.; Mi, M. Dihydromyricetin improves skeletal muscle insulin sensitivity by inducing autophagy via the AMPK-PGC-1α-Sirt3 signaling pathway. Endocrine 2015, 50, 378–389. [Google Scholar] [CrossRef]
- Oka, S.-I.; Sabry, A.D.; Cawley, K.M.; Warren, J.S. Multiple Levels of PGC-1α Dysregulation in Heart Failure. Front. Cardiovasc. Med. 2020, 7, 2. [Google Scholar] [CrossRef]
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of apoptosis in health and disease: The balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef]
- Morris, J.; Gillet, G.; Prudent, J.; Popgeorgiev, N. Bcl-2 Family of Proteins in the Control of Mitochondrial Calcium Signalling: An Old Chap with New Roles. Int. J. Mol. Sci. 2021, 22, 3730. [Google Scholar] [CrossRef] [PubMed]
- Krajewski, S.; Tanaka, S.; Takayama, S.; Schibler, M.J.; Fenton, W.; Reed, J.C. Investigation of the subcellular distribution of the bcl-2 oncoprotein: Residence in the nuclear envelope, endoplasmic reticulum, and outer mitochondrial membranes. Cancer Res. 1993, 53, 4701–4714. [Google Scholar] [PubMed]
- Chipuk, J.E.; Moldoveanu, T.; Llambi, F.; Parsons, M.J.; Green, D.R. The BCL-2 Family Reunion. Mol. Cell 2010, 37, 299–310. [Google Scholar] [CrossRef]
- Youle, R.J.; Strasser, A. The BCL-2 protein family: Opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 2008, 9, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; He, H.; Zhan, S.; Krajewski, S.; Reed, J.C.; Gottlieb, R.A. Bid Is Cleaved by Calpain to an Active Fragment in Vitro and during Myocardial Ischemia/Reperfusion. J. Biol. Chem. 2001, 276, 30724–30728. [Google Scholar] [CrossRef]
- Wang, H.-G.; Pathan, N.; Ethell, I.M.; Krajewski, S.; Yamaguchi, Y.; Shibasaki, F.; McKeon, F.; Bobo, T.; Franke, T.F.; Reed, J.C. Ca2+-Induced Apoptosis Through Calcineurin Dephosphorylation of BAD. Science 1999, 284, 339–343. [Google Scholar] [CrossRef]
- Rizzuto, R.; Brini, M.; Murgia, M.; Pozzan, T. Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science 1993, 262, 744–747. [Google Scholar] [CrossRef]
- Rizzuto, R.; Pinton, P.; Carrington, W.; Fay, F.S.; Fogarty, K.E.; Lifshitz, L.M.; Tuft, R.A.; Pozzan, T. Close Contacts with the Endoplasmic Reticulum as Determinants of Mitochondrial Ca2+ Responses. Science 1998, 280, 1763–1766. [Google Scholar] [CrossRef]
- Szabadkai, G.; Bianchi, K.; Várnai, P.; De Stefani, D.; Wieckowski, M.; Cavagna, D.; Nagy, A.I.; Balla, T.; Rizzuto, R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 2006, 175, 901–911. [Google Scholar] [CrossRef]
- Giorgi, C.; De Stefani, D.; Bononi, A.; Rizzuto, R.; Pinton, P. Structural and functional link between the mitochondrial network and the endoplasmic reticulum. Int. J. Biochem. Cell Biol. 2009, 41, 1817–1827. [Google Scholar] [CrossRef]
- Hajnóczky, G.; Csordás, G.; Das, S.; Garcia-Perez, C.; Saotome, M.; Roy, S.S.; Yi, M. Mitochondrial calcium signalling and cell death: Approaches for assessing the role of mitochondrial Ca2+ uptake in apoptosis. Cell Calcium 2006, 40, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Brustovetsky, N.; Brustovetsky, T.; Jemmerson, R.; Dubinsky, J.M. Calcium-induced cytochrome c release from CNS mitochondria is associated with the permeability transition and rupture of the outer membrane. J. Neurochem. 2002, 80, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Valencia, I.; Zhong, F.; McColl, K.S.; Roderick, H.; Bootman, M.; Berridge, M.J.; Conway, S.; Holmes, A.B.; Mignery, G.A.; et al. Bcl-2 functionally interacts with inositol 1,4,5-trisphosphate receptors to regulate calcium release from the ER in response to inositol 1,4,5-trisphosphate. J. Cell Biol. 2004, 166, 193–203. [Google Scholar] [CrossRef]
- Hanson, C.J.; Bootman, M.D.; Distelhorst, C.W.; Wojcikiewicz, R.J.; Roderick, H.L. Bcl-2 suppresses Ca2+ release through inositol 1,4,5-trisphosphate receptors and inhibits Ca2+ uptake by mitochondria without affecting ER calcium store content. Cell Calcium 2008, 44, 324–338. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Li, X.X.; Gottleib, E.; Hill, R.B.; Thompson, C.B.; Colombini, M. Bcl-xL promotes the open configuration of the voltage-dependent anion channel and metabolite passage through the outer mitochondrial membrane. J. Biol. Chem. 2001, 276, 19414–19419. [Google Scholar] [CrossRef] [PubMed]
