
Expression of Cancer Stem Cell Markers EpCAM and CD90 Is Correlated with Anti- and Pro-Oncogenic EphA2 Signaling in Hepatocellular Carcinoma

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
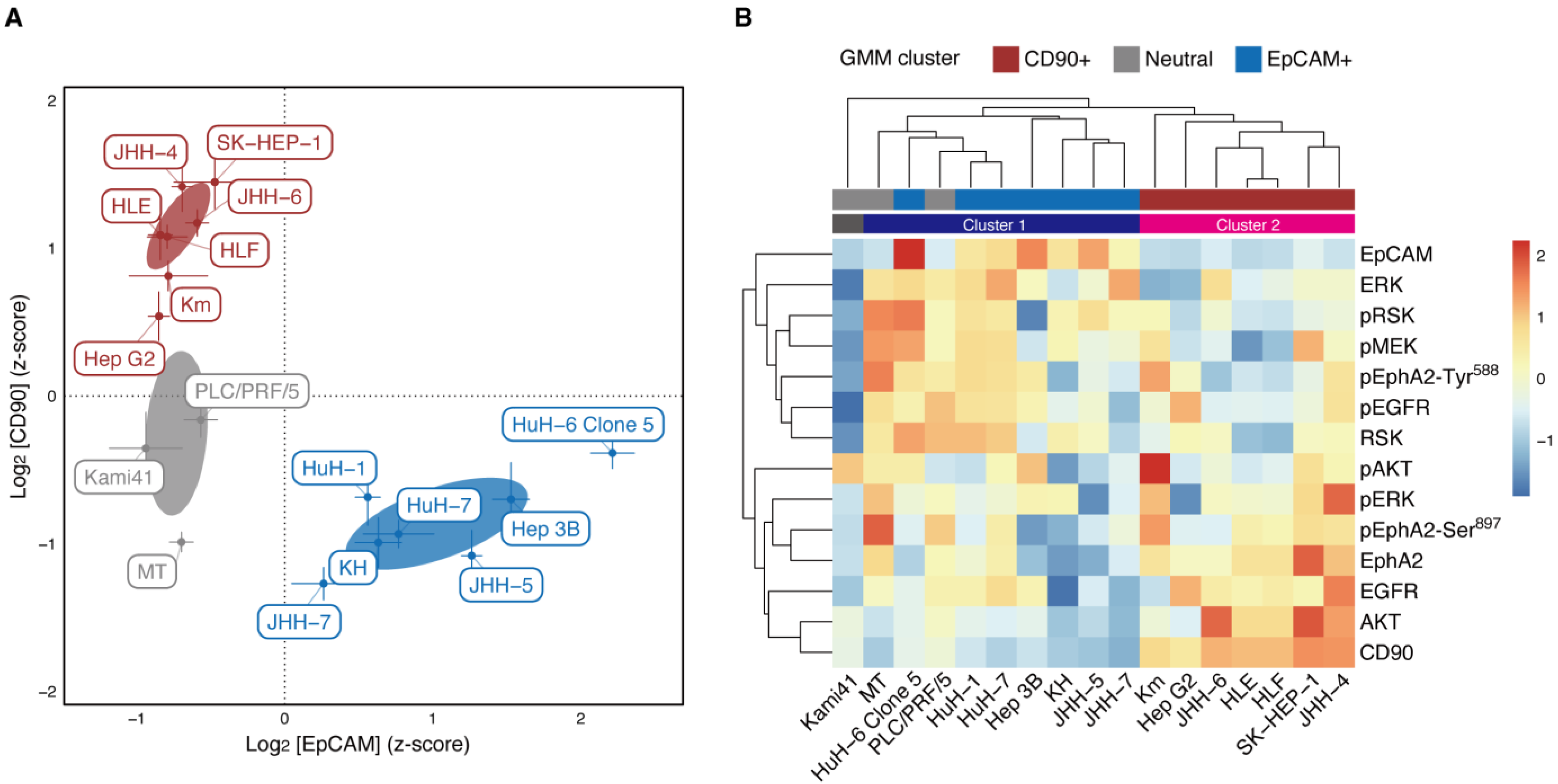
2.1. EpCAM and CD90 Expression Classify Hcc Cells into Functionally Distinct Clusters
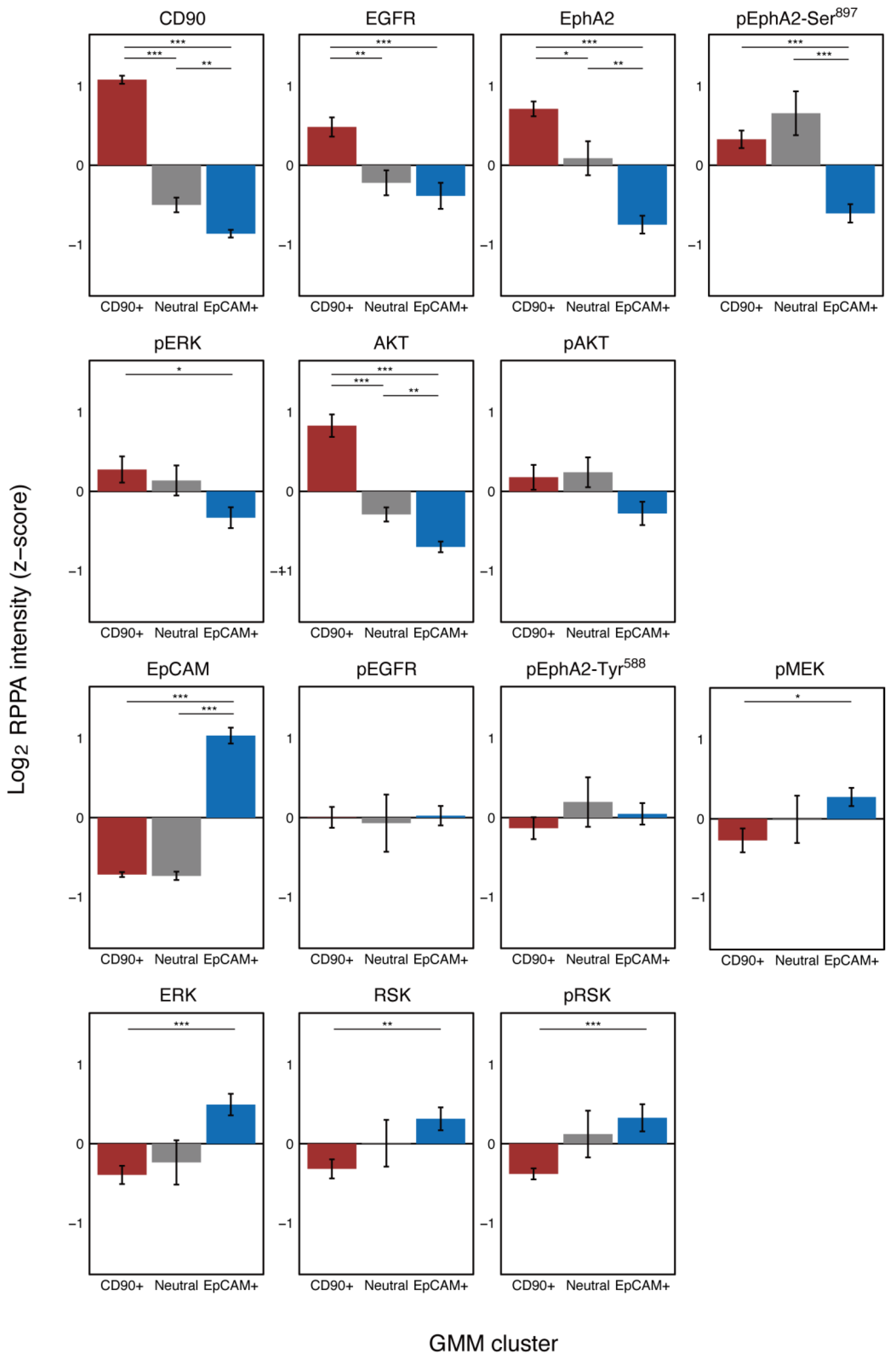
2.2. Protein Expression and Phosphorylation Specific to Cd90+ and Epcam Cell Clusters
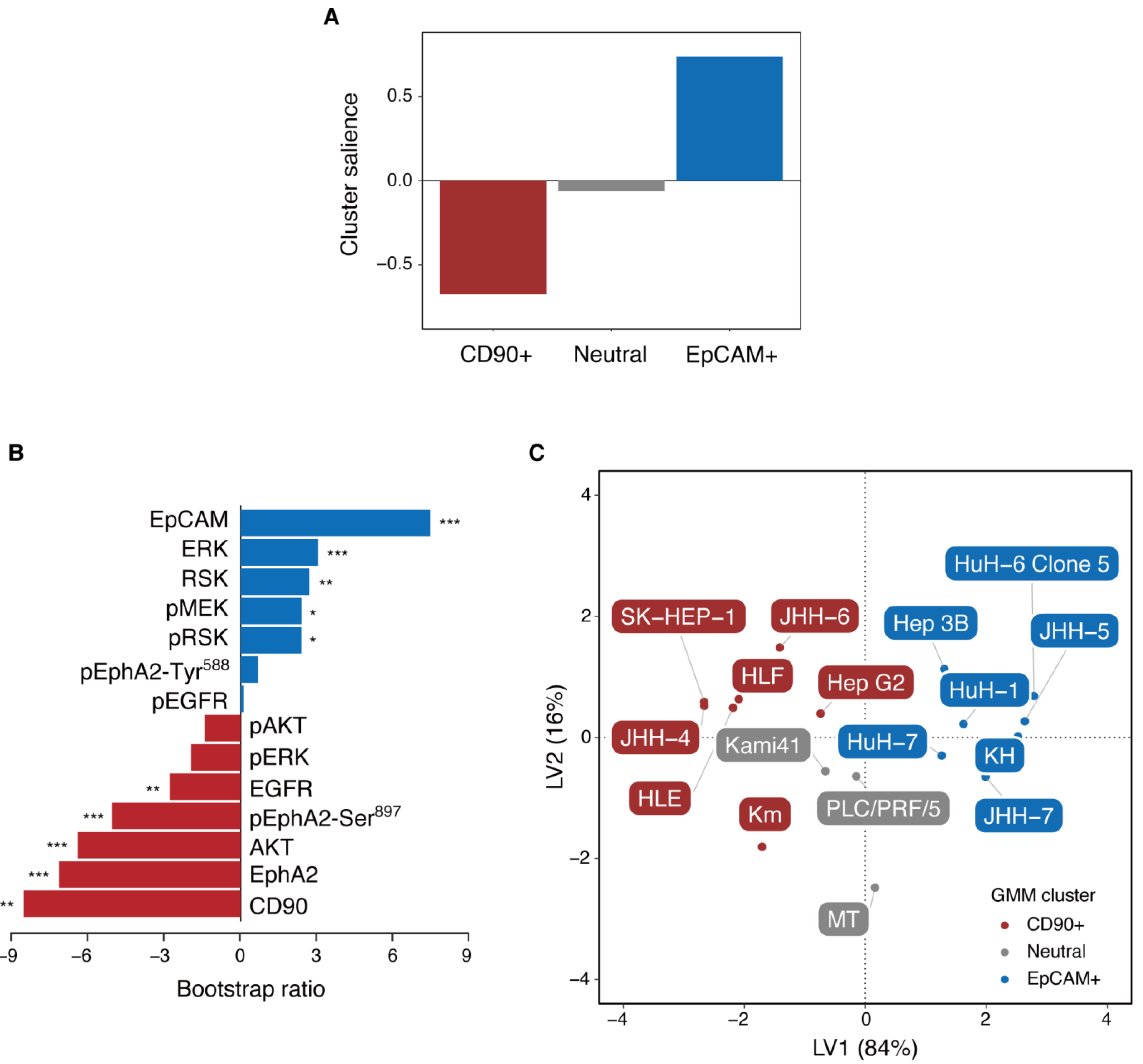
2.3. Partial Least Squares Analysis Reveals Anti- and Pro-Oncogenic Activity of Epcam+ and Cd90+ Cells
2.4. AKT Inhibitor Suppressed Cd90+ Cell Proliferation
