
Amaryllidaceae Alkaloids of Norbelladine-Type as Inspiration for Development of Highly Selective Butyrylcholinesterase Inhibitors: Synthesis, Biological Activity Evaluation, and Docking Studies

 ,
,  , , , ,
, , , ,  , , , , ,
, , , , ,  , ,
, ,  ,
,  and
and 
Abstract
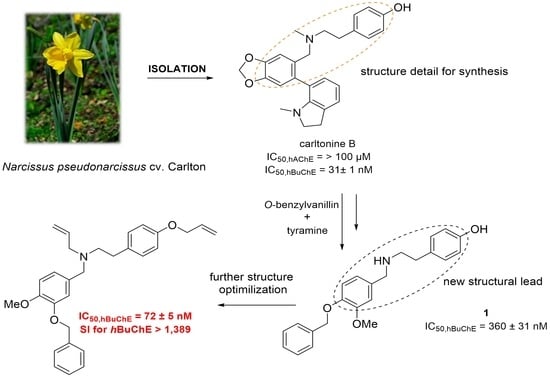
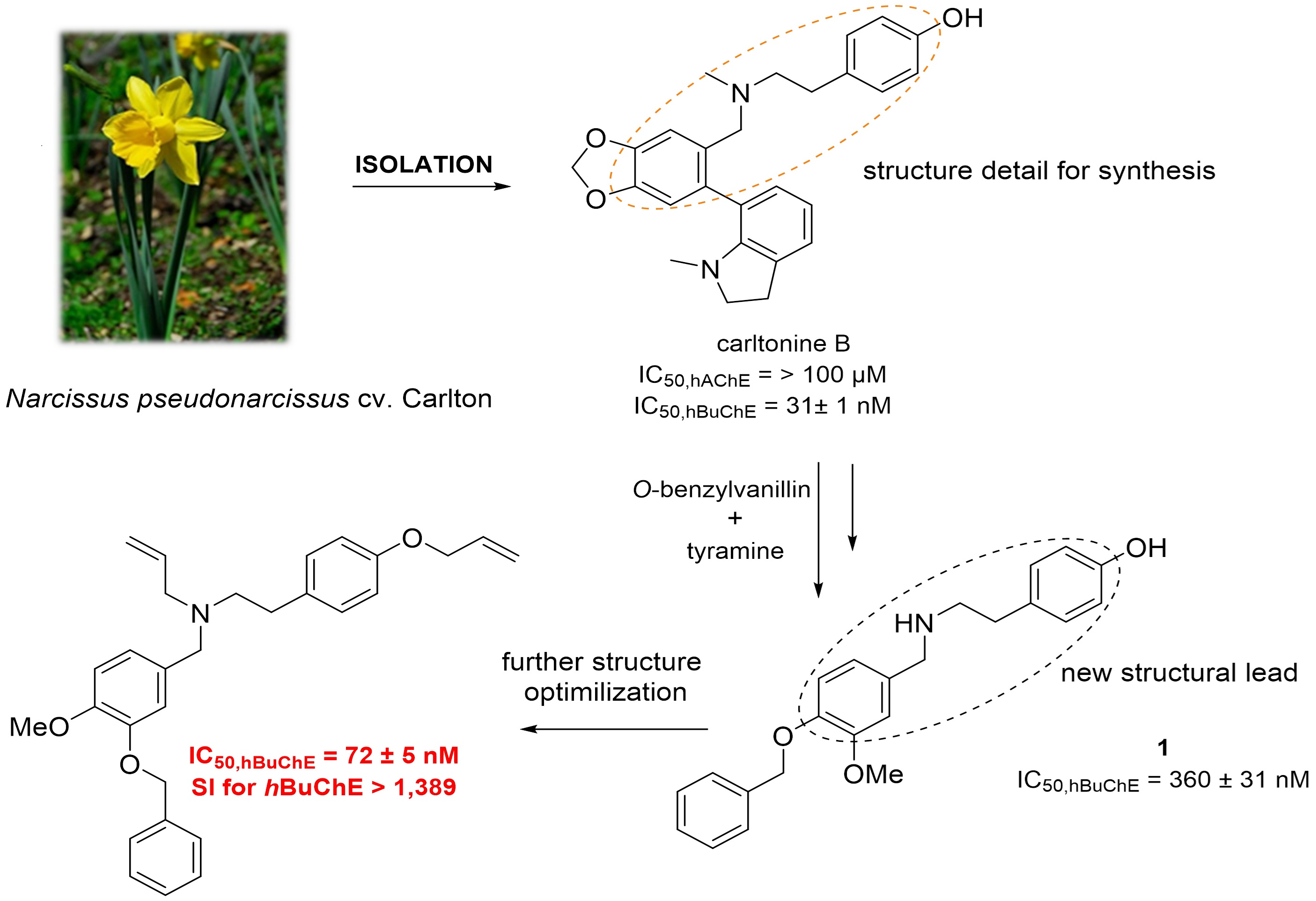
1. Introduction
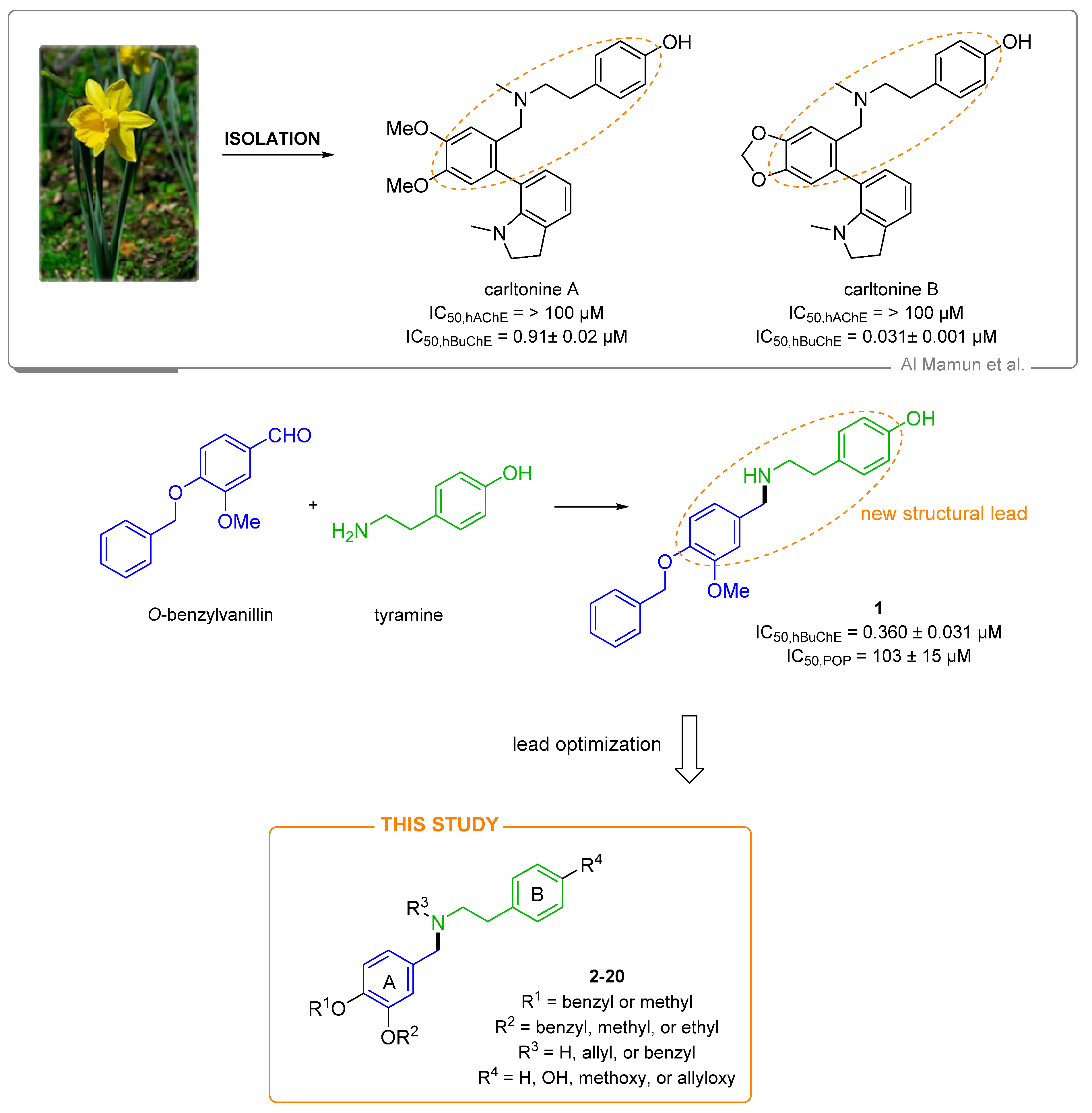
2. Design
3. Results and Discussion
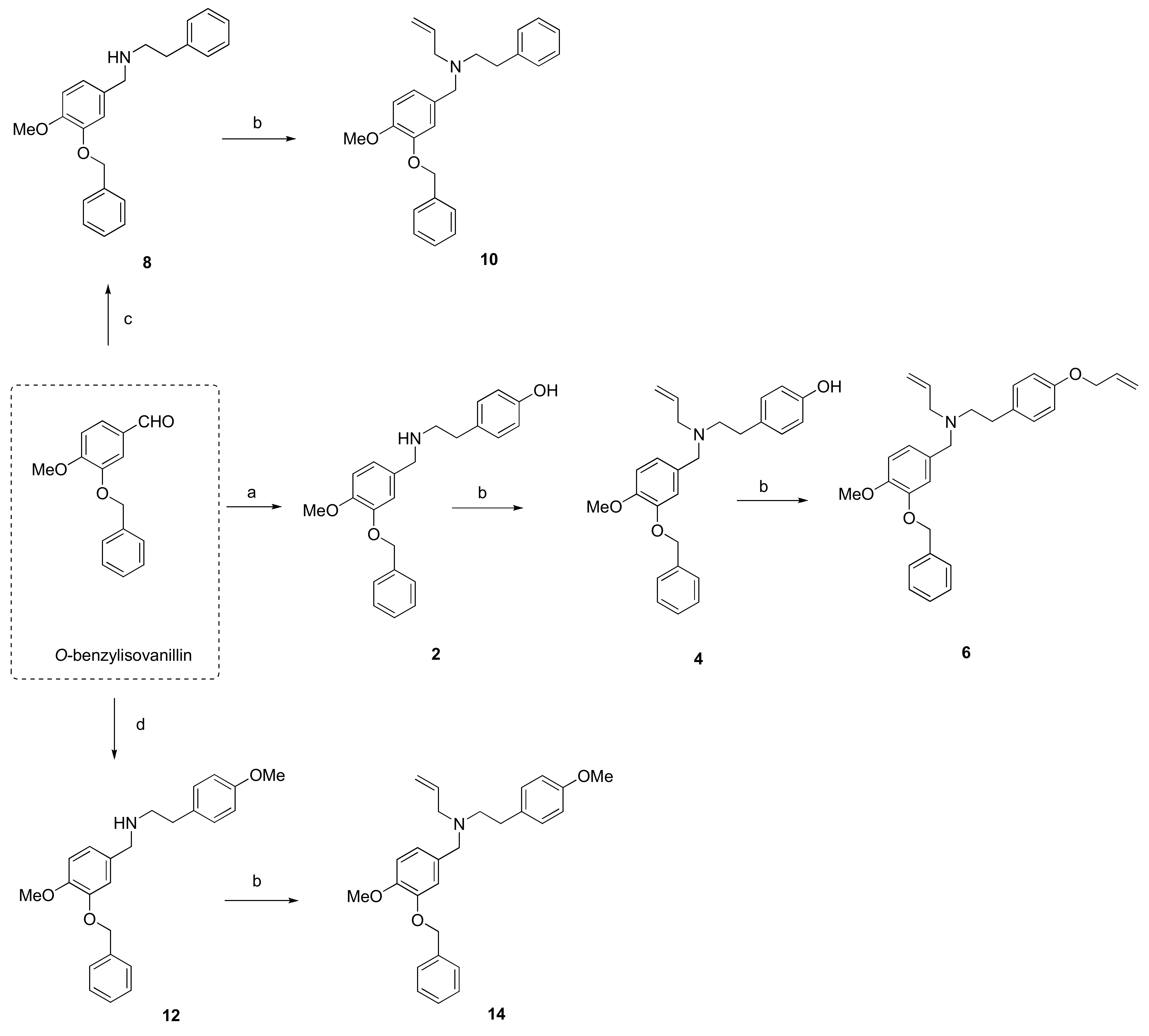
3.1. Synthesis of Novel hBuChE Inhibitors from O-benzylvanillin, O-benzylisovanillin, and 3-ethoxy-4-methoxybenzaldehyde (1–20)
3.2. In Vitro Cholinesterase Inhibitory Activities of New Norbelladine Derivatives (1–20)
3.3. POP Inhibition Activity of Selected Norbelladine Derivatives
3.4. Prediction of CNS Availability
3.5. Enzyme Kinetic Analysis of Compound 6
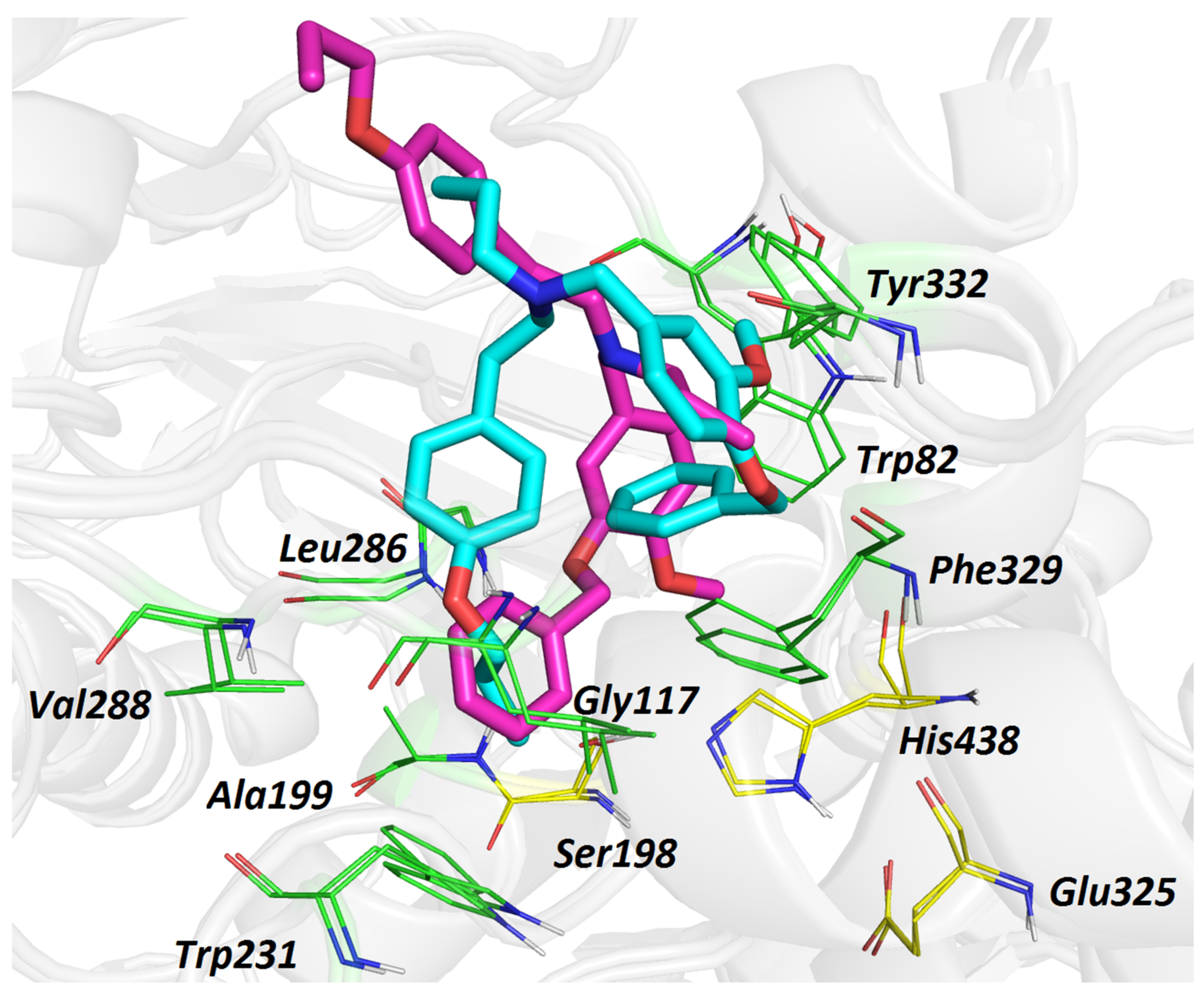
3.6. Molecular Dynamic Simulation for Compounds 5 and 6
3.7. Cytotoxicity of Compound 6