- Eno, C.O.; Eckenrode, E.F.; Olberding, K.E.; Zhao, G.; White, C.; Li, C. Distinct roles of mitochondria- and ER-localized Bcl-xL in apoptosis resistance and Ca2+ homeostasis. Mol. Biol. Cell 2012, 23, 2605–2618. [Google Scholar] [CrossRef]
- Rostovtseva, T.K.; Antonsson, B.; Suzuki, M.; Youle, R.J.; Colombini, M.; Bezrukov, S.M. Bid, but not Bax, regulates VDAC channels. J. Biol. Chem. 2004, 279, 13575–13583. [Google Scholar] [CrossRef]
- Roy, S.S.; Madesh, M.; Davies, E.; Antonsson, B.; Danial, N.; Hajnóczky, G. Bad Targets the Permeability Transition Pore Independent of Bax or Bak to Switch between Ca2+-Dependent Cell Survival and Death. Mol. Cell 2009, 33, 377–388. [Google Scholar] [CrossRef]
- Landes, T.; Emorine, L.J.; Courilleau, D.; Rojo, M.; Belenguer, P.; Arnauné-Pelloquin, L. The BH3-only Bnip3 binds to the dynamin Opa1 to promote mitochondrial fragmentation and apoptosis by distinct mechanisms. EMBO Rep. 2010, 11, 459–465. [Google Scholar] [CrossRef]
- Varanita, T.; Soriano, M.E.; Romanello, V.; Zaglia, T.; Quintana-Cabrera, R.; Semenzato, M.; Menabò, R.; Costa, V.; Civiletto, G.; Pesce, P.; et al. The Opa1-Dependent Mitochondrial Cristae Remodeling Pathway Controls Atrophic, Apoptotic, and Ischemic Tissue Damage. Cell Metab. 2015, 21, 834–844. [Google Scholar] [CrossRef]
- Rikka, S.; Quinsay, M.N.; Thomas, R.; Kubli, D.A.; Zhang, X.; Murphy, A.N.; Gustafsson, A.B. Bnip3 impairs mitochondrial bioenergetics and stimulates mitochondrial turnover. Cell Death Differ. 2010, 18, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Maundrell, K.; Antonsson, B.; Magnenat, E.; Camps, M.; Muda, M.; Chabert, C.; Gillieron, C.; Boschert, U.; Vial-Knecht, E.; Martinou, J.-C.; et al. Bcl-2 Undergoes Phosphorylation by c-Jun N-terminal Kinase/Stress-activated Protein Kinases in the Presence of the Constitutively Active GTP-binding Protein Rac1. J. Biol. Chem. 1997, 272, 25238–25242. [Google Scholar] [CrossRef] [PubMed]
- Harada, H.; Becknell, B.; Wilm, M.; Mann, M.; Huang, L.J.-S.; Taylor, S.S.; Scott, J.D.; Korsmeyer, S.J. Phosphorylation and Inactivation of BAD by Mitochondria-Anchored Protein Kinase A. Mol. Cell 1999, 3, 413–422. [Google Scholar] [CrossRef]
- Giorgianni, F.; Usman Khan, M.; Weber, K.T.; Gerling, I.C.; Beranova-Giorgianni, S. Phosphoproteome mapping of cardio-myocyte mitochondria in a rat model of heart failure. Mol. Cell. Biochem. 2014, 389, 159–167. [Google Scholar] [CrossRef]
- Kruse, R.; Højlund, K. Mitochondrial phosphoproteomics of mammalian tissues. Mitochondrion 2016, 33, 45–57. [Google Scholar] [CrossRef]
- Pagliarini, D.J.; Dixon, J.E. Mitochondrial modulation: Reversible phosphorylation takes center stage? Trends Biochem. Sci. 2006, 31, 26–34. [Google Scholar] [CrossRef]
- Triplett, J.C.; Zhang, Z.; Sultana, R.; Cai, J.; Klein, J.B.; Büeler, H.; Butterfield, D.A. Quantitative expression proteomics and phosphoproteomics profile of brain from PINK1 knockout mice: Insights into mechanisms of familial Parkinson’s disease. J. Neurochem. 2015, 133, 750–765. [Google Scholar] [CrossRef]
- Boone, A.N.; Rodrigues, B.; Brownsey, R.W. Multiple-site phosphorylation of the 280 kDa isoform of acetyl-CoA carboxylase in rat cardiac myocytes: Evidence that cAMP-dependent protein kinase mediates effects of beta-adrenergic stimulation. Biochem. J. 1999, 341, 347–354. [Google Scholar] [CrossRef]
- Cohen, P.; Hardie, D.G. The actions of cyclic AMP on biosynthetic processes are mediated indirectly by cyclic AMP-dependent protein kinase. Biochim. Biophys. Acta 1991, 1094, 292–299. [Google Scholar] [CrossRef]
- Dyck, J.R.; Kudo, N.; Barr, A.J.; Davies, S.P.; Hardie, D.G.; Lopaschuk, G.D. Phosphorylation control of cardiac acetyl-CoA carboxylase by cAMP-dependent protein kinase and 5’-AMP activated protein kinase. Eur. J. Biochem. 1999, 262, 184–190. [Google Scholar] [CrossRef]
