
Targeted and Non-Targeted Mechanisms for Killing Hypoxic Tumour Cells—Are There New Avenues for Treatment?

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
Background to Tumour Hypoxia
2. Current Approaches to Radiosensitizing Hypoxic Tumour Cells
2.1. Tumour Hypoxia Imaging via PET for Guiding Carbogen Breathing Therapy
2.1.1. Background
2.1.2. Mechanism of Action
2.1.3. Current Studies and Future Considerations
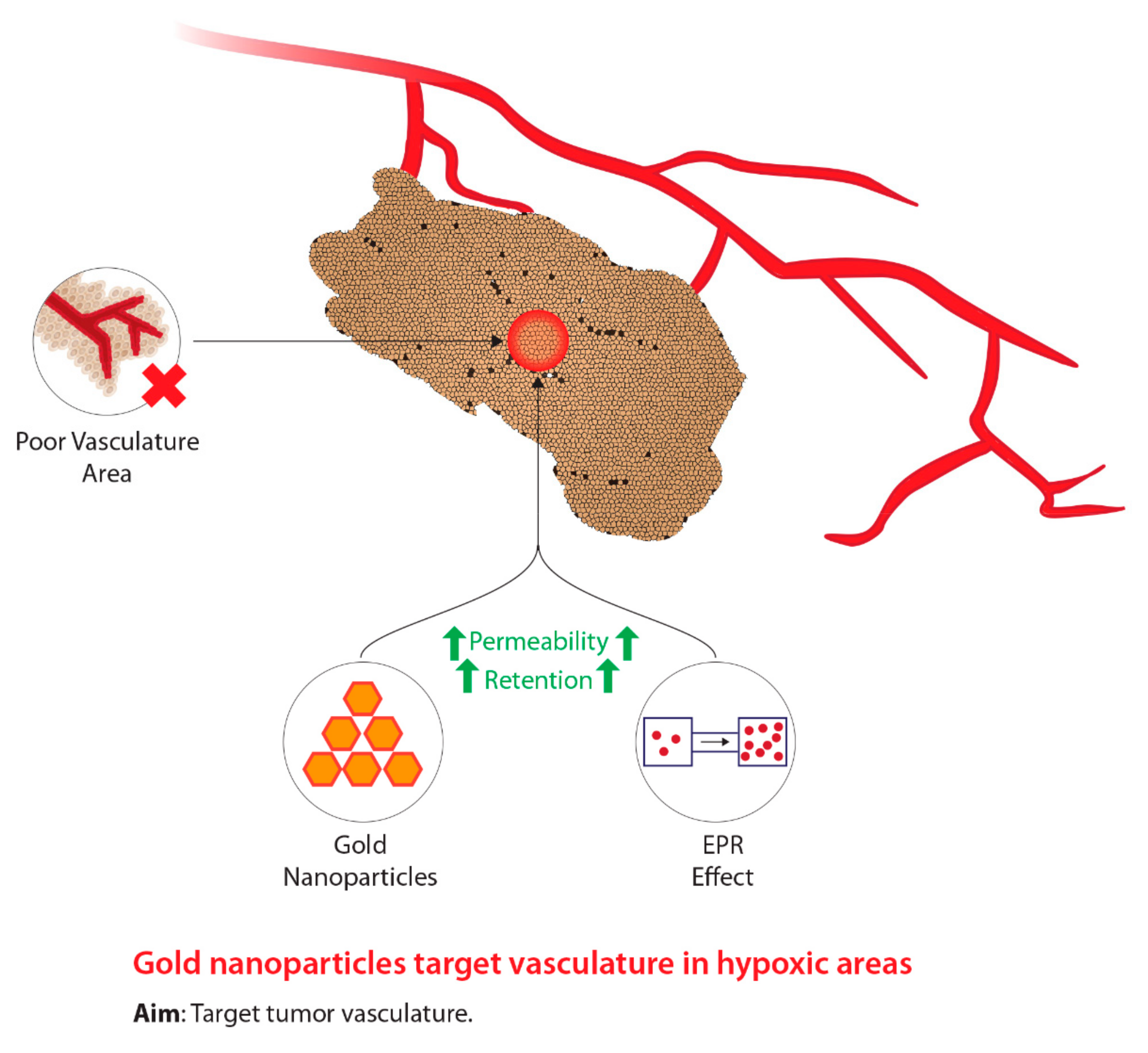
2.2. Gold Nanoparticles
2.2.1. Background
2.2.2. Mechanism of Action
2.2.3. Current Studies and Future Considerations
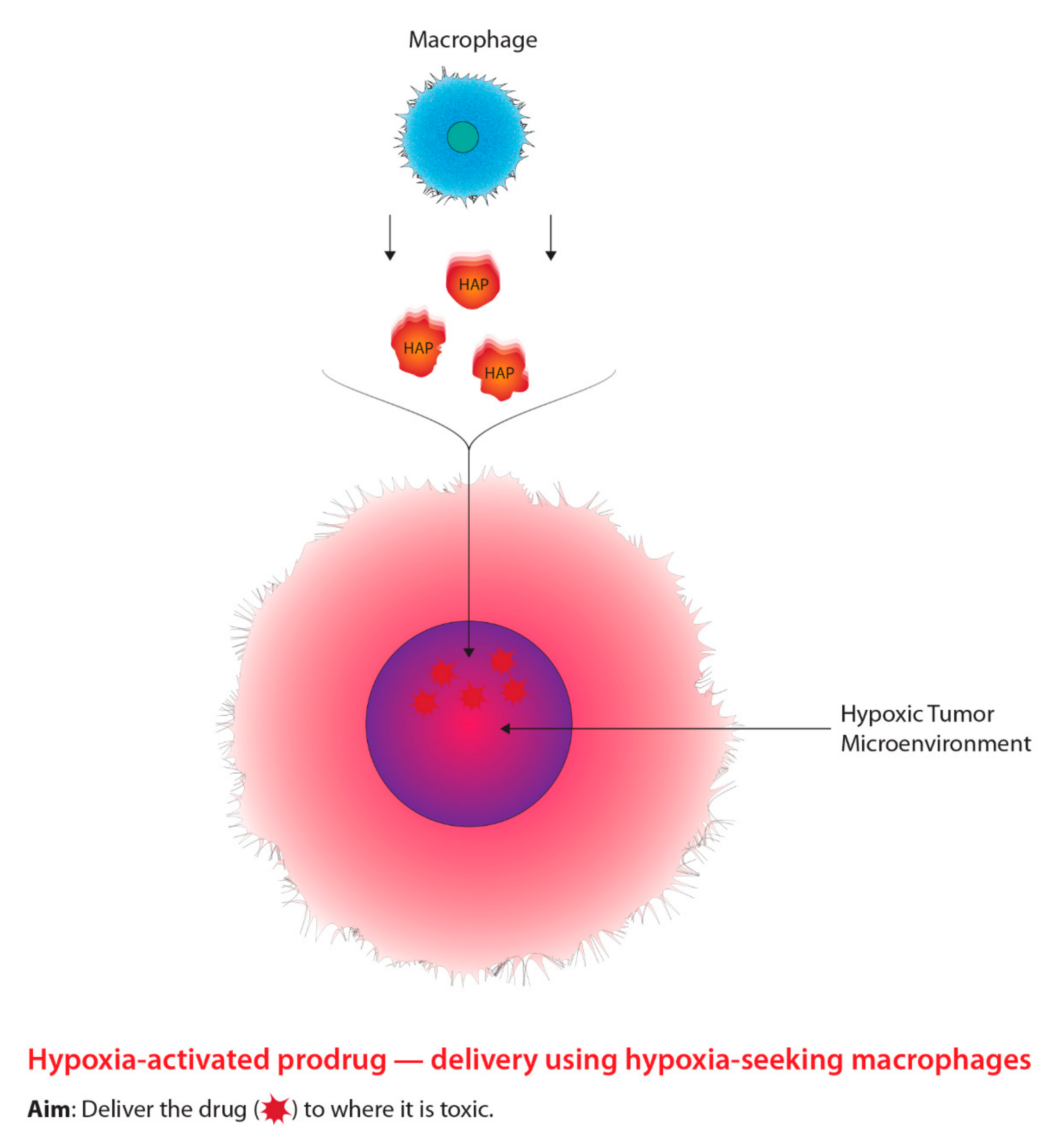
2.3. Macrophage-Mediated Drug Delivery: HAPs
2.3.1. Background
2.3.2. Mechanism of Action
2.3.3. Current Studies and Future Developments
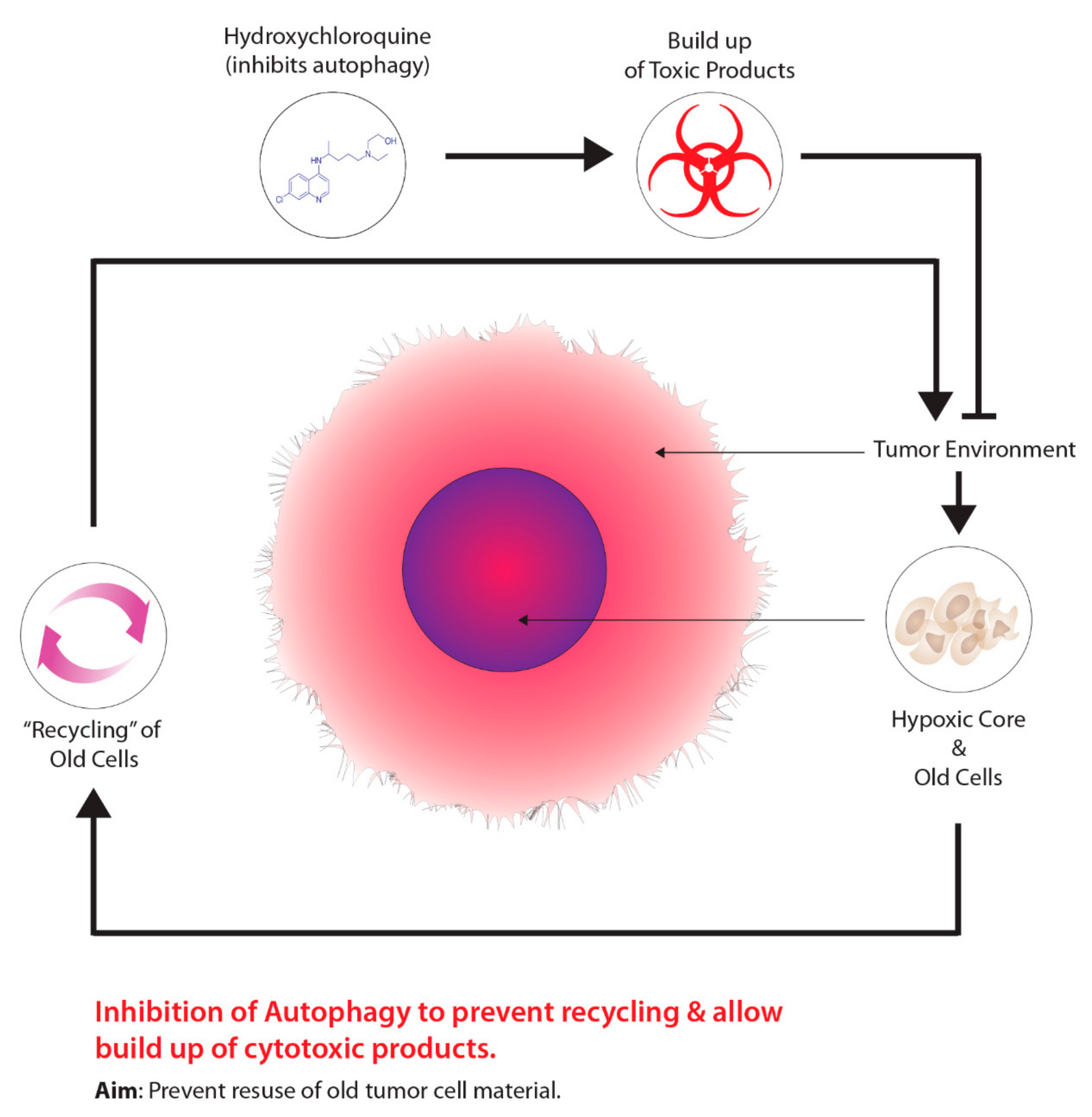
2.4. Autophagy and Tumour Metabolism
2.4.1. Background
2.4.2. Mechanisms of Action
2.4.3. Possible Involvement of Non-Targeted Effects
2.4.4. Current Studies and Future Developments
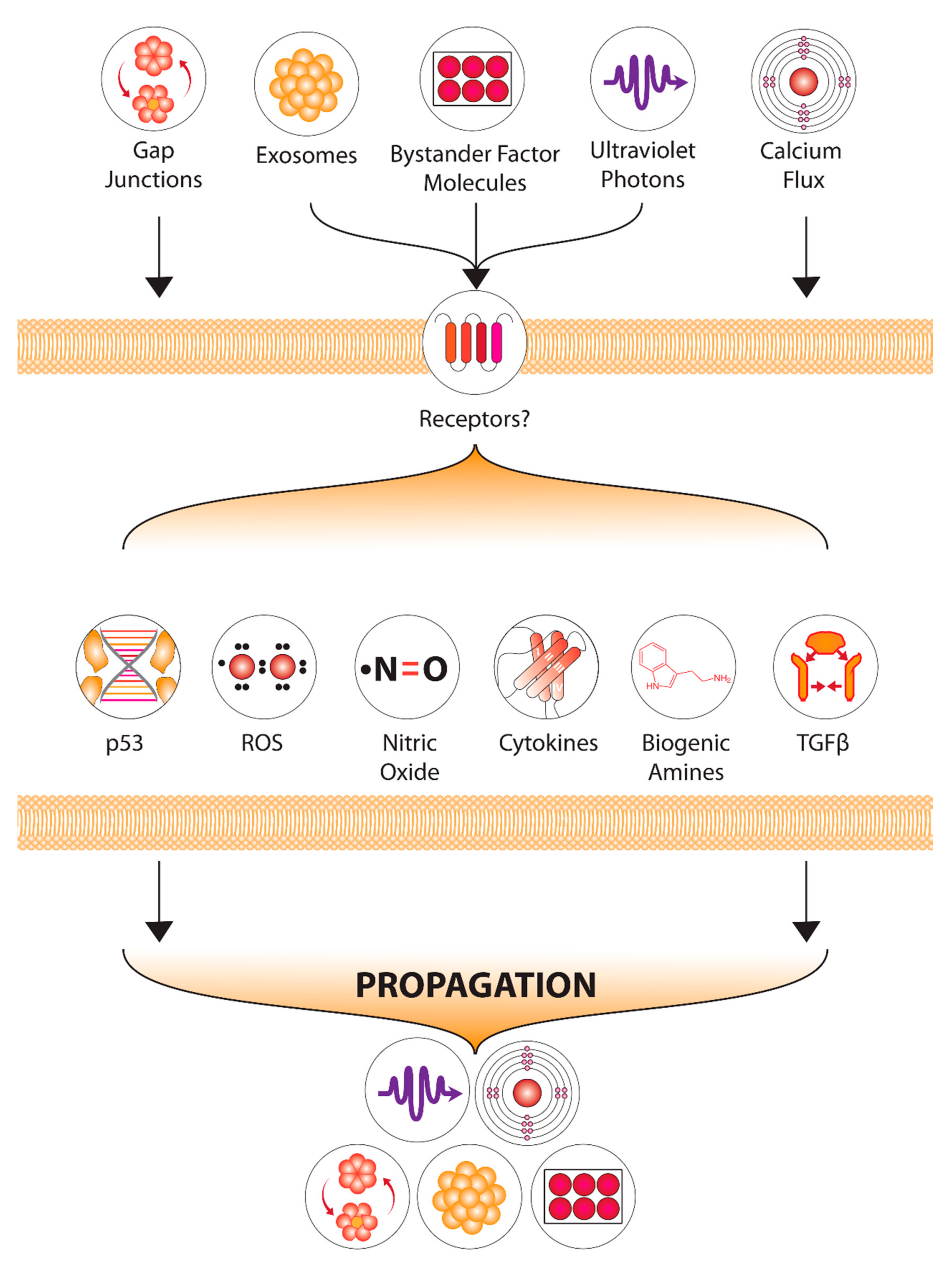
2.5. Non-Targeted Effects as a “Target”
2.5.1. Background
2.5.2. Non-Targeted Effects Mechanisms
2.5.3. Current Studies and Future Developments
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The Role of Hypoxia in Cancer Progression, Angiogenesis, Metastasis, and Resistance to Therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, B.S.; Horsman, M.R. Tumour Hypoxia: Impact on Radiation Therapy and Molecular Pathways. Front. Oncol. 2020, 10, 562. [Google Scholar] [CrossRef] [PubMed]
- Siemann, D.W.; Horsman, M.R. Modulation of the Tumour Vasculature and Oxygenation to Improve Therapy. Pharmacol. Ther. 2015, 153, 107–124. [Google Scholar] [CrossRef] [PubMed]
- Horsman, M.R.; Vaupel, P. Pathophysiological Basis for the Formation of the Tumour Microenvironment. Front. Oncol. 2016, 6, 66. [Google Scholar] [CrossRef]
- Folkman, J. How Is Blood Vessel Growth Regulated in Normal and Neoplastic Tissue? G.H.A. Clowes Memorial Award Lecture. Cancer Res. 1986, 46, 467–473. [Google Scholar]
- Bergers, G.; Benjamin, L.E. Tumourigenesis and the Angiogenic Switch. Nat. Rev. Cancer 2003, 3, 401–410. [Google Scholar] [CrossRef]
- Thomlinson, R.H.; Gray, L.H. The Histological Structure of Some Human Lung Cancers and the Possible Implications for Radiotherapy. Br. J. Cancer 1955, 9, 539–549. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia, Clonal Selection, and the Role of HIF-1 in Tumour Progression. Crit. Rev. Biochem. Mol. Biol. 2000, 35, 71–103. [Google Scholar] [CrossRef]
- Brown, J.M. The Hypoxic Cell: A Target for Selective Cancer Therapy--Eighteenth Bruce F. Cain Memorial Award Lecture. Cancer Res. 1999, 59, 5863–5870. [Google Scholar]
- Tannock, I.; Guttman, P. Response of Chinese Hamster Ovary Cells to Anticancer Drugs under Aerobic and Hypoxic Conditions. Br. J. Cancer 1981, 43, 245–248. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Teicher, B.A.; Holden, S.A.; al-Achi, A.; Herman, T.S. Classification of Antineoplastic Treatments by Their Differential Toxicity toward Putative Oxygenated and Hypoxic Tumour Subpopulations in Vivo in the FSaIIC Murine Fibrosarcoma. Cancer Res. 1990, 50, 3339–3344. [Google Scholar]
- Graham, K.; Unger, E. Overcoming Tumour Hypoxia as a Barrier to Radiotherapy, Chemotherapy and Immunotherapy in Cancer Treatment. Int. J. Nanomed. 2018, 13, 6049–6058. [Google Scholar] [CrossRef]
- Rockwell, S.; Dobrucki, I.T.; Kim, E.Y.; Marrison, S.T.; Vu, V.T. Hypoxia and Radiation Therapy: Past History, Ongoing Research, and Future Promise. Curr. Mol. Med. 2009, 9, 442–458. [Google Scholar] [CrossRef]
- Höckel, M.; Vaupel, P. Tumour Hypoxia: Definitions and Current Clinical, Biologic, and Molecular Aspects. J. Natl. Cancer Inst. 2001, 93, 266–276. [Google Scholar] [CrossRef]
- Hall, E.J.; Giaccia, A.J. Radiobiology for the Radiologist, 7th ed.; Wolters Kluwer Health/Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2012; ISBN 978-1-60831-193-4. [Google Scholar]
- Mothersill, C.; Bristow, R.G.; Harding, S.M.; Smith, R.W.; Mersov, A.; Seymour, C.B. A Role for P53 in the Response of Bystander Cells to Receipt of Medium Borne Signals from Irradiated Cells. Int. J. Radiat. Biol. 2011, 87, 1120–1125. [Google Scholar] [CrossRef] [PubMed]
- Le, M.; Mothersill, C.E.; Seymour, C.B.; Rainbow, A.J.; McNeill, F.E. An Observed Effect of P53 Status on the Bystander Response to Radiation-Induced Cellular Photon Emission. Radiat. Res. 2017, 187, 169–185. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, K.; Kohshi, K.; Ishiuchi, S.; Matsushita, M.; Yoshimi, N.; Murayama, S. Old but New Methods in Radiation Oncology: Hyperbaric Oxygen Therapy. Int. J. Clin. Oncol. 2013, 18, 364–370. [Google Scholar] [CrossRef]
- Jain, K.K. Textbook of Hyperbaric Medicine; Springer International Publishing: Cham, Switzerland, 1953; ISBN 978-3-319-47138-9. [Google Scholar]
- Hartmann, K.A.; van der Kleij, A.J.; Carl, U.M.; Hulshof, M.C.; Willers, R.; Sminia, P. Effects of Hyperbaric Oxygen and Normobaric Carbogen on the Radiation Response of the Rat Rhabdomyosarcoma R1H. Int. J. Radiat. Oncol. Biol. Phys. 2001, 51, 1037–1044. [Google Scholar] [CrossRef]
- Churchill-Davidson, I. The Oxygen Effect in Radiotherapy—Historical Review. Hyperb. Oxyg. Radiat. Ther. Cancer 1968, 1, 1–15. [Google Scholar] [CrossRef]
- Bennett, M.H.; Feldmeier, J.; Smee, R.; Milross, C. Hyperbaric Oxygenation for Tumour Sensitisation to Radiotherapy. Cochrane Database Syst. Rev. 2018, 2018, CD005007. [Google Scholar] [CrossRef] [PubMed]
- Kohshi, K.; Kinoshita, Y.; Imada, H.; Kunugita, N.; Abe, H.; Terashima, H.; Tokui, N.; Uemura, S. Effects of Radiotherapy after Hyperbaric Oxygenation on Malignant Gliomas. Br. J. Cancer 1999, 80, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Moen, I.; Stuhr, L.E.B. Hyperbaric Oxygen Therapy and Cancer—A Review. Target. Oncol. 2012, 7, 233–242. [Google Scholar] [CrossRef]
- Kjellen, E.; Joiner, M.C.; Collier, J.M.; Johns, H.; Rojas, A. A Therapeutic Benefit from Combining Normobaric Carbogen or Oxygen with Nicotinamide in Fractionated X-Ray Treatments. Radiother. Oncol. J. Eur. Soc. Ther. Radiol. Oncol. 1991, 22, 81–91. [Google Scholar] [CrossRef]
- Tran, L.-B.-A.; Bol, A.; Labar, D.; Karroum, O.; Bol, V.; Jordan, B.; Grégoire, V.; Gallez, B. Potential Role of Hypoxia Imaging Using 18F-FAZA PET to Guide Hypoxia-Driven Interventions (Carbogen Breathing or Dose Escalation) in Radiation Therapy. Radiother. Oncol. 2014, 113, 204–209. [Google Scholar] [CrossRef]
