
Rapid Transient Transcriptional Adaptation to Hypergravity in Jurkat T Cells Revealed by Comparative Analysis of Microarray and RNA-Seq Data

 ,
, 

Abstract
1. Introduction
2. Results
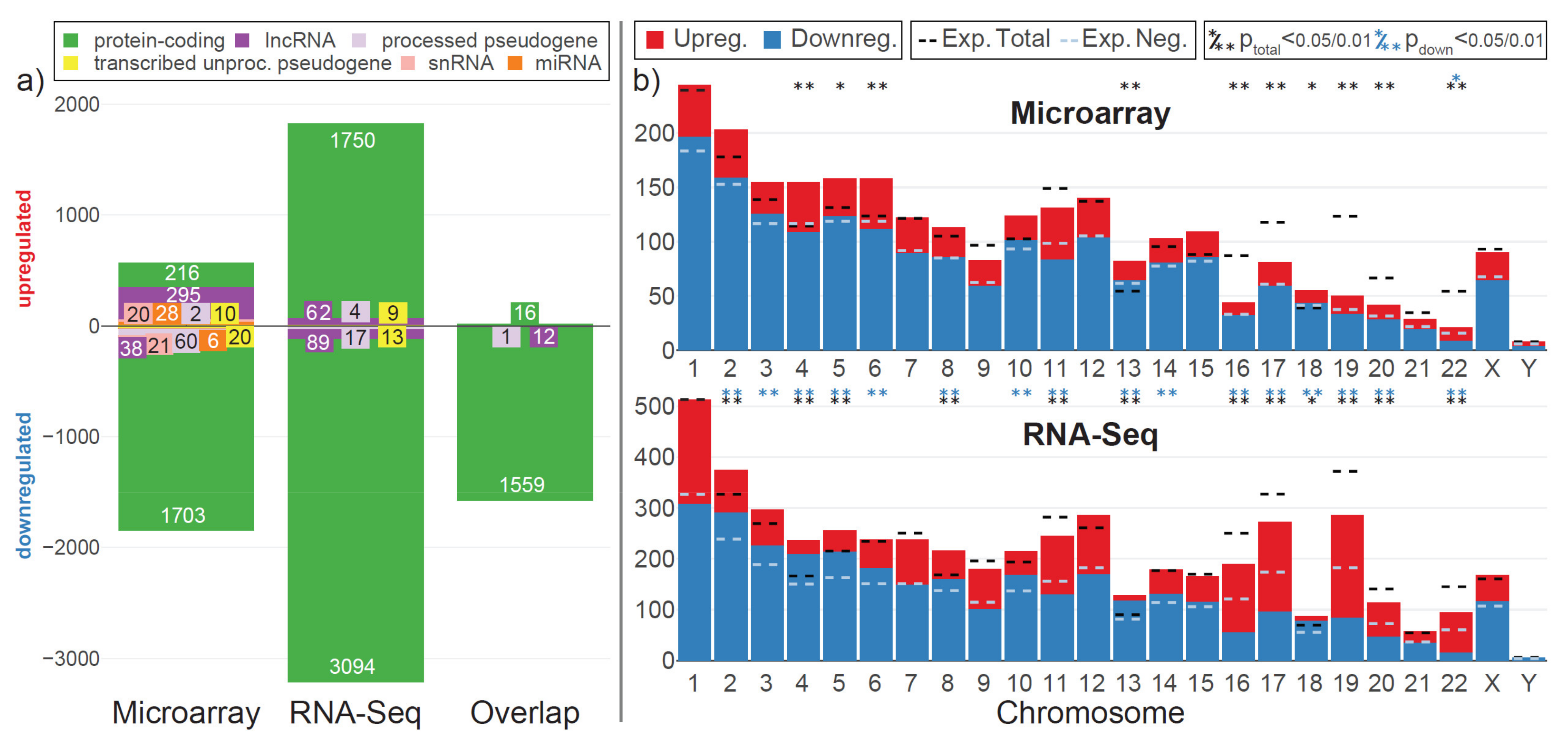
2.1. Comparison of RNA Microarray versus RNA-Seq Overall Distribution and Differential Gene Expression
2.2. A Comparison of Both Data Sets on an Aggregated Level Revealed Good Overall Agreeability
2.3. Characterization of Splicing Events
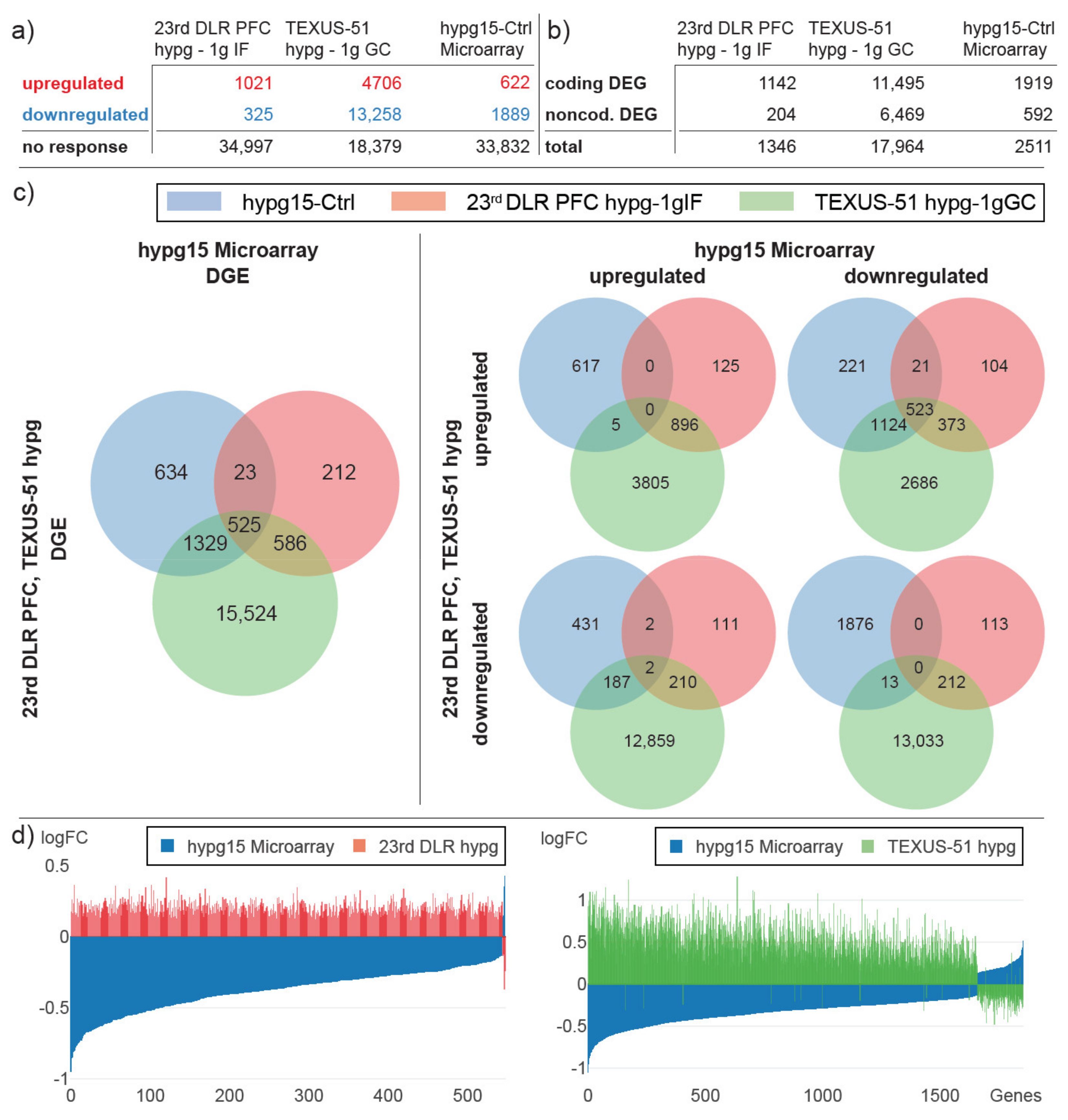
2.4. Differential Gene Expression Shows High Agreeability between Previous Short-Term Hypergravity Data and the Current Fifteen-Minute Study
2.5. Transient Transcriptional Response between Twenty-Second and Fifteen-Minute Hypergravity
2.6. Transcriptome Adaptation to Hypergravity
2.7. High Similarity in Gene Set Enrichment Analysis
2.8. A Cross-Correlation Analysis of Highly Validated Groups of Genes Revealed Temporal Consistency
3. Discussion
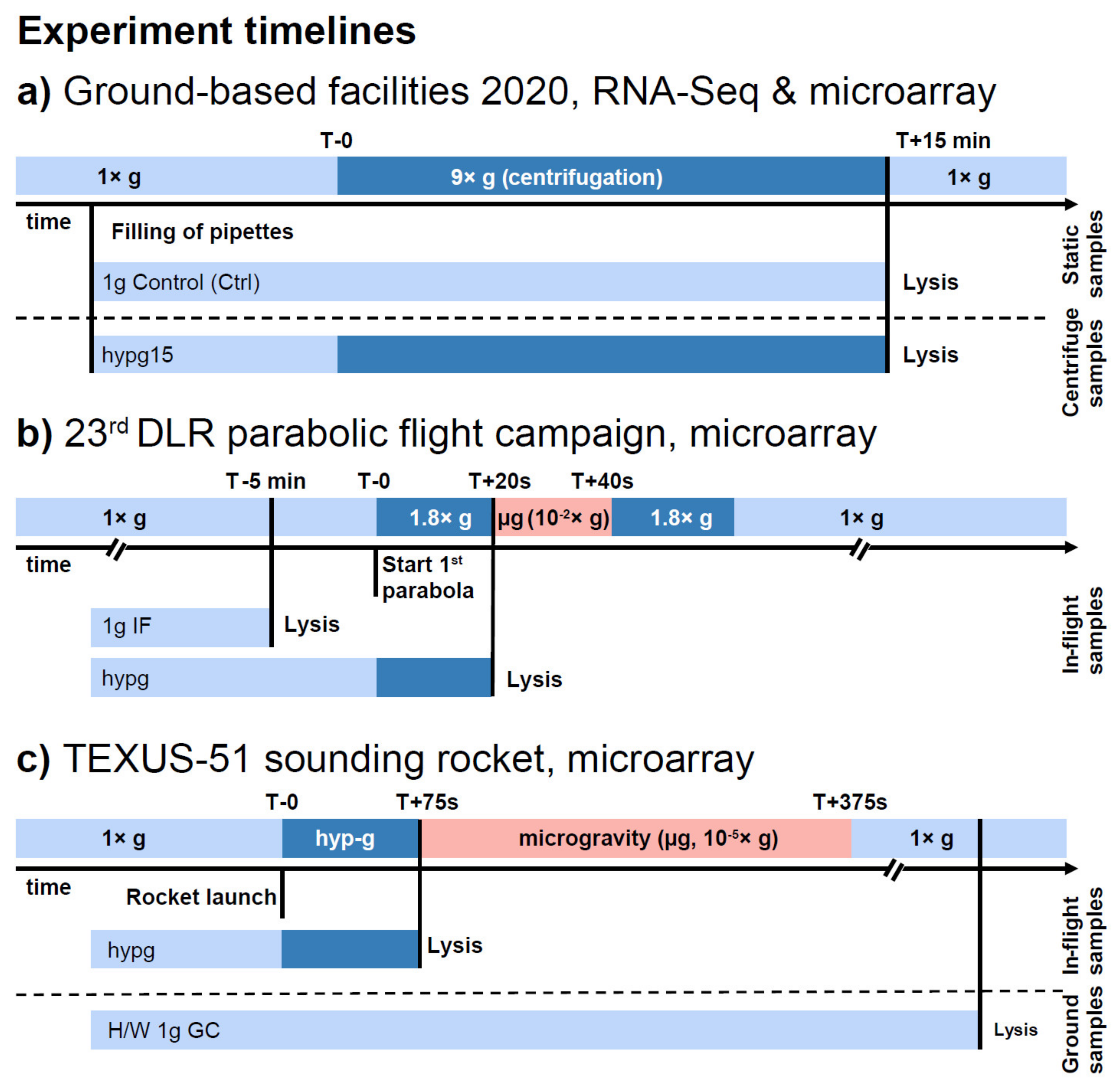
4. Materials and Methods
4.1. Preparation of Biological Specimen
4.2. Transcriptomics of Biological Specimen
4.3. RNA-Seq Sample Analysis
4.4. Microarray Sample Analysis
4.5. Previous and External Data Sets
4.6. Transcriptional Dynamics Analysis
4.7. Upset Plots
4.8. Set Inclusion Analysis
4.9. Spearman Correlation Coefficient
4.10. Differential Exon Usage (DEU)
4.11. Chromosomal Mapping
4.12. Gene Set Enrichment
4.13. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| FDR | False Discovery Rate |
| DEG | Differential Gene Expression |
| logFC | log2 fold change |
| DEU | Differential Exon Usage |
| NES | Normalized Enrichment Score |
| CD | Cluster of Differentiation protein |
References
- Barlow, P.W. Gravity perception in plants: A multiplicity of systems derived by evolution? Plant Cell Environ. 1995, 18, 951–962. [Google Scholar] [CrossRef]
- Morey, H. The Impact of gravity on life. In Evolution on Planet Earth: The Impact of the Physical Environment; Academic Press: Cambridge, MA, USA, 2003. [Google Scholar]
- Volkmann, D.; Baluska, F. Gravity: One of the driving forces for evolution. Protoplasma 2006, 229, 143–148. [Google Scholar] [CrossRef]
- Nasa Manned Spacecraft Center. Gemini Summary Conference; NASA: Washington, DC, USA, 1967. [Google Scholar]
- Konstantinova, I.V.; Antropova, Y.N.; Legenkov, V.I.; Zazhirey, V.D. Study of reactivity of blood lymphoid cells in crew members of the Soyuz-6, Soyuz-7 and Soyuz-8 spaceships before and after flight (blatogenesis delay of lymphocytes). Space Biol. Med. 1973, 7, 48–55. [Google Scholar]
- Stepanek, J.; Blue, R.S.; Parazynski, S. Space medicine in the era of civilian spaceflight. N. Engl. J. Med. 2019, 380, 1053–1060. [Google Scholar] [CrossRef]
- Cogoli, A.; Tschopp, A.; Fuchs-Bislin, P. Cell sensitivity to gravity. Science 1984, 225, 228–230. [Google Scholar] [CrossRef]
- Cogoli, A. Space flight and the immune system. Vaccine 1993, 11, 496–503. [Google Scholar] [CrossRef]
- Horneck, G.; Facius, R.; Reichert, M.; Rettberg, P.; Seboldt, W.; Manzey, D.; Comet, B.; Maillet, A.; Preiss, H.; Schauer, L.; et al. HUMEX, a study on the survivability and adaptation of humans to long-duration exploratory missions, part I: Lunar missions. Adv. Space Res. 2003, 31, 2389–2401. [Google Scholar] [CrossRef]
- Frippiat, J.-P.; Crucian, B.E.; de Quervain, D.J.F.; Grimm, D.; Montano, N.; Praun, S.; Roozendaal, B.; Schelling, G.; Thiel, M.; Ullrich, O.; et al. Towards human exploration of space: The THESEUS review series on immunology research priorities. NPJ Microgravity 2016, 2, 16040. [Google Scholar] [CrossRef]