- Nielsen, J.N.; Richter, E.A. Regulation of glycogen synthase in skeletal muscle during exercise. Acta Physiol. Scand. 2003, 178, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Winder, W.W.; Wilson, H.A.; Hardie, D.G.; Rasmussen, B.B.; Hutber, C.A.; Call, G.B.; Clayton, R.D.; Conley, L.M.; Yoon, S.; Zhou, B. Phosphorylation of rat muscle acetyl-CoA carboxylase by AMP-activated protein kinase and protein kinase A. J. Appl. Physiol. 1997, 82, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Gerhart-Hines, Z.; Dominy, J.E.; Blättler, S.M.; Jedrychowski, M.P.; Banks, A.S.; Lim, J.H.; Chim, H.; Gygi, S.P.; Puigserver, P. The cAMP/PKA pathway rapidly activates SIRT1 to promote fatty acid oxidation independently of changes in NAD(+). Mol. Cell 2011, 44, 851–863. [Google Scholar] [CrossRef] [PubMed]
- Canto, C.; Auwerx, J. PGC-1α, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr. Opin. Lipidol. 2009, 20, 98–105. [Google Scholar] [CrossRef]
- Parra, V.; Rothermel, B.A. Calcineurin signaling in the heart: The importance of time and place. J. Mol. Cell. Cardiol. 2016, 103, 121–136. [Google Scholar] [CrossRef] [PubMed]
- Santana, L.F.; Chase, E.G.; Votaw, V.S.; Nelson, M.; Greven, R. Functional coupling of calcineurin and protein kinase A in mouse ventricular myocytes. J. Physiol. 2002, 544, 57–69. [Google Scholar] [CrossRef]
- Shintani-Ishida, K.; Yoshida, K.-I. Ischemia induces phospholamban dephosphorylation via activation of calcineurin, PKC-α, and protein phosphatase 1, thereby inducing calcium overload in reperfusion. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2011, 1812, 743–751. [Google Scholar] [CrossRef]
- Burdyga, A.; Surdo, N.C.; Monterisi, S.; Di Benedetto, G.; Grisan, F.; Penna, E.; Pellegrini, L.; Zaccolo, M.; Bortolozzi, M.; Swietach, P.; et al. Phosphatases control PKA-dependent functional microdomains at the outer mi-tochondrial membrane. Proc. Natl. Acad. Sci. USA 2018, 115, e6497–e6506. [Google Scholar] [CrossRef]
- Prasad, A.M.; Inesi, G. Effects of thapsigargin and phenylephrine on calcineurin and protein kinase C signaling functions in cardiac myocytes. Am. J. Physiol. Physiol. 2009, 296, C992–C1002. [Google Scholar] [CrossRef][Green Version]
- Bers, D.M. Calcium Cycling and Signaling in Cardiac Myocytes. Annu. Rev. Physiol. 2008, 70, 23–49. [Google Scholar] [CrossRef]
- Müller, F.U.; Bokník, P.; Knapp, J.; Linck, B.; Lüss, H.; Neumann, J.; Schmitz, W. Activation and inactivation of cAMP-response element-mediated gene transcription in cardiac myocytes. Cardiovasc. Res. 2001, 52, 95–102. [Google Scholar] [CrossRef]
- Sassone-Corsi, P. The cyclic AMP pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011148. [Google Scholar] [CrossRef] [PubMed]
- Floras, J.S. Sympathetic activation in human heart failure: Diverse mechanisms, therapeutic opportunities. Acta Physiol. Scand. 2003, 177, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Corbin, J.D.; Sugden, P.H.; Lincoln, T.M.; Keely, S.L. Compartmentalization of adenosine 3’:5’-monophosphate and adenosine 3’:5’-monophosphate-dependent protein kinase in heart tissue. J. Biol. Chem. 1977, 252, 3854–3861. [Google Scholar] [CrossRef]
- Hayes, J.S.; Brunton, L.L.; Mayer, S.E. Selective activation of particulate cAMP-dependent protein kinase by isoproterenol and prostaglandin E1. J. Biol. Chem. 1980, 255, 5113–5119. [Google Scholar] [CrossRef]
- Hayes, J.S.; Brunton, L.L. Functional compartments in cyclic nucleotide action. J. Cycl. Nucleotide Res. 1982, 8, 1–16. [Google Scholar]
- Wong, W.; Scott, J.D. AKAP signalling complexes: Focal points in space and time. Nat. Rev. Mol. Cell Biol. 2004, 5, 959–970. [Google Scholar] [CrossRef]
- Zhang, J.; Ma, Y.; Taylor, S.S.; Tsien, R.Y. Genetically encoded reporters of protein kinase A activity reveal impact of substrate tethering. Proc. Natl. Acad. Sci. USA 2001, 98, 14997–15002. [Google Scholar] [CrossRef]
- Beene, D.L.; Scott, J.D. A-kinase anchoring proteins take shape. Curr. Opin. Cell Biol. 2007, 19, 192–198. [Google Scholar] [CrossRef]
- Ruehr, M.L.; Russell, M.A.; Bond, M. A-kinase anchoring protein targeting of protein kinase A in the heart. J. Mol. Cell. Cardiol. 2004, 37, 653–665. [Google Scholar] [CrossRef]