- Harrison, L.B.; Chadha, M.; Hill, R.J.; Hu, K.; Shasha, D. Impact of Tumour Hypoxia and Anemia on Radiation Therapy Outcomes. Oncologist 2002, 7, 492–508. [Google Scholar] [CrossRef] [PubMed]
- Fleming, I.N.; Manavaki, R.; Blower, P.J.; West, C.; Williams, K.J.; Harris, A.L.; Domarkas, J.; Lord, S.; Baldry, C.; Gilbert, F.J. Imaging Tumour Hypoxia with Positron Emission Tomography. Br. J. Cancer 2015, 112, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Unterrainer, M.; Eze, C.; Ilhan, H.; Marschner, S.; Roengvoraphoj, O.; Schmidt-Hegemann, N.S.; Walter, F.; Kunz, W.G.; af Rosenschöld, P.M.; Jeraj, R.; et al. Recent Advances of PET Imaging in Clinical Radiation Oncology. Radiat. Oncol. 2020, 15, 88. [Google Scholar] [CrossRef]
- Chapman, J.D. Hypoxic Sensitizers—Implications for Radiation Therapy. N. Engl. J. Med. 1979, 301, 1429–1432. [Google Scholar] [CrossRef]
- Nunn, A.; Linder, K.; Strauss, H.W. Nitroimidazoles and Imaging Hypoxia. Eur. J. Nucl. Med. 1995, 22, 265–280. [Google Scholar] [CrossRef]
- Edwards, D.I. Nitroimidazole Drugs--Action and Resistance Mechanisms. I. Mechanisms of Action. J. Antimicrob. Chemother. 1993, 31, 9–20. [Google Scholar] [CrossRef]
- Lopci, E.; Grassi, I.; Chiti, A.; Nanni, C.; Cicoria, G.; Toschi, L.; Fonti, C.; Lodi, F.; Mattioli, S.; Fanti, S. PET Radiopharmaceuticals for Imaging of Tumour Hypoxia: A Review of the Evidence. Am. J. Nucl. Med. Mol. Imaging 2014, 4, 365–384. [Google Scholar]
- Bourgeois, M.; Rajerison, H.; Guerard, F.; Mougin-Degraef, M.; Barbet, J.; Michel, N.; Cherel, M.; Faivre-Chauvet, A. Contribution of [64Cu]-ATSM PET in Molecular Imaging of Tumour Hypoxia Compared to Classical [18F]-MISO—A Selected Review. Nucl. Med. Rev. Cent. East. Eur. 2011, 14, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Hoigebazar, L.; Jeong, J.M. Hypoxia Imaging Agents Labeled with Positron Emitters. Recent Results Cancer Res. Fortschr. Krebsforsch. Prog. Dans Rech. Sur Cancer 2013, 194, 285–299. [Google Scholar] [CrossRef]
- Takasawa, M.; Moustafa, R.R.; Baron, J.-C. Applications of Nitroimidazole in Vivo Hypoxia Imaging in Ischemic Stroke. Stroke 2008, 39, 1629–1637. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Stypinski, D.; Xia, H.; McEwan, A.J.B.; Machulla, H.-J.; Wiebe, L.I. Fluoroazomycin Arabinoside (FAZA): Synthesis, 2H and 3H-Labelling and Preliminary Biological Evaluation of a Novel 2-Nitroimidazole Marker of Tissue Hypoxia. J. Label. Compd. Radiopharm. 1999, 42, 3–16. [Google Scholar] [CrossRef]
- Busk, M.; Horsman, M.R.; Jakobsen, S.; Bussink, J.; van der Kogel, A.; Overgaard, J. Cellular Uptake of PET Tracers of Glucose Metabolism and Hypoxia and Their Linkage. Eur. J. Nucl. Med. Mol. Imaging 2008, 35, 2294–2303. [Google Scholar] [CrossRef] [PubMed]
- Quartuccio, N.; Laudicella, R.; Mapelli, P.; Guglielmo, P.; Pizzuto, D.A.; Boero, M.; Arnone, G.; Picchio, M. Young AIMN Working Group. Hypoxia PET Imaging beyond 18F-FMISO in Patients with High-Grade Glioma: 18F-FAZA and Other Hypoxia Radiotracers. Clin. Transl. Imaging 2020, 8, 11–20. [Google Scholar] [CrossRef]
- Busk, M.; Mortensen, L.S.; Nordsmark, M.; Overgaard, J.; Jakobsen, S.; Hansen, K.V.; Theil, J.; Kallehauge, J.F.; D’Andrea, F.P.; Steiniche, T.; et al. PET Hypoxia Imaging with FAZA: Reproducibility at Baseline and during Fractionated Radiotherapy in Tumour-Bearing Mice. Eur. J. Nucl. Med. Mol. Imaging 2013, 40, 186–197. [Google Scholar] [CrossRef]
- Tran, L.-B.-A.; Bol, A.; Labar, D.; Jordan, B.; Magat, J.; Mignion, L.; Grégoire, V.; Gallez, B. Hypoxia Imaging with the Nitroimidazole 18F-FAZA PET Tracer: A Comparison with OxyLite, EPR Oximetry and 19F-MRI Relaxometry. Radiother. Oncol. J. Eur. Soc. Ther. Radiol. Oncol. 2012, 105, 29–35. [Google Scholar] [CrossRef]
- Adams, G.E.; Flockhart, I.R.; Smithen, C.E.; Stratford, I.J.; Wardman, P.; Watts, M.E. Electron-Affinic Sensitization. VII. A Correlation between Structures, One-Electron Reduction Potentials, and Efficiencies of Nitroimidazoles as Hypoxic Cell Radiosensitizers. Radiat. Res. 1976, 67, 9–20. [Google Scholar] [CrossRef]
- Wardman, P. Chemical Radiosensitizers for Use in Radiotherapy. Clin. Oncol. 2007, 19, 397–417. [Google Scholar] [CrossRef] [PubMed]
- Hall, E.J. Radiobiology for the Radiologist, 5th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2000; ISBN 978-0-7817-2649-8. [Google Scholar]
- Overgaard, J.; Sand Hansen, H.; Lindeløv, B.; Overgaard, M.; Jørgensen, K.; Rasmusson, B.; Berthelsen, A. Nimorazole as a Hypoxic Radiosensitizer in the Treatment of Supraglottic Larynx and Pharynx Carcinoma. First Report from the Danish Head and Neck Cancer Study (DAHANCA) Protocol 5-85. Radiother. Oncol. J. Eur. Soc. Ther. Radiol. Oncol. 1991, 20 (Suppl. S1), 143–149. [Google Scholar] [CrossRef]
- Overgaard, J.; Hansen, H.S.; Overgaard, M.; Bastholt, L.; Berthelsen, A.; Specht, L.; Lindeløv, B.; Jørgensen, K. A Randomized Double-Blind Phase III Study of Nimorazole as a Hypoxic Radiosensitizer of Primary Radiotherapy in Supraglottic Larynx and Pharynx Carcinoma. Results of the Danish Head and Neck Cancer Study (DAHANCA) Protocol 5-85. Radiother. Oncol. J. Eur. Soc. Ther. Radiol. Oncol. 1998, 46, 135–146. [Google Scholar] [CrossRef]
- Teicher, B.A.; Lazo, J.S.; Sartorelli, A.C. Classification of Antineoplastic Agents by Their Selective Toxicities toward Oxygenated and Hypoxic Tumour Cells. Cancer Res. 1981, 41, 73–81. [Google Scholar]
- Wardman, P. Nitroimidazoles as Hypoxic Cell Radiosensitizers and Hypoxia Probes: Misonidazole, Myths and Mistakes. Br. J. Radiol. 2019, 92, 20170915. [Google Scholar] [CrossRef]
- Wang, H.; Mu, X.; He, H.; Zhang, X.-D. Cancer Radiosensitizers. Trends Pharmacol. Sci. 2018, 39, 24–48. [Google Scholar] [CrossRef]
- Matsumura, Y.; Maeda, H. A New Concept for Macromolecular Therapeutics in Cancer Chemotherapy: Mechanism of Tumouritropic Accumulation of Proteins and the Antitumour Agent Smancs. Cancer Res. 1986, 46, 6387–6392. [Google Scholar]
- Wong, A.D.; Ye, M.; Ulmschneider, M.B.; Searson, P.C. Quantitative Analysis of the Enhanced Permeation and Retention (EPR) Effect. PLoS ONE 2015, 10, e0123461. [Google Scholar] [CrossRef]
- Minchinton, A.I.; Tannock, I.F. Drug Penetration in Solid Tumours. Nat. Rev. Cancer 2006, 6, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Golombek, S.K.; May, J.-N.; Theek, B.; Appold, L.; Drude, N.; Kiessling, F.; Lammers, T. Tumour Targeting via EPR: Strategies to Enhance Patient Responses. Adv. Drug Deliv. Rev. 2018, 130, 17–38. [Google Scholar] [CrossRef] [PubMed]
- Torchilin, V. Tumour Delivery of Macromolecular Drugs Based on the EPR Effect. Adv. Drug Deliv. Rev. 2011, 63, 131–135. [Google Scholar] [CrossRef]