3.8. MAO-A and MAO-B Inhibition Potency of Compound 6
4. Conclusions
5. Materials and Methods
5.1. General Experimental Procedures
5.2. Preparation of Carltonine Derivatives: General Procedure for Preparation of Compounds 1, 2, 7, 8, 11, 12, 15, and 18
5.2.1. N-(4-benzyloxy-3-methoxybenzyl)-2-phenylethan-1-amine (7)
5.2.2. N-(3-benzyloxy-4-methoxybenzyl)-2-phenylethan-1-amine (8)
5.2.3. {[4-(benzyloxy)-3-methoxyphenyl]methyl}[2-(4-methoxyphenyl)ethyl]amine (11)
5.2.4. N-(3-benzyloxy-4-methoxybenzyl)-2-(4-methoxyphenyl)ethan-1-amine (12)
5.2.5. N-(3-ethoxy-4-methoxybenzyl)-2-(4-hydroxyphenyl)ethan-1-amine (15)
5.2.6. N-(3-ethoxy-4-methoxybenzyl)-2-phenylethan-1-amine (18)
5.3. Preparation of Carltonine Derivatives: General Procedure for Acylation to Give Compounds 3–6, 9, 10, 13, 14, 16, 17, 19, 20
5.3.1. N-allyl-N-(4-benzyloxy-3-methoxybenzyl)-2-(4-hydroxyphenyl)ethan-1-amine (3)
5.3.2. N-allyl-N-(3-benzyloxy-4-methoxybenzyl)-2-(4-hydroxyphenyl)ethan-1-amine (4)