- Lygren, B.; Carlson, C.R.; Santamaria, K.; Lissandron, V.; McSorley, T.; Litzenberg, J.; Lorenz, D.; Wiesner, B.; Rosenthal, W.; Zaccolo, M.; et al. AKAP complex regulates Ca2+ re-uptake into heart sarcoplasmic reticulum. EMBO Rep. 2007, 8, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Lin, R.Y.; Rubin, C.S. Organelle-specific targeting of protein kinase AII (PKAII). Molecular and in situ characterization of murine A kinase anchor proteins that recruit regulatory subunits of PKAII to the cytoplasmic surface of mitochondria. J. Biol. Chem. 1997, 272, 15247–15257. [Google Scholar] [CrossRef] [PubMed]
- Feliciello, A.; Rubin, C.S.; Avvedimento, E.V.; Gottesman, M.E. Expression of A Kinase Anchor Protein 121 Is Regulated by Hormones in Thyroid and Testicular Germ Cells. J. Biol. Chem. 1998, 273, 23361–23366. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.J.-S.; Durick, K.; Weiner, J.; Chun, J.; Taylor, S.S. Identification of a Novel Protein Kinase A Anchoring Protein That Binds Both Type I and Type II Regulatory Subunits. J. Biol. Chem. 1997, 272, 8057–8064. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.-Y.; Moss, S.B.; Rubin, C.S. Characterization of S-AKAP84, a Novel Developmentally Regulated A Kinase Anchor Protein of Male Germ Cells. J. Biol. Chem. 1995, 270, 27804–27811. [Google Scholar] [CrossRef] [PubMed]
- Steen, R.L.; Martins, S.; Taskén, K.; Collas, P. Recruitment of Protein Phosphatase 1 to the Nuclear Envelope by a-Kinase Anchoring Protein Akap149 Is a Prerequisite for Nuclear Lamina Assembly. J. Cell Biol. 2000, 150, 1251–1262. [Google Scholar] [CrossRef] [PubMed]
- Zaccolo, M. cAMP signal transduction in the heart: Understanding spatial control for the development of novel therapeutic strategies. Br. J. Pharmacol. 2009, 158, 50–60. [Google Scholar] [CrossRef]
- Papa, S.; Sardanelli, A.M.; Scacco, S.; Technikova-Dobrova, Z. cAMP-dependent protein kinase and phosphoproteins in mammalian mitochondria. An extension of the cAMP-mediated intracellular signal transduction. FEBS Lett. 1999, 444, 245–249. [Google Scholar] [CrossRef]
- Merrill, R.; Dagda, R.; Dickey, A.; Cribbs, J.T.; Green, S.H.; Usachev, Y.; Strack, S. Mechanism of Neuroprotective Mitochondrial Remodeling by PKA/AKAP1. PLoS Biol. 2011, 9, e1000612. [Google Scholar] [CrossRef]
- Carlucci, A.; Lignitto, L.; Feliciello, A. Control of mitochondria dynamics and oxidative metabolism by cAMP, AKAPs and the proteasome. Trends Cell Biol. 2008, 18, 604–613. [Google Scholar] [CrossRef]
- Piccoli, C.; Scacco, S.; Bellomo, F.; Signorile, A.; Iuso, A.; Boffoli, D.; Scrima, R.; Capitanio, N.; Papa, S. cAMP controls oxygen metabolism in mammalian cells. FEBS Lett. 2006, 580, 4539–4543. [Google Scholar] [CrossRef]
- Livigni, A.; Scorziello, A.; Agnese, S.; Adornetto, A.; Carlucci, A.; Garbi, C.; Castaldo, I.; Annunziato, L.; Avvedimento, E.V.; Feliciello, A. Mitochondrial AKAP121 Links cAMP and src Signaling to Oxidative Metabolism. Mol. Biol. Cell 2006, 17, 263–271. [Google Scholar] [CrossRef]
- Bender, E.; Kadenbach, B. The allosteric ATP-inhibition of cytochrome c oxidase activity is reversibly switched on by cAMP-dependent phosphorylation. FEBS Lett. 2000, 466, 130–134. [Google Scholar] [CrossRef]
- De Rasmo, D.; Micelli, L.; Santeramo, A.; Signorile, A.; Lattanzio, P.; Papa, S. cAMP regulates the functional activity, coupling efficiency and structural organization of mammalian FOF1 ATP synthase. Biochim. Biophys. Acta 2016, 1857, 350–358. [Google Scholar] [CrossRef] [PubMed]
- García-Bermúdez, J.; Sánchez-Aragó, M.; Soldevilla, B.; del Arco, A.; Nuevo-Tapioles, C.; Cuezva, J.M. PKA Phosphorylates the ATPase Inhibitory Factor 1 and Inactivates Its Capacity to Bind and Inhibit the Mitochondrial H+-ATP Synthase. Cell Rep. 2015, 12, 2143–2155. [Google Scholar] [CrossRef]
- De Rasmo, D.; Panelli, D.; Sardanelli, A.M.; Papa, S. cAMP-dependent protein kinase regulates the mitochondrial import of the nuclear encoded NDUFS4 subunit of complex I. Cell. Signal. 2008, 20, 989–997. [Google Scholar] [CrossRef] [PubMed]