- Prabhakar, U.; Maeda, H.; Jain, R.K.; Sevick-Muraca, E.M.; Zamboni, W.; Farokhzad, O.C.; Barry, S.T.; Gabizon, A.; Grodzinski, P.; Blakey, D.C. Challenges and Key Considerations of the Enhanced Permeability and Retention (EPR) Effect for Nanomedicine Drug Delivery in Oncology. Cancer Res. 2013, 73, 2412–2417. [Google Scholar] [CrossRef]
- Maeda, H.; Nakamura, H.; Fang, J. The EPR Effect for Macromolecular Drug Delivery to Solid Tumours: Improvement of Tumour Uptake, Lowering of Systemic Toxicity, and Distinct Tumour Imaging in Vivo. Adv. Drug Deliv. Rev. 2013, 65, 71–79. [Google Scholar] [CrossRef]
- Hashizume, H.; Baluk, P.; Morikawa, S.; McLean, J.W.; Thurston, G.; Roberge, S.; Jain, R.K.; McDonald, D.M. Openings between Defective Endothelial Cells Explain Tumour Vessel Leakiness. Am. J. Pathol. 2000, 156, 1363–1380. [Google Scholar] [CrossRef]
- Peer, D.; Karp, J.M.; Hong, S.; Farokhzad, O.C.; Margalit, R.; Langer, R. Nanocarriers as an Emerging Platform for Cancer Therapy. Nat. Nanotechnol. 2007, 2, 751–760. [Google Scholar] [CrossRef]
- Cheng, Z.; Al Zaki, A.; Hui, J.Z.; Muzykantov, V.R.; Tsourkas, A. Multifunctional Nanoparticles: Cost versus Benefit of Adding Targeting and Imaging Capabilities. Science 2012, 338, 903–910. [Google Scholar] [CrossRef] [PubMed]
- Her, S.; Jaffray, D.A.; Allen, C. Gold Nanoparticles for Applications in Cancer Radiotherapy: Mechanisms and Recent Advancements. Adv. Drug Deliv. Rev. 2017, 109, 84–101. [Google Scholar] [CrossRef] [PubMed]
- Mi, Y.; Shao, Z.; Vang, J.; Kaidar-Person, O.; Wang, A.Z. Application of Nanotechnology to Cancer Radiotherapy. Cancer Nanotechnol. 2016, 7, 11. [Google Scholar] [CrossRef] [PubMed]
- Babaei, M.; Ganjalikhani, M. The Potential Effectiveness of Nanoparticles as Radio Sensitizers for Radiotherapy. BioImpacts BI 2014, 4, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yang, J.; Fu, S.; Wu, J. Gold Nanoparticles as Radiosensitizers in Cancer Radiotherapy. Int. J. Nanomed. 2020, 15, 9407–9430. [Google Scholar] [CrossRef]
- Dimitriou, N.M.; Tsekenis, G.; Balanikas, E.C.; Pavlopoulou, A.; Mitsiogianni, M.; Mantso, T.; Pashos, G.; Boudouvis, A.G.; Lykakis, I.N.; Tsigaridas, G.; et al. Gold Nanoparticles, Radiations and the Immune System: Current Insights into the Physical Mechanisms and the Biological Interactions of This New Alliance towards Cancer Therapy. Pharmacol. Ther. 2017, 178, 1–17. [Google Scholar] [CrossRef]
- Retif, P.; Pinel, S.; Toussaint, M.; Frochot, C.; Chouikrat, R.; Bastogne, T.; Barberi-Heyob, M. Nanoparticles for Radiation Therapy Enhancement: The Key Parameters. Theranostics 2015, 5, 1030–1044. [Google Scholar] [CrossRef]
- Yao, X.; Huang, C.; Chen, X.; Yi, Z.; Sanche, L. Chemical Radiosensitivity of DNA Induced by Gold Nanoparticles. J. Biomed. Nanotechnol. 2015, 11, 478–485. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Han, L.; Zhuang, J.; Yang, D.-P. Protein-Directed Gold Nanoparticles with Excellent Catalytic Activity for 4-Nitrophenol Reduction. Mater. Sci. Eng. C Mater. Biol. Appl. 2017, 78, 429–434. [Google Scholar] [CrossRef] [PubMed]
- Mateo, D.; Morales, P.; Ávalos, A.; Haza, A.I. Oxidative Stress Contributes to Gold Nanoparticle-Induced Cytotoxicity in Human Tumour Cells. Toxicol. Mech. Methods 2014, 24, 161–172. [Google Scholar] [CrossRef]
- Decrock, E.; Hoorelbeke, D.; Ramadan, R.; Delvaeye, T.; De Bock, M.; Wang, N.; Krysko, D.V.; Baatout, S.; Bultynck, G.; Aerts, A.; et al. Calcium, Oxidative Stress and Connexin Channels, a Harmonious Orchestra Directing the Response to Radiotherapy Treatment? Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 1099–1120. [Google Scholar] [CrossRef]
- Sinclair, W.K.; Morton, R.A. X-Ray Sensitivity during the Cell Generation Cycle of Cultured Chinese Hamster Cells. Radiat. Res. 1966, 29, 450–474. [Google Scholar] [CrossRef]
- Mackey, M.A.; Saira, F.; Mahmoud, M.A.; El-Sayed, M.A. Inducing Cancer Cell Death by Targeting Its Nucleus: Solid Gold Nanospheres versus Hollow Gold Nanocages. Bioconjug. Chem. 2013, 24, 897–906. [Google Scholar] [CrossRef]
- Cui, L.; Tse, K.; Zahedi, P.; Harding, S.M.; Zafarana, G.; Jaffray, D.A.; Bristow, R.G.; Allen, C. Hypoxia and Cellular Localization Influence the Radiosensitizing Effect of Gold Nanoparticles (AuNPs) in Breast Cancer Cells. Radiat. Res. 2014, 182, 475–488. [Google Scholar] [CrossRef]
- Pan, Y.; Leifert, A.; Ruau, D.; Neuss, S.; Bornemann, J.; Schmid, G.; Brandau, W.; Simon, U.; Jahnen-Dechent, W. Gold Nanoparticles of Diameter 1.4 Nm Trigger Necrosis by Oxidative Stress and Mitochondrial Damage. Small 2009, 5, 2067–2076. [Google Scholar] [CrossRef] [PubMed]
- Butterworth, K.T.; Coulter, J.A.; Jain, S.; Forker, J.; McMahon, S.J.; Schettino, G.; Prise, K.M.; Currell, F.J.; Hirst, D.G. Evaluation of Cytotoxicity and Radiation Enhancement Using 1.9 Nm Gold Particles: Potential Application for Cancer Therapy. Nanotechnology 2010, 21, 295101. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, P.; Li, F.; Jin, X.; Li, J.; Chen, W.; Li, Q. Metal-Based NanoEnhancers for Future Radiotherapy: Radiosensitizing and Synergistic Effects on Tumour Cells. Theranostics 2018, 8, 1824–1849. [Google Scholar] [CrossRef] [PubMed]
- Djuzenova, C.S.; Elsner, I.; Katzer, A.; Worschech, E.; Distel, L.V.; Flentje, M.; Polat, B. Radiosensitivity in Breast Cancer Assessed by the Histone γ-H2AX and 53BP1 Foci. Radiat. Oncol. 2013, 8, 98. [Google Scholar] [CrossRef] [PubMed]
- Vignard, J.; Mirey, G.; Salles, B. Ionizing-Radiation Induced DNA Double-Strand Breaks: A Direct and Indirect Lighting Up. Radiother. Oncol. J. Eur. Soc. Ther. Radiol. Oncol. 2013, 108, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Mothersill, C.; Crean, M.; Lyons, M.; McSweeney, J.; Mooney, R.; O’Reilly, J.; Seymour, C.B. Expression of Delayed Toxicity and Lethal Mutations in the Progeny of Human Cells Surviving Exposure to Radiation and other Environmental Mutagens. Int. J. Radiat. Biol. 1998, 74, 673–680. [Google Scholar] [CrossRef]
- Glaviano, A.; Nayak, V.; Cabuy, E.; Baird, D.M.; Yin, Z.; Newson, R.; Ladon, D.; Rubio, M.A.; Slijepcevic, P.; Lyng, F.; et al. Effects of HTERT on Metal Ion-Induced Genomic Instability. Oncogene 2006, 25, 3424–3435. [Google Scholar] [CrossRef]
- Glaviano, A.; Mothersill, C.; Case, C.P.; Rubio, M.A.; Newson, R.; Lyng, F. Effects of HTERT on Genomic Instability Caused by Either Metal or Radiation or Combined Exposure. Mutagenesis 2009, 24, 25–33. [Google Scholar] [CrossRef]