- Choukèr, A.; Ullrich, O. The Immune System in Space: Are We Prepared? Springer International Publishing: Basel, Switzerland, 2016. [Google Scholar]
- Adrian, A.; Schoppmann, K.; Sromicki, J.; Brungs, S.; von der Wiesche, M.; Hock, B.; Kolanus, W.; Hemmersbach, R.; Ullrich, O. The oxidative burst reaction in mammalian cells depends on gravity. Cell Commun. Signal. 2013, 11, 98. [Google Scholar] [CrossRef] [PubMed]
- Thiel, C.S.; de Zélicourt, D.; Tauber, S.; Adrian, A.; Franz, M.; Simmet, D.M.; Schoppmann, K.; Hauschild, S.; Krammer, S.; Christen, M.; et al. Rapid adaptation to microgravity in mammalian macrophage cells. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, K.; Thiel, C.; Timm, J.; Schmidt, P.M.; Huber, K.; Tauber, S.; Hemmersbach, R.; Seibt, D.; Kroll, H.; Grote, K.-H.; et al. Microgravity-induced alterations in signal transduction in cells of the immune system. Acta Astronaut. 2010, 67, 1116–1125. [Google Scholar] [CrossRef]
- Thiel, C.S.; Paulsen, K.; Bradacs, G.; Lust, K.; Tauber, S.; Dumrese, C.; Hilliger, A.; Schoppmann, K.; Biskup, J.; Golz, N.; et al. Rapid alterations of cell cycle control proteins in human T lymphocytes in microgravity. Cell Commun. Signal. 2012, 10, 1–16. [Google Scholar] [CrossRef]
- Mangala, L.S.; Zhang, Y.; He, Z.; Emami, K.; Ramesh, G.T.; Story, M.; Rohde, L.H.; Wu, H. Effects of simulated microgravity on expression profile of microRNA in human lymphoblastoid cells. J. Biol. Chem. 2011, 286, 32483–32490. [Google Scholar] [CrossRef]
- Tauber, S.; Hauschild, S.; Paulsen, K.; Gutewort, A.; Raig, C.; Breuer-Hürlimann, E.; Biskup, J.; Philpot, C.; Lier, H.; Engelmann, F.; et al. Signal transduction in primary human T lymphocytes in altered gravity during parabolic flight and clinostat experiments. Cell. Physiol. Biochem. 2015, 35, 1034–1051. [Google Scholar] [CrossRef] [PubMed]
- Thiel, C.S.; Hauschild, S.; Huge, A.; Tauber, S.; Lauber, B.A.; Polzer, J.; Paulsen, K.; Lier, H.; Engelmann, F.; Schmitz, B.; et al. Dynamic gene expression response to altered gravity in human T cells. Sci. Rep. 2017, 7, 1–22. [Google Scholar] [CrossRef]
- Thiel, C.S.; Huge, A.; Hauschild, S.; Tauber, S.; Lauber, B.A.; Polzer, J.; Paulsen, K.; Lier, H.; Engelmann, F.; Schmitz, B.; et al. Stability of gene expression in human T cells in different gravity environments is clustered in chromosomal region 11p15.4. NPJ Microgravity 2017, 3, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Thiel, C.S.; Tauber, S.; Christoffel, S.; Huge, A.; Lauber, B.A.; Polzer, J.; Paulsen, K.; Lier, H.; Engelmann, F.; Schmitz, B.; et al. Rapid coupling between gravitational forces and the transcriptome in human myelomonocytic U937 cells. Sci. Rep. 2018, 8, 1–24. [Google Scholar] [CrossRef]
- Choukér, A. Stress Challenges and Immunity in Space; Springer International Publishing: Basel, Switzerland, 2019. [Google Scholar]
- Albrecht-Buehler, G. Possible mechanisms of indirect gravity sensing by cells. ASGSB Bull. Publ. Am. Soc. Gravit. Space Biol. 1991, 4, 25–34. [Google Scholar]
- Klopp, E.; Graff, D.; Struckmeier, J.; Born, M.; Curtze, S.; Hofmann, M.; Jones, D. The osteoblast mechano-receptor, microgravity perception and thermodynamics. J. Gravit. Physiol. J. Int. Soc. Gravit. Physiol. 2002, 9, P269–P270. [Google Scholar]