- De Rasmo, D.; Palmisano, G.; Scacco, S.; Technikova-Dobrova, Z.; Panelli, D.; Cocco, T.M.; Sardanelli, A.M.; Gnoni, A.; Micelli, L.; Trani, A.; et al. Phosphorylation pattern of the NDUFS4 subunit of complex I of the mammalian respiratory chain. Mitochondrion 2010, 10, 464–471. [Google Scholar] [CrossRef] [PubMed]
- De Rasmo, D.; Signorile, A.; Santeramo, A.; Larizza, M.; Lattanzio, P.; Capitanio, G.; Papa, S. Intramitochondrial adenylyl cyclase controls the turnover of nuclear-encoded subunits and activity of mammalian complex I of the respiratory chain. Biochim. Biophys. Acta BBA Bioenerg. 2014, 1853, 183–191. [Google Scholar] [CrossRef]
- Robin, M.-A.; Prabu, S.K.; Raza, H.; Anandatheerthavarada, H.K.; Avadhani, N.G. Phosphorylation Enhances Mitochondrial Targeting of GSTA4-4 through Increased Affinity for Binding to Cytoplasmic Hsp70. J. Biol. Chem. 2003, 278, 18960–18970. [Google Scholar] [CrossRef]
- Dagda, R.K.; Gusdon, A.M.; Pien, I.; Strack, S.; Green, S.; Li, C.; Van Houten, B.; Cherra, S.J.; Chu, C.T. Mitochondrially localized PKA reverses mitochondrial pathology and dysfunction in a cellular model of Parkinson’s disease. Cell Death Differ. 2011, 18, 1914–1923. [Google Scholar] [CrossRef]
- Dickey, A.S.; Strack, S. PKA/AKAP1 and PP2A/Bβ2 regulate neuronal morphogenesis via Drp1 phosphorylation and mitochondrial bioenergetics. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 15716–15726. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, Y.; Kawaguchi, Y.; Fujimoto, T.; Kanayama, N.; Magari, M.; Tokumitsu, H. Differential AMP-activated Protein Kinase (AMPK) Recognition Mechanism of Ca2+/Calmodulin-dependent Protein Kinase Kinase Isoforms. J. Biol. Chem. 2016, 291, 13802–13808. [Google Scholar] [CrossRef] [PubMed]
- Gowans, G.J.; Hawley, S.A.; Ross, F.A.; Hardie, D.G. AMP Is a True Physiological Regulator of AMP-Activated Protein Kinase by Both Allosteric Activation and Enhancing Net Phosphorylation. Cell Metab. 2013, 18, 556–566. [Google Scholar] [CrossRef] [PubMed]
- Hawley, S.A.; Boudeau, J.; Reid, J.L.; Mustard, K.J.; Udd, L.; Mäkelä, T.P.; Alessi, D.R.; Hardie, D.G. Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J. Biol. 2003, 2, 28. [Google Scholar] [CrossRef] [PubMed]
- Hawley, S.A.; Pan, D.A.; Mustard, K.J.; Ross, L.; Bain, J.; Edelman, A.M.; Frenguelli, B.G.; Hardie, D.G. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005, 2, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Shaw, R.J.; Kosmatka, M.; Bardeesy, N.; Hurley, R.L.; Witters, L.A.; DePinho, R.; Cantley, L.C. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc. Natl. Acad. Sci. USA 2004, 101, 3329–3335. [Google Scholar] [CrossRef]
- Wu, S.; Zou, M.H. AMPK, Mitochondrial Function, and Cardiovascular Disease. Int. J. Mol. Sci. 2020, 21, 4987. [Google Scholar] [CrossRef]
- Toyama, E.Q.; Herzig, S.; Courchet, J.; Lewis, T.L.; Losón, O.C.; Hellberg, K.; Young, N.P.; Chen, H.; Polleux, F.; Chan, D.C.; et al. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science 2016, 351, 275–281. [Google Scholar] [CrossRef]
- Egan, D.F.; Shackelford, D.B.; Mihaylova, M.M.; Gelino, S.; Kohnz, R.A.; Mair, W.; Vasquez, D.S.; Joshi, A.; Gwinn, D.M.; Taylor, R.; et al. Phosphorylation of ULK1 (hATG1) by AMP-Activated Protein Kinase Connects Energy Sensing to Mitophagy. Science 2010, 331, 456–461. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nature 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Wang, B.; Nie, J.; Wu, L.; Hu, Y.; Wen, Z.; Dong, L.; Zou, M.H.; Chen, C.; Wang, D.W. AMPKα2 Protects Against the Development of Heart Failure by Enhancing Mitophagy via PINK1 Phosphorylation. Circ. Res. 2018, 122, 712–729. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, N.J.; Parker, B.L.; Chaudhuri, R.; Fisher-Wellman, K.H.; Kleinert, M.; Humphrey, S.J.; Yang, P.; Holliday, M.; Trefely, S.; Fazakerley, D.J.; et al. Global Phosphoproteomic Analysis of Human Skeletal Muscle Reveals a Network of Exercise-Regulated Kinases and AMPK Substrates. Cell Metab. 2015, 22, 922–935. [Google Scholar] [CrossRef] [PubMed]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2017, 19, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Rabinovitch, R.C.; Samborska, B.; Faubert, B.; Ma, E.H.; Gravel, S.-P.; Andrzejewski, S.; Raissi, T.C.; Pause, A.; St.-Pierre, J.; Jones, R.G. AMPK Maintains Cellular Metabolic Homeostasis through Regulation of Mitochondrial Reactive Oxygen Species. Cell Rep. 2017, 21, 1–9. [Google Scholar] [CrossRef]