5.3.3. N-allyl-N-(4-benzyloxy-3-methoxybenzyl)-2-(4-allyloxyphenyl)ethan-1-amine (5)
5.3.4. N-allyl-N-(3-benzyloxy-4-methoxybenzyl)-2-(4-allyloxyphenyl)ethan-1-amine (6)
5.3.5. N-allyl-N-(4-benzyloxy-3-methoxybenzyl)-2-phenylethan-1-amine (9)
5.3.6. N-allyl-N-(3-benzyloxy-4-methoxybenzyl)-2-phenylethan-1-amine (10)
5.3.7. N-allyl-N-(4-benzyloxy-3-methoxybenzyl)-2-(4-methoxyphenyl)ethan-1-amine (13)
5.3.8. N-allyl-N-(3-benzyloxy-4-methoxybenzyl)-2-(4-methoxyphenyl)ethan-1-amine (14)
5.3.9. N-allyl-N-(3-ethoxy-4-methoxybenzyl)-2-(4-hydroxyphenyl)ethan-1-amine (16)
5.3.10. N-allyl-N-(3-ethoxy-4-methoxybenzyl)-2-(4-allyloxyphenyl)ethan-1-amine (17)
5.3.11. N-allyl-N-(3-ethoxy-4-methoxybenzyl)-2-phenylethan-1-amine (19)
5.3.12. N-benzyl-N-(3-ethoxy-4-methoxybenzyl)-2-phenylethan-1-amine (20)
5.4. hAChE and hBuChE Inhibition Assay
5.5. Kinetic Study of hBuChE Inhibition
5.6. POP Inhibition Assay
5.7. MAOs Inhibition Assay
5.8. Cytotoxicity Assay
5.9. In-Silico Studies