- Häder, D.-P.; Braun, M.; Grimm, D.; Hemmersbach, R. Gravireceptors in eukaryotes—A comparison of case studies on the cellular level. NPJ Microgravity 2017, 3, 1–8. [Google Scholar] [CrossRef]
- Velculescu, V.E.; Zhang, L.; Vogelstein, B.; Kinzler, K.W. Serial analysis of gene expression. Science 1995, 270, 484–487. [Google Scholar] [CrossRef]
- Lowe, R.; Shirley, N.; Bleackley, M.; Dolan, S.; Shafee, T. Transcriptomics technologies. PLoS Comput. Biol. 2017, 13, e1005457. [Google Scholar] [CrossRef]
- Berrios, D.C.; Galazka, J.; Grigorev, K.; Gebre, S.; Costes, S.V. NASA GeneLab: Interfaces for the exploration of space omics data. Nucleic Acids Res. 2021, 49, D1515–D1522. [Google Scholar] [CrossRef]
- Ray, S.; Gebre, S.; Fogle, H.; Berrios, D.C.; Tran, P.B.; Galazka, J.M.; Costes, S.V. GeneLab: Omics database for spaceflight experiments. Bioinformatics 2019, 35, 1753–1759. [Google Scholar] [CrossRef]
- Baldi, P. The design, analysis, and interpretation of gene expression profiling experiments. In DNA Microarrays and Gene Expression; Cambridge University Press: Cambridge, UK, 2002; pp. 97–134. [Google Scholar]
- Groen, A.K. The pros and cons of gene expression analysis by microarrays. J. Hepatol. 2001, 35, 295–296. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Hung, J.-H.; Weng, Z. Analysis of microarray and RNA-seq expression profiling data. Cold Spring Harb. Protoc. 2017, 2017, pdb–top093104. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zhang, Y.; Williams, J.; Antoniou, E.; McCombie, W.R.; Wu, S.; Zhu, W.; Davidson, N.O.; Denoya, P.; Li, E. Parallel comparison of Illumina RNA-Seq and Affymetrix microarray platforms on transcriptomic profiles generated from 5-aza-deoxy-cytidine treated HT-29 colon cancer cells and simulated datasets. BMC Bioinform. 2013, 14, S1. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Gong, B.; Bushel, P.R.; Thierry-Mieg, J.; Thierry-Mieg, D.; Xu, J.; Fang, H.; Hong, H.; Shen, J.; Su, Z.; et al. The concordance between RNA-seq and microarray data depends on chemical treatment and transcript abundance. Nat. Biotechnol. 2014, 32, 926–932. [Google Scholar] [CrossRef]
- Thiel, C.S.; Christoffel, S.; Tauber, S.; Vahlensieck, C.; de Zélicourt, D.; Layer, L.E.; Lauber, B.; Polzer, J.; Ullrich, O. Rapid cellular perception of gravitational forces in human jurkat t cells and transduction into gene expression regulation. Int. J. Mol. Sci. 2020, 21, 514. [Google Scholar] [CrossRef]
- Sabbah, H.N.; Khaja, F.; Brymer, J.F.; McFarland, T.M.; Albert, D.E.; Snyder, J.E.; Goldstein, S.; Stein, P.D. Noninvasive evaluation of left ventricular performance based on peak aortic blood acceleration measured with a continuous-wave Doppler velocity meter. Circulation 1986, 74, 323–329. [Google Scholar] [CrossRef]
- Chemla, D.; Démolis, P.; Thyrault, M.; Annane, D.; Lecarpentier, Y.; Giudicelli, J.F. Blood flow acceleration in the carotid and brachial arteries of healthy volunteers: Respective contributions of cardiac performance and local resistance. Fundam. Clin. Pharmacol. 1996, 10, 393–399. [Google Scholar] [CrossRef]
- Kogenaru, S.; Qing, Y.; Guo, Y.; Wang, N. RNA-seq and microarray complement each other in transcriptome profiling. BMC Genom. 2012, 13, 629. [Google Scholar] [CrossRef]
- Mills, J.D.; Kawahara, Y.; Janitz, M. Strand-specific RNA-seq provides greater resolution of transcriptome profiling. Curr. Genom. 2013, 14, 173–181. [Google Scholar] [CrossRef]