- Coen, N.; Kadhim, M.A.; Wright, E.G.; Case, C.P.; Mothersill, C.E. Particulate Debris from a Titanium Metal Prosthesis Induces Genomic Instability in Primary Human Fibroblast Cells. Br. J. Cancer 2003, 88, 548–552. [Google Scholar] [CrossRef]
- Rosa, S.; Connolly, C.; Schettino, G.; Butterworth, K.T.; Prise, K.M. Biological Mechanisms of Gold Nanoparticle Radiosensitization. Cancer Nanotechnol. 2017, 8, 2. [Google Scholar] [CrossRef]
- Ghita, M.; McMahon, S.J.; Taggart, L.E.; Butterworth, K.T.; Schettino, G.; Prise, K.M. A Mechanistic Study of Gold Nanoparticle Radiosensitisation Using Targeted Microbeam Irradiation. Sci. Rep. 2017, 7, 44752. [Google Scholar] [CrossRef]
- Wang, Y.-F.; Liu, L.; Xue, X.; Liang, X.-J. Nanoparticle-Based Drug Delivery Systems: What Can They Really Do in Vivo? F1000Research 2017, 6, 681. [Google Scholar] [CrossRef] [PubMed]
- Koonce, N.A.; Quick, M.C.; Hardee, M.E.; Jamshidi-Parsian, A.; Dent, J.A.; Paciotti, G.F.; Nedosekin, D.; Dings, R.P.M.; Griffin, R.J. Combination of Gold Nanoparticle-Conjugated TNF-α and Radiation Therapy Results in a Synergistic Anti-Tumour Response in Murine Carcinoma Models. Int. J. Radiat. Oncol. Biol. Phys. 2015, 93, 588–596. [Google Scholar] [CrossRef] [PubMed]
- Kumthekar, P.; Rademaker, A.; Ko, C.; Dixit, K.; Schwartz, M.; Sonabend, A.; Sharp, L.; Lukas, R.; Stupp, R.; Horbinski, C.; et al. A Phase 0 First-in-Human Study Using NU-0129: A Gold Base Spherical Nucleic Acid (SNA) Nanoconjugate Targeting BCL2L12 in Recurrent Glioblastoma Patients. J. Clin. Oncol. 2019, 37, 3012. [Google Scholar] [CrossRef]
- Khoobchandani, M.; Katti, K.K.; Karikachery, A.R.; Thipe, V.C.; Srisrimal, D.; Dhurvas Mohandoss, D.K.; Darshakumar, R.D.; Joshi, C.M.; Katti, K.V. New Approaches in Breast Cancer Therapy through Green Nanotechnology and Nano-Ayurvedic Medicine—Preclinical and Pilot Human Clinical Investigations. Int. J. Nanomed. 2020, 15, 181–197. [Google Scholar] [CrossRef] [PubMed]
- Schuemann, J.; Berbeco, R.; Chithrani, B.D.; Cho, S.; Kumar, R.; McMahon, S.; Sridhar, S.; Krishnan, S. Roadmap to Clinical Use of Gold Nanoparticles for Radiosensitization. Int. J. Radiat. Oncol. Biol. Phys. 2016, 94, 189–205. [Google Scholar] [CrossRef]
- Brown, J.M.; Siim, B.G. Hypoxia-Specific Cytotoxins in Cancer Therapy. Semin. Radiat. Oncol. 1996, 6, 22–36. [Google Scholar] [CrossRef]
- Zeng, Y.; Ma, J.; Zhan, Y.; Xu, X.; Zeng, Q.; Liang, J.; Chen, X. Hypoxia-Activated Prodrugs and Redox-Responsive Nanocarriers. Int. J. Nanomed. 2018, 13, 6551–6574. [Google Scholar] [CrossRef]
- Rischin, D.; Peters, L.; Fisher, R.; Macann, A.; Denham, J.; Poulsen, M.; Jackson, M.; Kenny, L.; Penniment, M.; Corry, J.; et al. Tirapazamine, Cisplatin, and Radiation versus Fluorouracil, Cisplatin, and Radiation in Patients with Locally Advanced Head and Neck Cancer: A Randomized Phase II Trial of the Trans-Tasman Radiation Oncology Group (TROG 98.02). J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2005, 23, 79–87. [Google Scholar] [CrossRef]
- Thambi, T.; Park, J.H.; Lee, D.S. Hypoxia-Responsive Nanocarriers for Cancer Imaging and Therapy: Recent Approaches and Future Perspectives. Chem. Commun. 2016, 52, 8492–8500. [Google Scholar] [CrossRef]
- Gao, G.H.; Li, Y.; Lee, D.S. Environmental PH-Sensitive Polymeric Micelles for Cancer Diagnosis and Targeted Therapy. J. Control. Release 2013, 169, 180–184. [Google Scholar] [CrossRef]
- Visser, J.G.; Van Staden, A.D.P.; Smith, C. Harnessing Macrophages for Controlled-Release Drug Delivery: Lessons From Microbes. Front. Pharmacol. 2019, 10, 22. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Yang, Z.; Li, F.; Xu, L.; Sun, Y. Cell-Mediated Targeting Drugs Delivery Systems. Drug Deliv. 2020, 27, 1425–1437. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Chu, D.; Wang, Z. Leukocyte-Mediated Delivery of Nanotherapeutics in Inflammatory and Tumour Sites. Theranostics 2017, 7, 751–763. [Google Scholar] [CrossRef]
- Pierigè, F.; Serafini, S.; Rossi, L.; Magnani, M. Cell-Based Drug Delivery. Adv. Drug Deliv. Rev. 2008, 60, 286–295. [Google Scholar] [CrossRef]
- Yousefpour, P.; Chilkoti, A. Co-Opting Biology to Deliver Drugs. Biotechnol. Bioeng. 2014, 111, 1699–1716. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-F.; Liu, Y.; Yang, G.; Zhao, C.-X. Macrophage-Mediated Cancer Drug Delivery. Mater. Today Sustain. 2021, 11–12, 100055. [Google Scholar] [CrossRef]
- Liu, X.; Li, J.; Peng, X.; Lv, B.; Wang, P.; Zhao, X.; Yu, B. Geraniin Inhibits LPS-Induced THP-1 Macrophages Switching to M1 Phenotype via SOCS1/NF-ΚB Pathway. Inflammation 2016, 39, 1421–1433. [Google Scholar] [CrossRef]
- Zhu, Y.; Li, X.; Chen, J.; Chen, T.; Shi, Z.; Lei, M.; Zhang, Y.; Bai, P.; Li, Y.; Fei, X. The Pentacyclic Triterpene Lupeol Switches M1 Macrophages to M2 and Ameliorates Experimental Inflammatory Bowel Disease. Int. Immunopharmacol. 2016, 30, 74–84. [Google Scholar] [CrossRef]
- Ponnaiya, B.; Cornforth, M.N.; Ullrich, R.L. Radiation-Induced Chromosomal Instability in BALB/c and C57BL/6 Mice: The Difference Is as Clear as Black and White. Radiat. Res. 1997, 147, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Mothersill, C.; Rea, D.; Wright, E.G.; Lorimore, S.A.; Murphy, D.; Seymour, C.B.; O’Malley, K. Individual Variation in the Production of a “bystander Signal” Following Irradiation of Primary Cultures of Normal Human Urothelium. Carcinogenesis 2001, 22, 1465–1471. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, K.J.; Coates, P.J.; Lorimore, S.A.; Wright, E.G. The Genetic Basis of Tissue Responses to Ionizing Radiation. Br. J. Radiol. 2007, 80, S2–S6. [Google Scholar] [CrossRef]
- Lorimore, S.A.; Mukherjee, D.; Robinson, J.I.; Chrystal, J.A.; Wright, E.G. Long-Lived Inflammatory Signaling in Irradiated Bone Marrow Is Genome Dependent. Cancer Res. 2011, 71, 6485–6491. [Google Scholar] [CrossRef]
- Mukherjee, D.; Coates, P.J.; Lorimore, S.A.; Wright, E.G. Responses to Ionizing Radiation Mediated by Inflammatory Mechanisms. J. Pathol. 2014, 232, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Poh, A.R.; Ernst, M. Targeting Macrophages in Cancer: From Bench to Bedside. Front. Oncol. 2018, 8, 49. [Google Scholar] [CrossRef] [PubMed]
- Chanmee, T.; Ontong, P.; Konno, K.; Itano, N. Tumour-Associated Macrophages as Major Players in the Tumour Microenvironment. Cancers 2014, 6, 1670–1690. [Google Scholar] [CrossRef] [PubMed]
- Aras, S.; Zaidi, M.R. TAMeless Traitors: Macrophages in Cancer Progression and Metastasis. Br. J. Cancer 2017, 117, 1583–1591. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; He, Y.; Sun, X.; Li, Q.; Wang, W.; Zhao, A.; Di, W. A High M1/M2 Ratio of Tumour-Associated Macrophages Is Associated with Extended Survival in Ovarian Cancer Patients. J. Ovarian Res. 2014, 7, 19. [Google Scholar] [CrossRef]