2.5. AKT Inhibitors Suppress Cd90+ Cell Migration
3. Discussion
4. Materials and Methods
4.1. Antibodies
4.2. Cell Lines and Culture Conditions
4.3. Reverse Phase Protein Array (RPPA)
4.4. Cell Growth and Migration Assays
4.5. Clustering Analysis
4.6. Partial Least Squares Analysis
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Bahardoust, M.; Sarveazad, A.; Agah, S.; Babahajian, A.; Amini, N. Predictors of 5 year survival rate in hepatocellular carcinoma patients. J. Res. Med. Sci. 2019, 24, 86. [Google Scholar] [CrossRef]
- Villanueva, A. Hepatocellular Carcinoma. N. Engl. J. Med. 2019, 380, 1450–1462. [Google Scholar] [CrossRef] [Green Version]
- Wilhelm, S.M.; Carter, C.; Tang, L.; Wilkie, D.; McNabola, A.; Rong, H.; Chen, C.; Zhang, X.; Vincent, P.; McHugh, M.; et al. BAY 43-9006 Exhibits Broad Spectrum Oral Antitumor Activity and Targets the RAF/MEK/ERK Pathway and Receptor Ty-rosine Kinases Involved in Tumor Progression and Angiogenesis. Cancer Res. 2004, 64, 7099–7109. [Google Scholar] [CrossRef] [Green Version]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; De Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in Advanced Hepatocellular Carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.-L.; Kang, Y.-K.; Chen, Z.; Tsao, C.-J.; Qin, S.; Kim, J.S.; Luo, R.; Feng, J.; Ye, S.; Yang, T.-S.; et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: A phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009, 10, 25–34. [Google Scholar] [CrossRef]
- Zhu, Y.-J.; Zheng, B.; Wang, H.-Y.; Chen, L. New knowledge of the mechanisms of sorafenib resistance in liver cancer. Acta Pharmacol. Sin. 2017, 38, 614–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padda, I.S.; Parmar, M. Lenvatinib; StatPearls Publishing: Bethesda, MD, USA, 2021. Available online: https://pubmed.ncbi.nlm.nih.gov/33620845/ (accessed on 9 August 2021).