- Zhang, W.; Yu, Y.; Hertwig, F.; Thierry-Mieg, J.; Zhang, W.; Thierry-Mieg, D.; Wang, J.; Furlanello, C.; Devanarayan, V.; Cheng, J.; et al. Comparison of RNA-seq and microarray-based models for clinical endpoint prediction. Genome Biol. 2015, 16, 133. [Google Scholar] [CrossRef]
- Zhao, S.; Fung-Leung, W.-P.; Bittner, A.; Ngo, K.; Liu, X. Comparison of RNA-seq and microarray in transcriptome profiling of activated T cells. PLoS ONE 2014, 9, e78644. [Google Scholar] [CrossRef] [PubMed]
- Ujvari, A.; Chamnongpol, S.; Kourennaia, O.; Myers, K.; Bressler, V.; Hubbard, J.; Schweitzer, A.; Kubu, C. Concordance of Affymetrix GeneChip® Human Transcriptome Array 2.0 and USB® VeriQuest™ real-time PCR data (LB207). FASEB J. 2014, 28 (Suppl. S1), LB207. [Google Scholar] [CrossRef]
- Chang, T.T.; Walther, I.; Li, C.F.; Boonyaratanakornkit, J.; Galleri, G.; Meloni, M.A.; Pippia, P.; Cogoli, A.; Hughes-Fulford, M. The Rel/NF-κB pathway and transcription of immediate early genes in T cell activation are inhibited by microgravity. J. Leukoc. Biol. 2012, 92, 1133–1145. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, B.; Seetharam, A.; Wang, Z.; Liu, Y.; Lossie, A.C.; Thimmapuram, J.; Irudayaraj, J. A study of alterations in DNA epigenetic modifications (5mC and 5hmC) and gene expression influenced by simulated microgravity in human lymphoblastoid cells. PLoS ONE 2016, 11, e0147514. [Google Scholar] [CrossRef]
- Ziegler, S.F.; Ramsdell, F.; Alderson, M.R. The activation antigen CD69. Stem Cells 1994, 12, 456–465. [Google Scholar] [CrossRef] [PubMed]
- Ritz, B.W.; Lelkes, P.I.; Gardner, E.M. Functional recovery of peripheral blood mononuclear cells in modeled microgravity. FASEB J. 2006, 20, 305–307. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Morukov, B.; Rykova, M.; Antropova, E.; Berendeeva, T.; Ponomaryov, S.; Larina, I. T-cell immunity and cytokine production in cosmonauts after long-duration space flights. Acta Astronaut. 2011, 68, 739–746. [Google Scholar] [CrossRef]
- Hauschild, S.; Tauber, S.; Lauber, B.; Thiel, C.S.; Layer, L.E.; Ullrich, O. T cell regulation in microgravity—The current knowledge from in vitro experiments conducted in space, parabolic flights and ground-based facilities. Acta Astronaut. 2014, 104, 365–377. [Google Scholar] [CrossRef]
- Risso, A.; Tell, G.; Vascotto, C.; Costessi, A.; Arena, S.; Scaloni, A.; Cosulich, M.E. Activation of human T lymphocytes under conditions similar to those that occur during exposure to microgravity: A proteomics study. Proteomics 2005, 5, 1827–1837. [Google Scholar] [CrossRef] [PubMed]
- Howe, K.L.; Achuthan, P.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; Bhai, J.; et al. Ensembl 2021. Nucleic Acids Res. 2021, 49, D884–D891. [Google Scholar] [CrossRef] [PubMed]
- The UniProt Consortium. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards suite: From gene data mining to disease genome sequence analyses. Curr. Protoc. Bioinform. 2016, 54, 1–30. [Google Scholar] [CrossRef]
- Kalina, T.; Fišer, K.; Pérez-Andrés, M.; Kuzílková, D.; Cuenca, M.; Bartol, S.J.W.; Blanco, E.; Engel, P.; van Zelm, M.C. CD maps—Dynamic profiling of CD1–CD100 surface expression on human leukocyte and lymphocyte subsets. Front. Immunol. 2019, 10, 2434. [Google Scholar] [CrossRef]
- Afshinnekoo, E.; Scott, R.T.; MacKay, M.J.; Pariset, E.; Cekanaviciute, E.; Barker, R.; Gilroy, S.; Hassane, D.; Smith, S.M.; Zwart, S.R.; et al. Fundamental biological features of spaceflight: Advancing the field to enable deep-space exploration. Cell 2020, 183, 1162–1184. [Google Scholar] [CrossRef]