- Naruse, K.; King, G.L. Protein Kinase C and Myocardial Biology and Function. Circ. Res. 2000, 86, 1104–1106. [Google Scholar] [CrossRef]
- Malhotra, A.; Kang, B.P.S.; Opawumi, D.; Belizaire, W.; Meggs, L.G. Molecular biology of protein kinase C signaling in cardiac myocytes. Mol. Cell. Biochem. 2001, 225, 97–107. [Google Scholar] [CrossRef]
- Bogoyevitch, M.A.; Parker, P.J.; Sugden, P.H. Characterization of protein kinase C isotype expression in adult rat heart. Protein kinase C-epsilon is a major isotype present, and it is activated by phorbol esters, epinephrine, and endothelin. Circ. Res. 1993, 72, 757–767. [Google Scholar] [CrossRef]
- Rybin, V.O.; Steinberg, S.F. Protein kinase C isoform expression and regulation in the developing rat heart. Circ. Res. 1994, 74, 299–309. [Google Scholar] [CrossRef]
- Ping, P.; Zhang, J.; Qiu, Y.; Tang, X.L.; Manchikalapudi, S.; Cao, X.; Bolli, R. Ischemic preconditioning induces selective translocation of protein kinase C isoforms epsilon and eta in the heart of conscious rabbits without subcellular redistribution of total protein kinase C activity. Circ. Res. 1997, 81, 404–414. [Google Scholar] [CrossRef]
- Baines, C.P.; Song, C.X.; Zheng, Y.T.; Wang, G.W.; Zhang, J.; Wang, O.L.; Guo, Y.; Bolli, R.; Cardwell, E.M.; Ping, P. Protein kinase Cepsilon interacts with and inhibits the permeability transition pore in cardiac mitochondria. Circ. Res. 2003, 92, 873–880. [Google Scholar] [CrossRef]
- Scruggs, S.B.; Wang, D.; Ping, P. PRKCE gene encoding protein kinase C-epsilon—Dual roles at sarcomeres and mitochondria in cardiomyocytes. Gene 2016, 590, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Robia, S.L.; Ghanta, J.; Robu, V.G.; Walker, J.W. Localization and Kinetics of Protein Kinase C-Epsilon Anchoring in Cardiac Myocytes. Biophys. J. 2001, 80, 2140–2151. [Google Scholar] [CrossRef][Green Version]
- Mochly-Rosen, D. Localization of protein kinases by anchoring proteins: A theme in signal transduction. Science 1995, 268, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.O.; Karliner, J.S.; Mochly-Rosen, D. A selective epsilon-protein kinase C antagonist inhibits protection of cardiac myocytes from hypoxia-induced cell death. J. Biol. Chem. 1997, 272, 30945–30951. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.S.; Cohen, M.V.; Mochly-Rosen, D.; Downey, J.M. Protein kinase C-epsilon is responsible for the protection of preconditioning in rabbit cardiomyocytes. J. Mol. Cell. Cardiol. 1999, 31, 1937–1948. [Google Scholar] [CrossRef]
- Gregory, K.N.; Hahn, H.; Haghighi, K.; Marreez, Y.; Odley, A.; Dorn, G.W.; Kranias, E.G. Increased particulate partitioning of PKC epsilon reverses susceptibility of phospholamban knockout hearts to ischemic injury. J. Mol. Cell. Cardiol. 2004, 36, 313–318. [Google Scholar] [CrossRef]
- Ping, P.; Zhang, J.; Pierce, W.M.; Bolli, R. Functional proteomic analysis of protein kinase C epsilon signaling complexes in the normal heart and during cardioprotection. Circ. Res. 2001, 88, 59–62. [Google Scholar] [CrossRef]
- Edmondson, R.D.; Vondriska, T.M.; Biederman, K.J.; Zhang, J.; Jones, R.C.; Zheng, Y.; Allen, D.L.; Xiu, J.X.; Cardwell, E.M.; Pisano, M.R.; et al. Protein kinase C epsilon signaling complexes include metabolism- and transcription/translation-related proteins: Complimentary separation techniques with LC/MS/MS. Mol. Cell. Proteom. MCP 2002, 1, 421–433. [Google Scholar] [CrossRef]
- Chen, C.H.; Budas, G.R.; Churchill, E.N.; Disatnik, M.H.; Hurley, T.D.; Mochly-Rosen, D. Activation of aldehyde dehydro-genase-2 reduces ischemic damage to the heart. Science 2008, 321, 1493–1495. [Google Scholar] [CrossRef]