- Gustafson, H.H.; Holt-Casper, D.; Grainger, D.W.; Ghandehari, H. Nanoparticle Uptake: The Phagocyte Problem. Nano Today 2015, 10, 487–510. [Google Scholar] [CrossRef] [PubMed]
- Batrakova, E.V.; Gendelman, H.E.; Kabanov, A.V. Cell-Mediated Drugs Delivery. Expert Opin. Drug Deliv. 2011, 8, 415–433. [Google Scholar] [CrossRef]
- Nowacek, A.S.; Miller, R.L.; McMillan, J.; Kanmogne, G.; Kanmogne, M.; Mosley, R.L.; Ma, Z.; Graham, S.; Chaubal, M.; Werling, J.; et al. NanoART Synthesis, Characterization, Uptake, Release and Toxicology for Human Monocyte-Macrophage Drug Delivery. Nanomedicine 2009, 4, 903–917. [Google Scholar] [CrossRef]
- Söllner, T.; Bennett, M.K.; Whiteheart, S.W.; Scheller, R.H.; Rothman, J.E. A Protein Assembly-Disassembly Pathway in Vitro That May Correspond to Sequential Steps of Synaptic Vesicle Docking, Activation, and Fusion. Cell 1993, 75, 409–418. [Google Scholar] [CrossRef]
- Moriwaki, T.; Okamoto, S.; Sasanuma, H.; Nagasawa, H.; Takeda, S.; Masunaga, S.-I.; Tano, K. Cytotoxicity of Tirapazamine (3-Amino-1,2,4-Benzotriazine-1,4-Dioxide)-Induced DNA Damage in Chicken DT40 Cells. Chem. Res. Toxicol. 2017, 30, 699–704. [Google Scholar] [CrossRef]
- Guise, C.P.; Mowday, A.M.; Ashoorzadeh, A.; Yuan, R.; Lin, W.-H.; Wu, D.-H.; Smaill, J.B.; Patterson, A.V.; Ding, K. Bioreductive Prodrugs as Cancer Therapeutics: Targeting Tumour Hypoxia. Chin. J. Cancer 2014, 33, 80–86. [Google Scholar] [CrossRef]
- Mistry, I.N.; Thomas, M.; Calder, E.D.D.; Conway, S.J.; Hammond, E.M. Clinical Advances of Hypoxia-Activated Prodrugs in Combination With Radiation Therapy. Int. J. Radiat. Oncol. Biol. Phys. 2017, 98, 1183–1196. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, L.J.; Cazares-Körner, C.; Saha, J.; Evans, C.N.G.; Stratford, M.R.L.; Hammond, E.M.; Conway, S.J. Design, Synthesis and Evaluation of Molecularly Targeted Hypoxia-Activated Prodrugs. Nat. Protoc. 2016, 11, 781–794. [Google Scholar] [CrossRef]
- Stratford, I.J.; Workman, P. Bioreductive Drugs into the next Millennium. Anticancer. Drug Des. 1998, 13, 519–528. [Google Scholar]
- Phillips, R.M. Targeting the Hypoxic Fraction of Tumours Using Hypoxia-Activated Prodrugs. Cancer Chemother. Pharmacol. 2016, 77, 441–457. [Google Scholar] [CrossRef]
- Hay, M.P.; Hicks, K.O.; Pchalek, K.; Lee, H.H.; Blaser, A.; Pruijn, F.B.; Anderson, R.F.; Shinde, S.S.; Wilson, W.R.; Denny, W.A. Tricyclic [1,2,4]Triazine 1,4-Dioxides As Hypoxia Selective Cytotoxins. J. Med. Chem. 2008, 51, 6853–6865. [Google Scholar] [CrossRef]
- Hicks, K.O.; Myint, H.; Patterson, A.V.; Pruijn, F.B.; Siim, B.G.; Patel, K.; Wilson, W.R. Oxygen Dependence and Extravascular Transport of Hypoxia-Activated Prodrugs: Comparison of the Dinitrobenzamide Mustard PR-104A and Tirapazamine. Int. J. Radiat. Oncol. Biol. Phys. 2007, 69, 560–571. [Google Scholar] [CrossRef]
- Rischin, D.; Hicks, R.J.; Fisher, R.; Binns, D.; Corry, J.; Porceddu, S.; Peters, L.J. Trans-Tasman Radiation Oncology Group Study 98.02 Prognostic Significance of [18F]-Misonidazole Positron Emission Tomography-Detected Tumour Hypoxia in Patients with Advanced Head and Neck Cancer Randomly Assigned to Chemoradiation with or without Tirapazamine: A Substudy of Trans-Tasman Radiation Oncology Group Study 98.02. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2006, 24, 2098–2104. [Google Scholar] [CrossRef]
- Rischin, D.; Peters, L.J.; O’Sullivan, B.; Giralt, J.; Fisher, R.; Yuen, K.; Trotti, A.; Bernier, J.; Bourhis, J.; Ringash, J.; et al. Tirapazamine, Cisplatin, and Radiation versus Cisplatin and Radiation for Advanced Squamous Cell Carcinoma of the Head and Neck (TROG 02.02, HeadSTART): A Phase III Trial of the Trans-Tasman Radiation Oncology Group. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2010, 28, 2989–2995. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.; Carter, K.A.; Miranda, D.; Lovell, J.F. Chemophototherapy: An Emerging Treatment Option for Solid Tumours. Adv. Sci. 2017, 4, 1600106. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.A.; Shields, C.W.; Krishnan, V.; Wang, L.L.-W.; Zhao, Z.; Ukidve, A.; Lewandowski, M.; Gao, Y.; Mitragotri, S. Macrophage-Mediated Delivery of Hypoxia-Activated Prodrug Nanoparticles. Adv. Ther. 2020, 3, 1900162. [Google Scholar] [CrossRef]
- Papandreou, I.; Cairns, R.A.; Fontana, L.; Lim, A.L.; Denko, N.C. HIF-1 Mediates Adaptation to Hypoxia by Actively Downregulating Mitochondrial Oxygen Consumption. Cell Metab. 2006, 3, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Cairns, R.A.; Papandreou, I.; Sutphin, P.D.; Denko, N.C. Metabolic Targeting of Hypoxia and HIF1 in Solid Tumours Can Enhance Cytotoxic Chemotherapy. Proc. Natl. Acad. Sci. USA 2007, 104, 9445–9450. [Google Scholar] [CrossRef] [PubMed]
- White, E. The Role for Autophagy in Cancer. J. Clin. Investig. 2015, 125, 42–46. [Google Scholar] [CrossRef]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of Cells and Tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef]
- Aita, V.M.; Liang, X.H.; Murty, V.V.; Pincus, D.L.; Yu, W.; Cayanis, E.; Kalachikov, S.; Gilliam, T.C.; Levine, B. Cloning and Genomic Organization of Beclin 1, a Candidate Tumour Suppressor Gene on Chromosome 17q21. Genomics 1999, 59, 59–65. [Google Scholar] [CrossRef]
- Choi, A.M.K.; Ryter, S.W.; Levine, B. Autophagy in Human Health and Disease. N. Engl. J. Med. 2013, 368, 651–662. [Google Scholar] [CrossRef]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of Autophagy and Inhibition of Tumourigenesis by Beclin 1. Nature 1999, 402, 672–676. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Yun, C.W.; Lee, S.H. The Roles of Autophagy in Cancer. Int. J. Mol. Sci. 2018, 19, 3466. [Google Scholar] [CrossRef] [PubMed]
- Rabinowitz, J.D.; White, E. Autophagy and Metabolism. Science 2010, 330, 1344–1348. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.Y.; Ryan, K.M. Autophagy and Cancer--Issues We Need to Digest. J. Cell Sci. 2012, 125, 2349–2358. [Google Scholar] [CrossRef]
- Verbaanderd, C.; Maes, H.; Schaaf, M.B.; Sukhatme, V.P.; Pantziarka, P.; Sukhatme, V.; Agostinis, P.; Bouche, G. Repurposing Drugs in Oncology (ReDO)—Chloroquine and Hydroxychloroquine as Anti-Cancer Agents. Ecancermedicalscience 2017, 11, 781. [Google Scholar] [CrossRef]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting Autophagy in Cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef]