- Farhadian, M.; Rafat, S.A.; Hasanpur, K.; Ebrahimi, M.; Ebrahimie, E. Cross-species meta-analysis of transcriptomic data in combination with supervised machine learning models identifies the common gene signature of lactation process. Front. Genet. 2018, 9, 235. [Google Scholar] [CrossRef]
- Roux, J.; Rosikiewicz, M.; Robinson-Rechavi, M. What to compare and how: Comparative transcriptomics for Evo-Devo. J. Exp. Zool. Part B Mol. Dev. Evol. 2015, 324, 372–382. [Google Scholar] [CrossRef]
- da Silveira, W.A.; Fazelinia, H.; Rosenthal, S.B.; Laiakis, E.C.; Kim, M.S.; Meydan, C.; Kidane, Y.; Rathi, K.S.; Smith, S.M.; Stear, B.; et al. Comprehensive multi-omics analysis reveals mitochondrial stress as a central biological hub for spaceflight impact. Cell 2020, 183, 1185–1201.e20. [Google Scholar] [CrossRef]
- McDonald, J.T.; Stainforth, R.; Miller, J.; Cahill, T.; da Silveira, W.A.; Rathi, K.S.; Hardiman, G.; Taylor, D.; Costes, S.V.; Chauhan, V.; et al. NASA GeneLab platform utilized for biological response to space radiation in animal models. Cancers 2020, 12, 381. [Google Scholar] [CrossRef]
- Marioni, J.C.; Mason, C.E.; Mane, S.M.; Stephens, M.; Gilad, Y. RNA-seq: An assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 2008, 18, 1509–1517. [Google Scholar] [CrossRef] [PubMed]
- Mantione, K.J.; Kream, R.M.; Kuzelova, H.; Ptacek, R.; Raboch, J.; Samuel, J.M.; Stefano, G.B. Comparing bioinformatic gene expression profiling methods: Microarray and RNA-Seq. Med. Sci. Monit. Basic Res. 2014, 20, 138–142. [Google Scholar] [PubMed]
- Black, M.B.; Parks, B.B.; Pluta, L.; Chu, T.-M.; Allen, B.C.; Wolfinger, R.D.; Thomas, R.S. Comparison of microarrays and RNA-seq for gene expression analyses of dose-response experiments. Toxicol. Sci. Off. J. Soc. Toxicol. 2014, 137, 385–403. [Google Scholar] [CrossRef] [PubMed]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-Seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef]
- Romero, J.P.; Ortiz-Estévez, M.; Muniategui, A.; Carrancio, S.; de Miguel, F.J.; Carazo, F.; Montuenga, L.M.; Loos, R.; Pío, R.; Trotter, M.W.B.; et al. Comparison of RNA-seq and microarray platforms for splice event detection using a cross-platform algorithm. BMC Genom. 2018, 19, 703. [Google Scholar] [CrossRef]
- Mehmood, A.; Laiho, A.; Venäläinen, M.S.; McGlinchey, A.J.; Wang, N.; Elo, L.L. Systematic evaluation of differential splicing tools for RNA-seq studies. Brief. Bioinform. 2020, 21, 2052–2065. [Google Scholar] [CrossRef]
- Forgac, M. Vacuolar ATPases: Rotary proton pumps in physiology and pathophysiology. Nat. Rev. Mol. Cell Biol. 2007, 8, 917–929. [Google Scholar] [CrossRef]
- Inoue, T.; Wang, Y.; Jefferies, K.; Qi, J.; Hinton, A.; Forgac, M. Structure and regulation of the V-ATPases. J. Bioenerg. Biomembr. 2005, 37, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Holliday, L.S. Vacuolar H+-ATPase: An essential multitasking enzyme in physiology and pathophysiology. New J. Sci. 2014, 2014, e675430. [Google Scholar] [CrossRef]
- Huynh, K.K.; Grinstein, S. Regulation of vacuolar pH and its modulation by some microbial species. Microbiol. Mol. Biol. Rev. 2007, 71, 452–462. [Google Scholar] [CrossRef] [PubMed]
- Mellman, I.; Fuchs, R.; Helenius, A. Acidification of the endocytic and exocytic pathways. Annu. Rev. Biochem. 1986, 55, 663–700. [Google Scholar] [CrossRef]
- Nishi, T.; Forgac, M. The vacuolar (H+)-ATPases–nature’s most versatile proton pumps. Nat. Rev. Mol. Cell Biol. 2002, 3, 94–103. [Google Scholar] [CrossRef]