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.-H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.-W.; Han, G.; Jassem, J.; et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef] [Green Version]
- Suyama, K.; Iwase, H. Lenvatinib. Cancer Control 2018, 25, 1073274818789361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilhelm, S.M.; Dumas, J.; Adnane, L.; Lynch, M.; Carter, C.A.; Schütz, G.; Thierauch, K.-H.; Zopf, D. Regorafenib (BAY 73-4506): A new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int. J. Cancer 2011, 129, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.-H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, T.; Wang, X.W. Cancer stem cells in the development of liver cancer. J. Clin. Investig. 2013, 123, 1911–1918. [Google Scholar] [CrossRef]
- Yamashita, T.; Honda, M.; Nakamoto, Y.; Baba, M.; Nio, K.; Hara, Y.; Zeng, S.S.; Hayashi, T.; Kondo, M.; Takatori, H.; et al. Discrete nature of EpCAM+and CD90+cancer stem cells in human hepatocellular carcinoma. Hepatology 2013, 57, 1484–1497. [Google Scholar] [CrossRef]
- Gao, Q.; Zhu, H.; Dong, L.; Shi, W.; Chen, R.; Song, Z.; Huang, C.; Li, J.; Dong, X.; Zhou, Y.; et al. Integrated Proteogenomic Characterization of HBV-Related Hepatocellular Carcinoma. Cell 2019, 179, 561–577.e22. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhao, G.-D.; Shi, Z.; Qi, L.-L.; Zhou, L.-Y.; Fu, Z.-X. The Ras/Raf/MEK/ERK signaling pathway and its role in the occurrence and development of HCC. Oncol. Lett. 2016, 12, 3045–3050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.-C.; Yeh, C.-T.; Lin, K.-H. Cancer Stem Cell Functions in Hepatocellular Carcinoma and Comprehensive Therapeutic Strategies. Cells 2020, 9, 1331. [Google Scholar] [CrossRef]
- Yoshida, M.; Yamashita, T.; Okada, H.; Oishi, N.; Nio, K.; Hayashi, T.; Nomura, Y.; Hayashi, T.; Asahina, Y.; Ohwada, M.; et al. Sorafenib suppresses extrahepatic metastasis de novo in hepatocellular carcinoma through inhibition of mesenchymal cancer stem cells characterized by the expression of CD90. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Dimri, M.; Satyanarayana, A. Molecular Signaling Pathways and Therapeutic Targets in Hepatocellular Carcinoma. Cancers 2020, 12, 491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whittaker, S.; Marais, R.; Zhu, A.X. The role of signaling pathways in the development and treatment of hepatocellular carcinoma. Oncogene 2010, 29, 4989–5005. [Google Scholar] [CrossRef]
- Komposch, K.; Sibilia, M. EGFR Signaling in Liver Diseases. Int. J. Mol. Sci. 2015, 17, 30. [Google Scholar] [CrossRef]
- Zhou, Q.; Huang, T.; Wang, Y.-F.; Zhou, X.-B.; Liang, L.-J.; Peng, B.-G. Role of tissue factor in hepatocellular carcinoma genesis, invasion and metastasis. Chin. Med. J. 2011, 124, 3746–3751. [Google Scholar]
- Fan, M.; Liu, Y.; Xia, F.; Wang, Z.; Huang, Y.; Li, J.; Wang, Z.; Li, X. Increased expression of EphA2 and E-N cadherin switch in primary hepatocellular carcinoma. Tumori J. 2014, 99, 689–696. [Google Scholar] [CrossRef]
- Cui, X.-D.; Lee, M.-J.; Yu, G.-R.; Kim, I.-H.; Yu, H.-C.; Song, E.-Y.; Kim, D.-G. EFNA1 ligand and its receptor EphA2: Potential biomarkers for hepatocellular carcinoma. Int. J. Cancer 2009, 126, 940–949. [Google Scholar] [CrossRef] [PubMed]