- Townsend, K.N.; Hughson, L.R.K.; Schlie, K.; Poon, V.I.; Westerback, A.; Lum, J.J. Autophagy Inhibition in Cancer Therapy: Metabolic Considerations for Antitumour Immunity. Immunol. Rev. 2012, 249, 176–194. [Google Scholar] [CrossRef]
- Ben-Zvi, I.; Kivity, S.; Langevitz, P.; Shoenfeld, Y. Hydroxychloroquine: From Malaria to Autoimmunity. Clin. Rev. Allergy Immunol. 2012, 42, 145–153. [Google Scholar] [CrossRef]
- Viry, E.; Paggetti, J.; Baginska, J.; Mgrditchian, T.; Berchem, G.; Moussay, E.; Janji, B. Autophagy: An Adaptive Metabolic Response to Stress Shaping the Antitumour Immunity. Biochem. Pharmacol. 2014, 92, 31–42. [Google Scholar] [CrossRef]
- Cicchini, M.; Karantza, V.; Xia, B. Molecular Pathways: Autophagy in Cancer—A Matter of Timing and Context. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 498–504. [Google Scholar] [CrossRef]
- Cheong, H. Integrating Autophagy and Metabolism in Cancer. Arch. Pharm. Res. 2015, 38, 358–371. [Google Scholar] [CrossRef] [PubMed]
- Yim, W.W.-Y.; Mizushima, N. Lysosome Biology in Autophagy. Cell Discov. 2020, 6, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Tasdemir, E.; Galluzzi, L.; Maiuri, M.C.; Criollo, A.; Vitale, I.; Hangen, E.; Modjtahedi, N.; Kroemer, G. Methods for Assessing Autophagy and Autophagic Cell Death. Methods Mol. Biol. 2008, 445, 29–76. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-H.; Yang, P.-M.; Chuah, Q.-Y.; Lee, Y.-J.; Hsieh, Y.-F.; Peng, C.-W.; Chiu, S.-J. Autophagy Promotes Radiation-Induced Senescence but Inhibits Bystander Effects in Human Breast Cancer Cells. Autophagy 2014, 10, 1212–1228. [Google Scholar] [CrossRef]
- Kong, E.Y.; Cheng, S.H.; Yu, K.N. Induction of Autophagy and Interleukin 6 Secretion in Bystander Cells: Metabolic Cooperation for Radiation-Induced Rescue Effect? J. Radiat. Res. 2018, 59, 129–140. [Google Scholar] [CrossRef]
- Song, M.; Wang, Y.; Shang, Z.-F.; Liu, X.-D.; Xie, D.-F.; Wang, Q.; Guan, H.; Zhou, P.-K. Bystander Autophagy Mediated by Radiation-Induced Exosomal MiR-7-5p in Non-Targeted Human Bronchial Epithelial Cells. Sci. Rep. 2016, 6, 30165. [Google Scholar] [CrossRef]
- Mothersill, C.; Stamato, T.D.; Perez, M.L.; Cummins, R.; Mooney, R.; Seymour, C.B. Involvement of Energy Metabolism in the Production of ‘Bystander Effects’ by Radiation. Br. J. Cancer 2000, 82, 1740–1746. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, J.; Fu, J.; Wang, J.; Ye, S.; Liu, W.; Shao, C. Role of ROS-Mediated Autophagy in Radiation-Induced Bystander Effect of Hepatoma Cells. Int. J. Radiat. Biol. 2015, 91, 452–458. [Google Scholar] [CrossRef]
- Sharma, N.K.; Stone, S.; Kumar, V.P.; Biswas, S.; Aghdam, S.Y.; Holmes-Hampton, G.P.; Fam, C.M.; Cox, G.N.; Ghosh, S.P. Mitochondrial Degeneration and Autophagy Associated With Delayed Effects of Radiation in the Mouse Brain. Front. Aging Neurosci. 2019, 11, 357. [Google Scholar] [CrossRef]
- Mukherjee, S.; Chakraborty, A. Radiation-Induced Bystander Phenomenon: Insight and Implications in Radiotherapy. Int. J. Radiat. Biol. 2019, 95, 243–263. [Google Scholar] [CrossRef]
- Briceño, E.; Reyes, S.; Sotelo, J. Therapy of Glioblastoma Multiforme Improved by the Antimutagenic Chloroquine. Neurosurg. Focus 2003, 14, e3. [Google Scholar] [CrossRef] [PubMed]
- Briceño, E.; Calderon, A.; Sotelo, J. Institutional Experience with Chloroquine as an Adjuvant to the Therapy for Glioblastoma Multiforme. Surg. Neurol. 2007, 67, 388–391. [Google Scholar] [CrossRef] [PubMed]
- Sotelo, J.; Briceño, E.; López-González, M.A. Adding Chloroquine to Conventional Treatment for Glioblastoma Multiforme: A Randomized, Double-Blind, Placebo-Controlled Trial. Ann. Intern. Med. 2006, 144, 337–343. [Google Scholar] [CrossRef]
- Rangwala, R.; Leone, R.; Chang, Y.C.; Fecher, L.A.; Schuchter, L.M.; Kramer, A.; Tan, K.-S.; Heitjan, D.F.; Rodgers, G.; Gallagher, M.; et al. Phase I Trial of Hydroxychloroquine with Dose-Intense Temozolomide in Patients with Advanced Solid Tumours and Melanoma. Autophagy 2014, 10, 1369–1379. [Google Scholar] [CrossRef]
- Vogl, D.T.; Stadtmauer, E.A.; Tan, K.-S.; Heitjan, D.F.; Davis, L.E.; Pontiggia, L.; Rangwala, R.; Piao, S.; Chang, Y.C.; Scott, E.C.; et al. Combined Autophagy and Proteasome Inhibition: A Phase 1 Trial of Hydroxychloroquine and Bortezomib in Patients with Relapsed/Refractory Myeloma. Autophagy 2014, 10, 1380–1390. [Google Scholar] [CrossRef]
- Rangwala, R.; Chang, Y.C.; Hu, J.; Algazy, K.M.; Evans, T.L.; Fecher, L.A.; Schuchter, L.M.; Torigian, D.A.; Panosian, J.T.; Troxel, A.B.; et al. Combined MTOR and Autophagy Inhibition: Phase I Trial of Hydroxychloroquine and Temsirolimus in Patients with Advanced Solid Tumours and Melanoma. Autophagy 2014, 10, 1391–1402. [Google Scholar] [CrossRef]
- Mahalingam, D.; Mita, M.; Sarantopoulos, J.; Wood, L.; Amaravadi, R.K.; Davis, L.E.; Mita, A.C.; Curiel, T.J.; Espitia, C.M.; Nawrocki, S.T.; et al. Combined Autophagy and HDAC Inhibition: A Phase I Safety, Tolerability, Pharmacokinetic, and Pharmacodynamic Analysis of Hydroxychloroquine in Combination with the HDAC Inhibitor Vorinostat in Patients with Advanced Solid Tumours. Autophagy 2014, 10, 1403–1414. [Google Scholar] [CrossRef]
- Chude, C.I.; Amaravadi, R.K. Targeting Autophagy in Cancer: Update on Clinical Trials and Novel Inhibitors. Int. J. Mol. Sci. 2017, 18, 1279. [Google Scholar] [CrossRef]
- Loos, B.; du Toit, A.; Hofmeyr, J.-H.S. Defining and Measuring Autophagosome Flux—Concept and Reality. Autophagy 2014, 10, 2087–2096. [Google Scholar] [CrossRef]
- Barnard, R.A.; Wittenburg, L.A.; Amaravadi, R.K.; Gustafson, D.L.; Thorburn, A.; Thamm, D.H. Phase I Clinical Trial and Pharmacodynamic Evaluation of Combination Hydroxychloroquine and Doxorubicin Treatment in Pet Dogs Treated for Spontaneously Occurring Lymphoma. Autophagy 2014, 10, 1415–1425. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Takabatake, Y.; Takahashi, A.; Isaka, Y. Chloroquine in Cancer Therapy: A Double-Edged Sword of Autophagy. Cancer Res. 2013, 73, 3–7. [Google Scholar] [CrossRef]
- de Jonge, M.J.A.; Verweij, J. Renal Toxicities of Chemotherapy. Semin. Oncol. 2006, 33, 68–73. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdalla, F.C.; Abeliovich, H.; Abraham, R.T.; Acevedo-Arozena, A.; Adeli, K.; Agholme, L.; Agnello, M.; Agostinis, P.; Aguirre-Ghiso, J.A.; et al. Guidelines for the Use and Interpretation of Assays for Monitoring Autophagy. Autophagy 2012, 8, 445–544. [Google Scholar] [CrossRef] [PubMed]
- Mothersill, C.; Rusin, A.; Fernandez-Palomo, C.; Seymour, C. History of Bystander Effects Research 1905-Present; What Is in a Name? Int. J. Radiat. Biol. 2018, 94, 696–707. [Google Scholar] [CrossRef]