- Maxson, M.E.; Grinstein, S. The vacuolar-type H?-ATPase at a glance—More than a proton pump. J. Cell Sci. 2014, 127 Pt 23, 4987–4993. [Google Scholar] [CrossRef]
- Holliday, L.S.; Bubb, M.R.; Jiang, J.; Hurst, I.R.; Zuo, J. Interactions between vacuolar H+-ATPases and microfilaments in osteoclasts. J. Bioenerg. Biomembr. 2005, 37, 419–423. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, I.; Takahashi, N.; Udagawa, N.; Moriyama, Y.; Kurokawa, T.; Jimi, E.; Sasaki, T.; Suda, T. Lack of vacuolar proton ATPase association with the cytoskeleton in osteoclasts of osteosclerotic (oc/oc) mice. FEBS Lett. 1997, 401, 207–212. [Google Scholar] [CrossRef]
- Vitavska, O.; Merzendorfer, H.; Wieczorek, H. The V-ATPase subunit C binds to polymeric F-actin as well as to monomeric G-actin and induces cross-linking of actin filaments. J. Biol. Chem. 2005, 280, 1070–1076. [Google Scholar] [CrossRef]
- Lewis, M.L.; Reynolds, J.L.; Cubano, L.A.; Hatton, J.P.; Lawless, B.D.; Piepmeier, E.H. Spaceflight alters microtubules and increases apoptosis in human lymphocytes (Jurkat). FASEB J. 1998, 12, 1007–1018. [Google Scholar] [CrossRef] [PubMed]
- Sciola, L.; Cogoli-Greuter, M.; Cogoli, A.; Spano, A.; Pippia, P. Influence of microgravity on mitogen binding and cytoskeleton in Jurkat cells. Adv. Space Res. 1999, 24, 801–805. [Google Scholar] [CrossRef]
- Leclair, P.; Lim, C.J. CD47 (Cluster of differentiation 47): An anti-phagocytic receptor with a multitude of signaling functions. Anim. Cells Syst. 2020, 24, 243–252. [Google Scholar] [CrossRef]
- Wuest, S.L.; Gantenbein, B.; Ille, F.; Egli, M. Electrophysiological experiments in microgravity: Lessons learned and future challenges. NPJ Microgravity 2018, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, A.; Colonna, M. The role of NK cell recognition of nectin and nectin-like proteins in tumor immunosurveillance. Semin. Cancer Biol. 2006, 16, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Hashemi, B.B.; Penkala, J.E.; Vens, C.; Huls, H.; Cubbage, M.; Sams, C.F. T cell activation responses are differentially regulated during clinorotation and in spaceflight. FASEB J. 1999, 13, 2071–2082. [Google Scholar] [CrossRef] [PubMed]
- Bezouska, K.; Nepovim, A.; Horvath, O.; Pospisil, M.; Hamann, J.; Feizi, T. CD69 antigen of human lymphocytes is a calcium-dependent carbohydrate-binding protein. Biochem. Biophys. Res. Commun. 1995, 208, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Transcriptome Analysis Console (TAC) 4.0.1 User Guide. Affymetrix Inc.: Santa Clara, CA, USA, 2017.
- Lovén, J.; Orlando, D.A.; Sigova, A.A.; Lin, C.Y.; Rahl, P.B.; Burge, C.B.; Levens, D.L.; Lee, T.I.; Young, R.A. Revisiting Global Gene Expression Analysis. Cell 2012, 151, 476–482. [Google Scholar] [CrossRef]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef]
- Pelz, C.R.; Kulesz-Martin, M.; Bagby, G.; Sears, R.C. Global rank-invariant set normalization (GRSN) to reduce systematic distortions in microarray data. BMC Bioinf. 2008, 9, 520. [Google Scholar] [CrossRef]
- Landfors, M.; Philip, P.; Rydén, P.; Stenberg, P. Normalization of high dimensional genomics data where the distribution of the altered variables is skewed. PLoS ONE 2011, 6, e27942. [Google Scholar] [CrossRef]