- Iida, H.; Honda, M.; Kawai, H.F.; Yamashita, T.; Shirota, Y.; Wang, B.-C.; Miao, H.; Kaneko, S. Ephrin-A1 expression contributes to the malignant characteristics of -fetoprotein producing hepatocellular carcinoma. Gut 2005, 54, 843–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cioce, M.; Fazio, V. EphA2 and EGFR: Friends in Life, Partners in Crime. Can EphA2 Be a Predictive Biomarker of Response to Anti-EGFR Agents? Cancers 2021, 13, 700. [Google Scholar] [CrossRef]
- Koshikawa, N.; Hoshino, D.; Taniguchi, H.; Minegishi, T.; Tomari, T.; Nam, S.-O.; Aoki, M.; Sueta, T.; Nakagawa, T.; Miyamoto, S.; et al. Proteolysis of EphA2 Converts It from a Tumor Suppressor to an Oncoprotein. Cancer Res. 2015, 75, 3327–3339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, H.; Li, D.-Q.; Mukherjee, A.; Guo, H.; Petty, A.; Cutter, J.; Basilion, J.P.; Sedor, J.; Wu, J.; Danielpour, D.; et al. EphA2 Mediates Ligand-Dependent Inhibition and Ligand-Independent Promotion of Cell Migration and Invasion via a Reciprocal Regulatory Loop with Akt. Cancer Cell 2009, 16, 9–20. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Sakurai, H. Emerging and Diverse Functions of the EphA2 Noncanonical Pathway in Cancer Progression. Biol. Pharm. Bull. 2017, 40, 1616–1624. [Google Scholar] [CrossRef] [Green Version]
- Ding, Q.; Xia, W.; Liu, J.-C.; Yang, J.-Y.; Lee, D.-F.; Xia, J.; Bartholomeusz, G.; Li, Y.; Pan, Y.; Li, Z.; et al. Erk Associates with and Primes GSK-3β for Its Inactivation Resulting in Upregulation of β-Catenin. Mol. Cell 2005, 19, 159–170. [Google Scholar] [CrossRef]
- Zhang, S.; Gao, W.; Tang, J.; Zhang, H.; Zhou, Y.; Liu, J.; Chen, K.; Liu, F.; Li, W.; To, S.K.Y.; et al. The Roles of GSK-3β in Regulation of Retinoid Signaling and Sorafenib Treatment Response in Hepatocellular Carcinoma. Theranostics 2020, 10, 1230–1244. [Google Scholar] [CrossRef]
- Garten, A.; Grohmann, T.; Kluckova, K.; Lavery, G.; Kiess, W.; Penke, M. Sorafenib-Induced Apoptosis in Hepatocellular Carcinoma Is Reversed by SIRT1. Int. J. Mol. Sci. 2019, 20, 4048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnan, A.; Williams, L.; McIntosh, A.R.; Abdi, H. Partial Least Squares (PLS) methods for neuroimaging: A tutorial and review. NeuroImage 2011, 56, 455–475. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, A.R.; Lobaugh, N.J. Partial least squares analysis of neuroimaging data: Applications and advances. NeuroImage 2004, 23, S250–S263. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, A.R.; Bookstein, F.L.; Haxby, J.V.; Grady, C.L. Spatial Pattern Analysis of Functional Brain Images Using Partial Least Squares. NeuroImage 1996, 3, 143–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harada, K.; Hiramoto-Yamaki, N.; Negishi, M.; Katoh, H. Ephexin4 and EphA2 mediate resistance to anoikis through RhoG and phosphatidylinositol 3-kinase. Exp. Cell Res. 2011, 317, 1701–1713. [Google Scholar] [CrossRef]
- Simioni, C.; Martelli, A.M.; Cani, A.; Cetin-Atalay, R.; McCubrey, J.; Capitani, S.; Neri, L.M. The AKT Inhibitor MK-2206 is Cytotoxic in Hepatocarcinoma Cells Displaying Hyperphosphorylated AKT-1 and Synergizes with Conventional Chemotherapy. Oncotarget 2013, 4, 1496–1506. [Google Scholar] [CrossRef]
- Wilson, J.; Kunnimalaiyaan, S.; Gamblin, T. MK2206 inhibits hepatocellular carcinoma cellular proliferation via induction of apoptosis and cell cycle arrest. J. Surg. Res. 2014, 191, 280–285. [Google Scholar] [CrossRef]