- Ogbi, M.; Johnson, J.A. Protein kinase Cepsilon interacts with cytochrome c oxidase subunit IV and enhances cytochrome c oxidase activity in neonatal cardiac myocyte preconditioning. Biochem. J. 2006, 393, 191–199. [Google Scholar] [CrossRef]
- Budas, G.; Costa, H.M.; Ferreira, J.C.; Teixeira da Silva Ferreira, A.; Perales, J.; Krieger, J.E.; Mochly-Rosen, D.; Schechtman, D. Identification of εPKC targets during cardiac ischemic injury. Circ. J. Off. J. Jpn. Circ. Soc. 2012, 76, 1476–1485. [Google Scholar]
- Budas, G.R.; Churchill, E.N.; Disatnik, M.H.; Sun, L.; Mochly-Rosen, D. Mitochondrial import of PKCepsilon is mediated by HSP90: A role in cardioprotection from ischaemia and reperfusion injury. Cardiovasc. Res. 2010, 88, 83–92. [Google Scholar] [CrossRef]
- Yang, Z.; Sun, W.; Hu, K. Molecular mechanism underlying adenosine receptor-mediated mitochondrial targeting of protein kinase C. Biochim. Biophys. Acta BBA Bioenerg. 2011, 1823, 950–958. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Sivaprasad, U.; Huang, J.; Shankar, E.; Morrow, S.; Basu, A. Protein kinase C-epsilon protects MCF-7 cells from TNF-mediated cell death by inhibiting Bax translocation. Apoptosis Int. J. Program. Cell Death 2007, 12, 1893–1900. [Google Scholar] [CrossRef]
- McJilton, M.A.; Van Sikes, C.; Wescott, G.G.; Wu, D.; Foreman, T.L.; Gregory, C.W.; Weidner, D.A.; Harris Ford, O.; Morgan Lasater, A.; Mohler, J.L.; et al. Protein kinase Cepsilon interacts with Bax and promotes survival of human prostate cancer cells. Oncogene 2003, 22, 7958–7968. [Google Scholar] [CrossRef] [PubMed]
- Tsai, K.-L.; Liang, H.-J.; Yang, Z.-D.; Lue, S.-I.; Yang, S.-L.; Hsu, C. Early inactivation of PKCε associates with late mitochondrial translocation of Bad and apoptosis in ventricle of septic rat. J. Surg. Res. 2013, 186, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Lam, P.Y.; Han, D.; Cadenas, E. c-Jun N-terminal kinase regulates mitochondrial bioenergetics by modulating pyruvate dehydrogenase activity in primary cortical neurons. J. Neurochem. 2008, 104, 325–335. [Google Scholar] [CrossRef]
- Zhou, Q.; Lam, P.Y.; Han, D.; Cadenas, E. Activation of c-Jun-N-terminal kinase and decline of mitochondrial pyruvate de-hydrogenase activity during brain aging. FEBS Lett. 2009, 583, 1132–1140. [Google Scholar] [CrossRef]
- Chambers, J.; LoGrasso, P.V. Mitochondrial c-Jun N-terminal Kinase (JNK) Signaling Initiates Physiological Changes Resulting in Amplification of Reactive Oxygen Species Generation. J. Biol. Chem. 2011, 286, 16052–16062. [Google Scholar] [CrossRef]
- Win, S.; Than, T.A.; Fernández-Checa, J.C.; Kaplowitz, N. JNK interaction with Sab mediates ER stress induced inhibition of mitochondrial respiration and cell death. Cell Death Dis. 2014, 5, e989. [Google Scholar] [CrossRef]
- Jang, S.; Javadov, S. Inhibition of JNK Aggravates the Recovery of Rat Hearts after Global Ischemia: The Role of Mitochondrial JNK. PLoS ONE 2014, 9, e113526. [Google Scholar] [CrossRef] [PubMed]
- Win, S.; Than, T.A.; Min, R.W.M.; Aghajan, M.; Kaplowitz, N. c-Jun N-terminal kinase mediates mouse liver injury through a novel Sab (SH3BP5)-dependent pathway leading to inactivation of intramitochondrial Src. Hepatology 2016, 63, 1987–2003. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Li, X.; Chang, Y.; Zhao, L.; Liu, B.; Wei, S.; Xu, F.; Zhang, Y.; Chen, Y. Aldehyde Dehydrogenase-2 Attenuates Myocardial Remodeling and Contractile Dysfunction Induced by a High-Fat Diet. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2018, 48, 1843–1853. [Google Scholar] [CrossRef] [PubMed]
- Schulz, E.; Dopheide, J.; Schuhmacher, S.; Thomas, S.R.; Chen, K.; Daiber, A.; Wenzel, P.; Münzel, T.; Keaney, J.F. Suppression of the JNK pathway by induction of a metabolic stress response prevents vascular injury and dysfunction. Circulation 2008, 118, 1347–1357. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, O.S.; Deindl, S.; Sung, R.-J.; Nairn, A.; Kuriyan, J. Structure of the Autoinhibited Kinase Domain of CaMKII and SAXS Analysis of the Holoenzyme. Cell 2005, 123, 849–860. [Google Scholar] [CrossRef]