- Cheng, L.; Lo, L.-Y.; Tang, N.L.S.; Wang, D.; Leung, K.-S. CrossNorm: A novel normalization strategy for microarray data in cancers. Sci. Rep. 2016, 6, 18898. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Hu, Z.; Xia, Z.; Zhao, D.; Li, W.; Tyler, J.K. The Overlooked Fact: Fundamental Need for Spike-In Control for Virtually All Genome-Wide Analyses. Mol. Cell. Biol. 2016, 36, 662–667. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, N.; Liu, S.; Wang, J.; Zhang, N.; Zheng, X.; Leung, K.-S.; Cheng, L. Normalization Methods for the Analysis of Unbalanced Transcriptome Data: A Review. Front. Bioeng. Biotechnol. 2019, 7, 358. [Google Scholar] [CrossRef]
- Evans, C.; Hardin, J.; Stoebel, D.M. Selecting between-sample RNA-Seq normalization methods from the perspective of their assumptions. Brief. Bioinf. 2017, 19, 776–792. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experiment | Cell Type | Platform | Altered Gravity | Exposure Time | Transcriptomics |
|---|---|---|---|---|---|
| Hypg15_microarray | Jurkat T cell | Pipette centrifuge | Hyper-gravity 9× g | 15 min | HTA2.0 microarray |
| Hypg15_RNA-Seq | Jurkat T cell | Pipette centrifuge | Hyper-gravity 9× g | 15 min | Paired end poly-A RNA-Seq |
| 23rd DLR PFC [18] | Jurkat T cell | Parabolic flight | Hyper-gravity 1.8× g | 20 s | HTA2.0 microarray |
| TEXUS-51 [18] | Jurkat T cell | Sounding rocket | Hyper-gravity ~9× g | 75 s | HTA2.0 microarray |
| GLDS-13 [43] | Activated primary human T cells | ISS, reference centrifuge | Micro-gravity, 1× g | 3 days µg, activation in µg and 1× g for 1.5 h | Human U133 Plus 2.0 |
| GLDS-91 [44] | TK6 lymphoblastoid cells | HARV Rotary Cell Culture System | Simulated micro-gravity | 48 h | Paired-end total RNA-Seq |
| Gene Name | Chromosome Location [50] | UniProt Subcellular Localization [51] | Human Gene Database Summary, Abbreviated [52] | T Cell Bearing [53] |
|---|---|---|---|---|
| CD53 molecule | 1p13.3 | Cell membrane, cell junction | Transmembrane 4 superfamily, signal transduction events that play a role in the regulation of cell development, activation, growth, and motility. Cell surface glycoprotein that is known to complex with integrins. It contributes to the transduction of CD2-generated signals in T cells and natural killer cells and has been suggested to play a role in growth regulation. | + |
| CD1E molecule | 1q23.1 | Golgi apparatus membrane, early and late endosome | Transmembrane glycoproteins, T cell surface glycoprotein CD1e, soluble, binds diacetylated lipids, including phosphatidyl inositides and diacylated sulfoglycolipids, and is required for the presentation of glycolipid antigens on the cell surface. | ? |
| CD96 molecule | 3q13.13 | Membranes | Immunoglobulin superfamily type I membrane protein with role in the adhesive interactions of activated T cells during the late phase of the immune response. | + |
| CD47 molecule | 3q13.12 | Membranes | Membrane protein, involved in the increase in intracellular calcium concentration that occurs upon cell adhesion to extracellular matrix. | + |
| CD46 molecule | 1q32.2 | Membranes | Type I membrane protein, regulatory part of the complement system, cofactor activity for inactivation of complement components C3b and C4b by serum factor I, protects the host cell from damage by complement. | + |
| CD226 molecule | 18q22.2 | Cell membrane | Glycoprotein expressed on the surface of a subset of T cells. Involved in intercellular adhesion, lymphocyte signaling. | + |
| CD164 molecule | 6q21 | Membranes | Encodes a transmembrane sialomucin and cell adhesion molecule that regulates the proliferation, adhesion, and migration of hematopoietic progenitor cells. | + |