- Grabinski, N.; Ewald, F.; Hofmann, B.T.; Staufer, K.; Schumacher, U.; Nashan, B.; Jücker, M. Combined targeting of AKT and mTOR synergistically inhibits proliferation of hepatocellular carcinoma cells. Mol. Cancer 2012, 11, 85. [Google Scholar] [CrossRef] [Green Version]
- Ewald, F.; Nörz, D.; Grottke, A.; Bach, J.; Herzberger, C.; Hofmann, B.T.; Nashan, B.; Jücker, M. Vertical Targeting of AKT and mTOR as Well as Dual Targeting of AKT and MEK Signaling Is Synergistic in Hepatocellular Carcinoma. J. Cancer 2015, 6, 1195–1205. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, T.; Koshikawa, N.; Shimakami, T.; Terashima, T.; Nakagawa, M.; Nio, K.; Horii, R.; Iida, N.; Kawaguchi, K.; Arai, K.; et al. Serum Laminin γ2 Monomer as a Diagnostic and Predictive Biomarker for Hepatocellular Carcinoma. Hepatology 2021. [Google Scholar] [CrossRef]
- Hashiba, T.; Yamashita, T.; Okada, H.; Nio, K.; Hayashi, T.; Asahina, Y.; Hayashi, T.; Terashima, T.; Iida, N.; Takatori, H.; et al. Inactivation of Transcriptional Repressor Capicua Confers Sorafenib Resistance in Human Hepatocellular Carcinoma. Cell. Mol. Gastroenterol. Hepatol. 2020, 10, 269–285. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Yamashita, T.; Okada, H.; Nio, K.; Hara, Y.; Nomura, Y.; Hayashi, T.; Asahina, Y.; Yoshida, M.; Oishi, N.; et al. Sporadic PCDH18 somatic mutations in EpCAM-positive hepatocellular carcinoma. Cancer Cell Int. 2017, 17, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamashita, T.; Kaneko, S. Liver cancer stem cells: Recent progress in basic and clinical research. Regen. Ther. 2021, 17, 34–37. [Google Scholar] [CrossRef]
- Nagano, M.; Hoshino, D.; Toshima, J.; Seiki, M.; Koshikawa, N. NH 2 -terminal fragment of ZF21 protein suppresses tumor invasion via inhibiting the interaction of ZF21 with FAK. Cancer Sci. 2020, 111, 4393–4404. [Google Scholar] [CrossRef]
- Scrucca, L.; Fop, M.; Murphy, T.B.; Raftery, A.E. mclust 5: Clustering, Classification and Density Estimation Using Gaussian Finite Mixture Models. R J. 2016, 8, 289–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Asakura, N.; Nakamura, N.; Muroi, A.; Nojima, Y.; Yamashita, T.; Kaneko, S.; Ikeda, K.; Koshikawa, N.; Suzuki, T. Expression of Cancer Stem Cell Markers EpCAM and CD90 Is Correlated with Anti- and Pro-Oncogenic EphA2 Signaling in Hepatocellular Carcinoma. Int. J. Mol. Sci. 2021, 22, 8652. https://doi.org/10.3390/ijms22168652
Asakura N, Nakamura N, Muroi A, Nojima Y, Yamashita T, Kaneko S, Ikeda K, Koshikawa N, Suzuki T. Expression of Cancer Stem Cell Markers EpCAM and CD90 Is Correlated with Anti- and Pro-Oncogenic EphA2 Signaling in Hepatocellular Carcinoma. International Journal of Molecular Sciences. 2021; 22(16):8652. https://doi.org/10.3390/ijms22168652
Chicago/Turabian StyleAsakura, Nobuhiko, Naotoshi Nakamura, Atsushi Muroi, Yosui Nojima, Taro Yamashita, Shuichi Kaneko, Kazuki Ikeda, Naohiko Koshikawa, and Takashi Suzuki. 2021. "Expression of Cancer Stem Cell Markers EpCAM and CD90 Is Correlated with Anti- and Pro-Oncogenic EphA2 Signaling in Hepatocellular Carcinoma" International Journal of Molecular Sciences 22, no. 16: 8652. https://doi.org/10.3390/ijms22168652
APA StyleAsakura, N., Nakamura, N., Muroi, A., Nojima, Y., Yamashita, T., Kaneko, S., Ikeda, K., Koshikawa, N., & Suzuki, T. (2021). Expression of Cancer Stem Cell Markers EpCAM and CD90 Is Correlated with Anti- and Pro-Oncogenic EphA2 Signaling in Hepatocellular Carcinoma. International Journal of Molecular Sciences, 22(16), 8652. https://doi.org/10.3390/ijms22168652