- Grimm, M.; Brown, J.H. Beta-adrenergic receptor signaling in the heart: Role of CaMKII. J. Mol. Cell. Cardiol. 2010, 48, 322–330. [Google Scholar] [CrossRef]
- Strack, S.; Barban, M.A.; Wadzinski, B.E.; Colbran, R.J. Differential inactivation of postsynaptic density-associated and soluble Ca2+/calmodulin-dependent protein kinase II by protein phosphatases 1 and 2A. J. Neurochem. 2002, 68, 2119–2128. [Google Scholar] [CrossRef]
- Erickson, J.R.; Joiner, M.-L.A.; Guan, X.; Kutschke, W.; Yang, J.; Oddis, C.V.; Bartlett, R.K.; Lowe, J.S.; O’Donnell, S.E.; Aykin-Burns, N.; et al. A Dynamic Pathway for Calcium-Independent Activation of CaMKII by Methionine Oxidation. Cell 2008, 133, 462–474. [Google Scholar] [CrossRef]
- Erickson, J.R.; Nichols, C.B.; Uchinoumi, H.; Stein, M.L.; Bossuyt, J.; Bers, D.M. S-Nitrosylation Induces Both Autonomous Activation and Inhibition of Calcium/Calmodulin-dependent Protein Kinase II δ. J. Biol. Chem. 2015, 290, 25646–25656. [Google Scholar] [CrossRef]
- Erickson, J.R.; Pereira, L.; Wang, L.; Han, G.; Ferguson, A.; Dao, K.; Copeland, R.J.; Despa, F.; Hart, G.W.; Ripplinger, C.; et al. Diabetic hyperglycaemia activates CaMKII and arrhythmias by O-linked glycosylation. Nature 2013, 502, 372–376. [Google Scholar] [CrossRef]
- Backs, J.; Backs, T.; Neef, S.; Kreusser, M.M.; Lehmann, L.H.; Patrick, D.M.; Grueter, C.E.; Qi, X.; Richardson, J.A.; Hill, J.A.; et al. The delta isoform of CaM kinase II is required for pathological cardiac hypertrophy and remodeling after pressure overload. Proc. Natl. Acad. Sci. USA 2009, 106, 2342–2347. [Google Scholar] [CrossRef] [PubMed]
- Kirchhefer, U.; Schmitz, W.; Scholz, H.; Neumann, J. Activity of cAMP-dependent protein kinase and Ca2+/calmodulin-dependent protein kinase in failing and nonfailing human hearts. Cardiovasc. Res. 1999, 42, 254–261. [Google Scholar] [CrossRef]
- Hoch, B.; Meyer, R.; Hetzer, R.; Krause, E.G.; Karczewski, P. Identification and expression of delta-isoforms of the multi-functional Ca2+/calmodulin-dependent protein kinase in failing and nonfailing human myocardium. Circ. Res. 1999, 84, 713–721. [Google Scholar] [CrossRef] [PubMed]
- Rajtik, T.; Goncalvesova, E.; Varga, Z.V.; Leszek, P.; Kusmierczyk, M.; Hulman, M.; Kyselovic, J.; Ferdinandy, P.; Adameova, A. Posttranslational modifications of calcium/calmodulin-dependent protein kinase IIδ and its downstream signaling in human failing hearts. Am. J. Transl. Res. 2017, 9, 3573–3585. [Google Scholar] [PubMed]
- Westenbrink, B.D.; Ling, H.; Divakaruni, A.S.; Gray, C.B.; Zambon, A.C.; Dalton, N.D.; Peterson, K.L.; Gu, Y.; Matkovich, S.J.; Murphy, A.N.; et al. Mitochondrial reprogramming induced by CaMKIIδ mediates hypertrophy decompensation. Circ. Res. 2015, 116, e28–e39. [Google Scholar] [CrossRef]
- Luczak, E.D.; Wu, Y.; Granger, J.M.; Joiner, M.-L.A.; Wilson, N.R.; Gupta, A.; Umapathi, P.; Murphy, K.R.; Gaido, O.E.R.; Sabet, A.; et al. Mitochondrial CaMKII causes adverse metabolic reprogramming and dilated cardiomyopathy. Nat. Commun. 2020, 11, 4416. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chaanine, A.H. Metabolic Remodeling and Implicated Calcium and Signal Transduction Pathways in the Pathogenesis of Heart Failure. Int. J. Mol. Sci. 2021, 22, 10579. https://doi.org/10.3390/ijms221910579
Chaanine AH. Metabolic Remodeling and Implicated Calcium and Signal Transduction Pathways in the Pathogenesis of Heart Failure. International Journal of Molecular Sciences. 2021; 22(19):10579. https://doi.org/10.3390/ijms221910579
Chicago/Turabian StyleChaanine, Antoine H. 2021. "Metabolic Remodeling and Implicated Calcium and Signal Transduction Pathways in the Pathogenesis of Heart Failure" International Journal of Molecular Sciences 22, no. 19: 10579. https://doi.org/10.3390/ijms221910579
APA StyleChaanine, A. H. (2021). Metabolic Remodeling and Implicated Calcium and Signal Transduction Pathways in the Pathogenesis of Heart Failure. International Journal of Molecular Sciences, 22(19), 10579. https://doi.org/10.3390/ijms221910579