- Mothersill, C.; Seymour, C.B. Radiation-Induced Bystander Effects and the DNA Paradigm: An “out of Field” Perspective. Mutat. Res. 2006, 597, 5–10. [Google Scholar] [CrossRef]
- Heeran, A.B.; Berrigan, H.P.; O’Sullivan, J. The Radiation-Induced Bystander Effect (RIBE) and Its Connections with the Hallmarks of Cancer. Radiat. Res. 2019, 192, 668–679. [Google Scholar] [CrossRef]
- Watson, G.E.; Lorimore, S.A.; Macdonald, D.A.; Wright, E.G. Chromosomal Instability in Unirradiated Cells Induced in Vivo by a Bystander Effect of Ionizing Radiation. Cancer Res. 2000, 60, 5608–5611. [Google Scholar]
- Lyng, F.M.; Seymour, C.B.; Mothersill, C. Initiation of Apoptosis in Cells Exposed to Medium from the Progeny of Irradiated Cells: A Possible Mechanism for Bystander-Induced Genomic Instability? Radiat. Res. 2002, 157, 365–370. [Google Scholar] [CrossRef]
- Nugent, S.M.E.; Mothersill, C.E.; Seymour, C.; McClean, B.; Lyng, F.M.; Murphy, J.E.J. Increased Mitochondrial Mass in Cells with Functionally Compromised Mitochondria after Exposure to Both Direct Gamma Radiation and Bystander Factors. Radiat. Res. 2007, 168, 134–142. [Google Scholar] [CrossRef]
- Maguire, P.; Mothersill, C.; Seymour, C.; Lyng, F.M. Medium from Irradiated Cells Induces Dose-Dependent Mitochondrial Changes and BCL2 Responses in Unirradiated Human Keratinocytes. Radiat. Res. 2005, 163, 384–390. [Google Scholar] [CrossRef]
- Pouget, J.-P.; Georgakilas, A.G.; Ravanat, J.-L. Targeted and Off-Target (Bystander and Abscopal) Effects of Radiation Therapy: Redox Mechanisms and Risk/Benefit Analysis. Antioxid. Redox Signal. 2018, 29, 1447–1487. [Google Scholar] [CrossRef] [PubMed]
- Lyng, F.M.; Howe, O.L.; McClean, B. Reactive Oxygen Species-Induced Release of Signalling Factors in Irradiated Cells Triggers Membrane Signalling and Calcium Influx in Bystander Cells. Int. J. Radiat. Biol. 2011, 87, 683–695. [Google Scholar] [CrossRef] [PubMed]
- Rugo, R.E.; Mutamba, J.T.; Mohan, K.N.; Yee, T.; Chaillet, J.R.; Greenberger, J.S.; Engelward, B.P. Methyltransferases Mediate Cell Memory of a Genotoxic Insult. Oncogene 2011, 30, 751–756. [Google Scholar] [CrossRef] [PubMed]
- Mothersill, C.; Smith, R.W.; Saroya, R.; Denbeigh, J.; Rowe, B.; Banevicius, L.; Timmins, R.; Moccia, R.; Seymour, C.B. Irradiation of Rainbow Trout at Early Life Stages Results in Legacy Effects in Adults. Int. J. Radiat. Biol. 2010, 86, 817–828. [Google Scholar] [CrossRef]
- Shields, L.; Vega-Carrascal, I.; Singleton, S.; Lyng, F.M.; McClean, B. Cell Survival and DNA Damage in Normal Prostate Cells Irradiated Out-of-Field. Radiat. Res. 2014, 182, 499–506. [Google Scholar] [CrossRef]
- Jabbari, N.; Nawaz, M.; Rezaie, J. Bystander Effects of Ionizing Radiation: Conditioned Media from X-Ray Irradiated MCF-7 Cells Increases the Angiogenic Ability of Endothelial Cells. Cell Commun. Signal. 2019, 17, 165. [Google Scholar] [CrossRef]
- Zhou, H.; Randers-Pehrson, G.; Waldren, C.A.; Hei, T.K. Radiation-Induced Bystander Effect and Adaptive Response in Mammalian Cells. Adv. Space Res. Off. J. Comm. Space Res. 2004, 34, 1368–1372. [Google Scholar] [CrossRef]
- Maguire, P.; Mothersill, C.; McClean, B.; Seymour, C.; Lyng, F.M. Modulation of Radiation Responses by Pre-Exposure to Irradiated Cell Conditioned Medium. Radiat. Res. 2007, 167, 485–492. [Google Scholar] [CrossRef]
- Tang, H.; Chen, L.; Chen, L.; Chen, B.; Wang, T.; Yang, A.; Zhan, F.; Wu, L.; Bian, P. Interaction between Radioadaptive Response and Radiation-Induced Bystander Effect in Caenorhabditis Elegans: A Unique Role of the DNA Damage Checkpoint. Radiat. Res. 2016, 186, 662–668. [Google Scholar] [CrossRef]
- Seymour, C.B.; Mothersill, C. Relative Contribution of Bystander and Targeted Cell Killing to the Low-Dose Region of the Radiation Dose-Response Curve. Radiat. Res. 2000, 153, 508–511. [Google Scholar] [CrossRef]
- Butterworth, K.T.; McGarry, C.K.; Trainor, C.; O’Sullivan, J.M.; Hounsell, A.R.; Prise, K.M. Out-of-Field cell survival following exposure to intensity-modulated radiation fields. Int. J. Radiat. Oncol. Biol. Phys. 2011, 79, 1516–1522. [Google Scholar] [CrossRef]
- Liu, Z.; Mothersill, C.E.; McNeill, F.E.; Lyng, F.M.; Byun, S.H.; Seymour, C.B.; Prestwich, W.V. A Dose Threshold for a Medium Transfer Bystander Effect for a Human Skin Cell Line. Radiat. Res. 2006, 166, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Schettino, G.; Folkard, M.; Michael, B.D.; Prise, K.M. Low-Dose Binary Behavior of Bystander Cell Killing after Microbeam Irradiation of a Single Cell with Focused c(k) x Rays. Radiat. Res. 2005, 163, 332–336. [Google Scholar] [CrossRef]
- Buonanno, M.; de Toledo, S.M.; Pain, D.; Azzam, E.I. Long-Term Consequences of Radiation-Induced Bystander Effects Depend on Radiation Quality and Dose and Correlate with Oxidative Stress. Radiat. Res. 2011, 175, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Azzam, E.I.; Jay-Gerin, J.-P.; Pain, D. Ionizing Radiation-Induced Metabolic Oxidative Stress and Prolonged Cell Injury. Cancer Lett. 2012, 327, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Clutton, S.M.; Townsend, K.M.; Walker, C.; Ansell, J.D.; Wright, E.G. Radiation-Induced Genomic Instability and Persisting Oxidative Stress in Primary Bone Marrow Cultures. Carcinogenesis 1996, 17, 1633–1639. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Palomo, C.; Bräuer-Krisch, E.; Laissue, J.; Vukmirovic, D.; Blattmann, H.; Seymour, C.; Schültke, E.; Mothersill, C. Use of Synchrotron Medical Microbeam Irradiation to Investigate Radiation-Induced Bystander and Abscopal Effects in Vivo. Phys. Med. 2015, 31, 584–595. [Google Scholar] [CrossRef] [PubMed]
- Swati; Chadha, V.D. Role of Epigenetic Mechanisms in Propagating Off-Targeted Effects Following Radiation Based Therapies—A Review. Mutat. Res. Mutat. Res. 2021, 787, 108370. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, Y.; Mo, F.; Patel, G.; Butterworth, K.; Shao, C.; Prise, K.M. The Roles of HIF-1α in Radiosensitivity and Radiation-Induced Bystander Effects under Hypoxia. Front. Cell Dev. Biol. 2021, 9, 637454. [Google Scholar] [CrossRef]
- Le, M.; Fernandez-Palomo, C.; McNeill, F.E.; Seymour, C.B.; Rainbow, A.J.; Mothersill, C.E. Exosomes Are Released by Bystander Cells Exposed to Radiation-Induced Biophoton Signals: Reconciling the Mechanisms Mediating the Bystander Effect. PLoS ONE 2017, 12, e0173685. [Google Scholar] [CrossRef] [PubMed]
- Mothersill, C.; Seymour, R.J.; Seymour, C.B. Increased Radiosensitivity in Cells of Two Human Cell Lines Treated with Bystander Medium from Irradiated Repair-Deficient Cells. Radiat. Res. 2006, 165, 26–34. [Google Scholar] [CrossRef]