| CD2AP (associated protein) | 6p12.3 | Membrane ruffle, Cell junction, Cytoskeleton | Directly interacts with filamentous actin and a variety of cell membrane proteins through multiple actin binding sites, SH3 domains, proline-rich region containing binding sites for SH3 domains, adapter protein between membrane proteins, and the actin cytoskeleton. | + |
| CD69 molecule | 12p13.31 | Membranes | Calcium-dependent lectin superfamily of type II transmembrane receptors. Expression of the encoded protein is induced upon activation of T lymphocytes, may play a role in proliferation. Involved in lymphocyte proliferation and functions as a signal transmitting receptor in lymphocytes, natural killer (NK) cells, and platelets. | + |
| Previously Known | Novel Insights | |
|---|---|---|
| Transcriptional response over time | Rapid adaptation, no continuous response [18]. Stable region 11p15.4 [19]. | Complex dynamics with counter-response between 75 s and 15 min (Figure 5, Figure 6 and Figure 7) |
| Cross-validation of effects | Hyper- and microgravity cross-validation at time points 20 s, 75 s, 5 min [18]. | 15 min time point validation by independent transcriptomics analysis technologies (RNA microarray, RNA-Seq) |
| Gene set effects | GO “Histone binding function” enriched for TEXUS-54 µg [18]. GO “Transport”, “Cytosol”, “Nucleotide binding”, “Poly(A) RNA binding”, “Nuclear speck”, “RNA binding”, “Intracellular membrane-bound organelle”, “Regulation of alternative mRNA splicing via spliceosome” enriched for hyp-g in TEXUS-51 and GBF study [35]. GO “GPCR signaling”, “zing finger-involved DNA binding”, “bacterial defense”, “sensory perception genes” enriched in stable genes [19]. | Counter-response between 20 s/75 s and 15 min (Figure 9) |
| ATP6V | Initially described: ATP6V1A significantly upregulated in hyp-g for TEXUS-51, ATP6V1D for 23rd DLR PFC [18]. | Counter-response between 20 s/75 s and 15 min (Figure 10), long-term effects (Figure 10) |
| CD molecules | - | Strongly downregulated after 15 min, counter-response and long-term effects (Figure 10) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vahlensieck, C.; Thiel, C.S.; Adelmann, J.; Lauber, B.A.; Polzer, J.; Ullrich, O. Rapid Transient Transcriptional Adaptation to Hypergravity in Jurkat T Cells Revealed by Comparative Analysis of Microarray and RNA-Seq Data. Int. J. Mol. Sci. 2021, 22, 8451. https://doi.org/10.3390/ijms22168451
Vahlensieck C, Thiel CS, Adelmann J, Lauber BA, Polzer J, Ullrich O. Rapid Transient Transcriptional Adaptation to Hypergravity in Jurkat T Cells Revealed by Comparative Analysis of Microarray and RNA-Seq Data. International Journal of Molecular Sciences. 2021; 22(16):8451. https://doi.org/10.3390/ijms22168451
Chicago/Turabian StyleVahlensieck, Christian, Cora S. Thiel, Jan Adelmann, Beatrice A. Lauber, Jennifer Polzer, and Oliver Ullrich. 2021. "Rapid Transient Transcriptional Adaptation to Hypergravity in Jurkat T Cells Revealed by Comparative Analysis of Microarray and RNA-Seq Data" International Journal of Molecular Sciences 22, no. 16: 8451. https://doi.org/10.3390/ijms22168451
APA StyleVahlensieck, C., Thiel, C. S., Adelmann, J., Lauber, B. A., Polzer, J., & Ullrich, O. (2021). Rapid Transient Transcriptional Adaptation to Hypergravity in Jurkat T Cells Revealed by Comparative Analysis of Microarray and RNA-Seq Data. International Journal of Molecular Sciences, 22(16), 8451. https://doi.org/10.3390/ijms22168451