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Cummings, J.L. Alzheimer’s disease. N. Engl. J. Med. 2004, 351, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Nichols, E.; Szoeke, C.E.I.; Vollset, S.E.; Abbasi, N.; Abd-Allah, F.; Abdela, J.; Aichour, M.T.E.; Akinyemi, R.O.; Alahdab, F.; Asgedom, S.W.; et al. Global, Regional, and National Burden of Alzheimer’s Disease and Other Dementias, 1990–2016: A Systematic Analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 88–106. [Google Scholar] [CrossRef]
- Davies, P.; Maloney, A.J. Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet 1976, 308, 1403. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Liu, Y.; Nguyen, M.; Robert, A.; Meunier, B. Metal Ions in Alzheimer’s Disease: A Key Role or Not? Acc. Chem. Res. 2019, 52, 2026–2035. [Google Scholar] [CrossRef]
- Rukhsana, S.; Patrizia, M.; Francesca, M.; Roberta, C.; Maurob, B.; Allan, B.D. Increased protein and lipid oxidative damage in mitochondria isolated from lymphocytes from patients with Alzheimer’s disease: Insights into the role of oxidative stress in Alzheimer’s disease and initial investigations into a potential biomarker for this dementing disorder. J. Alzheimer’s Dis. 2011, 24, 77–84. [Google Scholar] [CrossRef]
- Hampel, H.; Mesulam, M.; Cuello, A.C.; Farlow, M.R.; Giacobini, E.; Grossberg, G.T.; Khachaturian, A.S.; Vergallo, A.; Cavedo, E.; Snyder, P.J.; et al. The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain 2018, 141, 1917–1933. [Google Scholar] [CrossRef]
- Zemek, F.; Drtinova, L.; Nepovimova, E.; Sepsova, V.; Korabecny, J.; Klimes, J.; Zemek, K.K. Outcomes of Alzheimer’s disease therapy with acetylcholinesterase inhibitors and memantine. Expert Opin. Drug Saf. 2014, 13, 759–774. [Google Scholar] [CrossRef]
- Reid, G.A.; Chilukuri, N.; Darvesh, S. Butyrylcholinesterase and the cholinergic system. Neuroscience 2013, 234, 53–68. [Google Scholar] [CrossRef]