- Poon, R.C.C.; Agnihotri, N.; Seymour, C.; Mothersill, C. Bystander Effects of Ionizing Radiation Can Be Modulated by Signaling Amines. Environ. Res. 2007, 105, 200–211. [Google Scholar] [CrossRef]
- Mothersill, C.; Saroya, R.; Smith, R.W.; Singh, H.; Seymour, C.B. Serum Serotonin Levels Determine the Magnitude and Type of Bystander Effects in Medium Transfer Experiments. Radiat. Res. 2010, 174, 119–123. [Google Scholar] [CrossRef]
- Fazzari, J.; Mersov, A.; Smith, R.; Seymour, C.; Mothersill, C. Effect of 5-Hydroxytryptamine (Serotonin) Receptor Inhibitors on the Radiation-Induced Bystander Effect. Int. J. Radiat. Biol. 2012, 88, 786–790. [Google Scholar] [CrossRef]
- Curtis, J.J.; Vo, N.T.K.; Seymour, C.B.; Mothersill, C.E. 5-HT2A and 5-HT3 Receptors Contribute to the Exacerbation of Targeted and Non-Targeted Effects of Ionizing Radiation-Induced Cell Death in Human Colon Carcinoma Cells. Int. J. Radiat. Biol. 2020, 96, 482–490. [Google Scholar] [CrossRef]
- Lyng, F.M.; Seymour, C.B.; Mothersill, C. Production of a Signal by Irradiated Cells Which Leads to a Response in Unirradiated Cells Characteristic of Initiation of Apoptosis. Br. J. Cancer 2000, 83, 1223–1230. [Google Scholar] [CrossRef] [PubMed]
- Curtis, J.J.; Vo, N.T.K.; Seymour, C.B.; Mothersill, C.E. Serotonin and 5-HT3 Receptors Sensitize Human Skin Cells to Direct Irradiation Cell Death but Not to Soluble Radiation-Induced Bystander Signals. Environ. Res. 2020, 180, 108807. [Google Scholar] [CrossRef] [PubMed]
- Zambetti, G.P.; Levine, A.J. A Comparison of the Biological Activities of Wild-Type and Mutant P53. FASEB J. 1993, 7, 855–865. [Google Scholar] [CrossRef] [PubMed]
- Al-Mayah, A.H.J.; Irons, S.L.; Pink, R.C.; Carter, D.R.F.; Kadhim, M.A. Possible Role of Exosomes Containing RNA in Mediating Nontargeted Effect of Ionizing Radiation. Radiat. Res. 2012, 177, 539–545. [Google Scholar] [CrossRef]
- Jella, K.K.; Rani, S.; O’Driscoll, L.; McClean, B.; Byrne, H.J.; Lyng, F.M. Exosomes Are Involved in Mediating Radiation Induced Bystander Signaling in Human Keratinocyte Cells. Radiat. Res. 2014, 181, 138–145. [Google Scholar] [CrossRef]
- Mo, L.-J.; Song, M.; Huang, Q.-H.; Guan, H.; Liu, X.-D.; Xie, D.-F.; Huang, B.; Huang, R.-X.; Zhou, P.-K. Exosome-Packaged MiR-1246 Contributes to Bystander DNA Damage by Targeting LIG4. Br. J. Cancer 2018, 119, 492–502. [Google Scholar] [CrossRef]
- Du, Y.; Du, S.; Liu, L.; Gan, F.; Jiang, X.; Wangrao, K.; Lyu, P.; Gong, P.; Yao, Y. Radiation-Induced Bystander Effect Can Be Transmitted through Exosomes Using MiRNAs as Effector Molecules. Radiat. Res. 2020, 194, 89–100. [Google Scholar] [CrossRef]
- Ahmad, S.B.; McNeill, F.E.; Byun, S.H.; Prestwich, W.V.; Mothersill, C.; Seymour, C.; Armstrong, A.; Fernandez, C. Ultra-violet light emission from HPV-G cells irradiated with low let radiation from 90Y; consequences for radiation induced bystander effects. Dose-Response Publ. Int. Hormesis Soc. 2013, 11, 498–516. [Google Scholar] [CrossRef]
- Le, M.; McNeill, F.E.; Seymour, C.; Rainbow, A.J.; Mothersill, C.E. An Observed Effect of Ultraviolet Radiation Emitted from Beta-Irradiated HaCaT Cells upon Non-Beta-Irradiated Bystander Cells. Radiat. Res. 2015, 183, 279–290. [Google Scholar] [CrossRef]
- Le, M.; Mothersill, C.E.; Seymour, C.B.; Ahmad, S.B.; Armstrong, A.; Rainbow, A.J.; McNeill, F.E. Factors Affecting Ultraviolet-A Photon Emission from β-Irradiated Human Keratinocyte Cells. Phys. Med. Biol. 2015, 60, 6371–6389. [Google Scholar] [CrossRef] [PubMed]
- Mothersill, C.E.; Moriarty, M.J.; Seymour, C.B. Radiotherapy and the Potential Exploitation of Bystander Effects. Int. J. Radiat. Oncol. Biol. Phys. 2004, 58, 575–579. [Google Scholar] [CrossRef] [PubMed]
- Marín, A.; Martín, M.; Liñán, O.; Alvarenga, F.; López, M.; Fernández, L.; Büchser, D.; Cerezo, L. Bystander Effects and Radiotherapy. Rep. Pract. Oncol. Radiother. 2014, 20, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Massaccesi, M.; Boldrini, L.; Piras, A.; Stimato, G.; Quaranta, F.; Azario, L.; Mattiucci, G.C.; Valentini, V. Spatially Fractionated Radiotherapy (SFRT) Targeting the Hypoxic Tumour Segment for the Intentional Induction of Non-Targeted Effects: An in Silico Study to Exploit a New Treatment Paradigm. Tech. Innov. Patient Support Radiat. Oncol. 2020, 14, 11–14. [Google Scholar] [CrossRef]
- Tubin, S.; Khan, M.K.; Salerno, G.; Mourad, W.F.; Yan, W.; Jeremic, B. Mono-Institutional Phase 2 Study of Innovative Stereotactic Body RadioTherapy Targeting PArtial Tumour HYpoxic (SBRT-PATHY) Clonogenic Cells in Unresectable Bulky Non-Small Cell Lung Cancer: Profound Non-Targeted Effects by Sparing Peri-Tumoural Immune Microenvironment. Radiat. Oncol. 2019, 14, 212. [Google Scholar] [CrossRef] [PubMed]
- Foehrenbacher, A.; Patel, K.; Abbattista, M.R.; Guise, C.P.; Secomb, T.W.; Wilson, W.R.; Hicks, K.O. The Role of Bystander Effects in the Antitumour Activity of the Hypoxia-Activated Prodrug PR-104. Front. Oncol. 2013, 3, 263. [Google Scholar] [CrossRef]
- Hong, C.R.; Bogle, G.; Wang, J.; Patel, K.; Pruijn, F.B.; Wilson, W.R.; Hicks, K.O. Bystander Effects of Hypoxia-Activated Prodrugs: Agent-Based Modeling Using Three Dimensional Cell Cultures. Front. Pharmacol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.R.; Wilson, W.R.; Hicks, K.O. An Intratumour Pharmacokinetic/Pharmacodynamic Model for the Hypoxia-Activated Prodrug Evofosfamide (TH-302): Monotherapy Activity Is not Dependent on a Bystander Effect. Neoplasia 2019, 21, 159–171. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Apilan, A.G.; Mothersill, C. Targeted and Non-Targeted Mechanisms for Killing Hypoxic Tumour Cells—Are There New Avenues for Treatment? Int. J. Mol. Sci. 2021, 22, 8651. https://doi.org/10.3390/ijms22168651
Apilan AG, Mothersill C. Targeted and Non-Targeted Mechanisms for Killing Hypoxic Tumour Cells—Are There New Avenues for Treatment? International Journal of Molecular Sciences. 2021; 22(16):8651. https://doi.org/10.3390/ijms22168651
Chicago/Turabian StyleApilan, Alyssa Gabrielle, and Carmel Mothersill. 2021. "Targeted and Non-Targeted Mechanisms for Killing Hypoxic Tumour Cells—Are There New Avenues for Treatment?" International Journal of Molecular Sciences 22, no. 16: 8651. https://doi.org/10.3390/ijms22168651
APA StyleApilan, A. G., & Mothersill, C. (2021). Targeted and Non-Targeted Mechanisms for Killing Hypoxic Tumour Cells—Are There New Avenues for Treatment? International Journal of Molecular Sciences, 22(16), 8651. https://doi.org/10.3390/ijms22168651