- Ramos-Rodriguez, J.J.; Pacheco-Herrero, M.; Thyssen, D.; Murillo-Carretero, M.I.; Berrocoso, E.; Spires-Jones, T.L.; Bacskai, B.J.; Garcia-Alloza, M. Rapid β-amyloid deposition and cognitive impairment after cholinergic denervation in APP/PS1 mice. J. Neuropathol. Exp. Neurol. 2013, 72, 272–285. [Google Scholar] [CrossRef]
- Mori, F.; Lai, C.C.; Fusi, F.; Giacobini, E. Cholinesterase inhibitors increase secretion of APPs in rat brain cortex. Neuroreport 1995, 6, 633–636. [Google Scholar] [CrossRef]
- Inestrosa, N.C.; Alvarez, A.; Pérez, C.A.; Moreno, R.D.; Vicente, M.; Linker, C.; Casanueva, O.I.; Soto, C.; Garrido, J. Acetylcholinesterase accelerates assembly of amyloid-beta-peptides into Alzheimer’s fibrils: Possible role of the peripheral site of the enzyme. Neuron 1996, 16, 881–891. [Google Scholar] [CrossRef]
- Bartolini, M.; Bertucci, C.; Cavrini, V.; Andrisano, V. Beta-Amyloid aggregation induced by human acetylcholinesterase: Inhibition studies. Biochem. Pharmacol. 2003, 65, 407–416. [Google Scholar] [CrossRef]
- Darvesh, S.; Cash, M.K.; Reid, G.A.; Martin, E.; Mitnitski, A.; Geula, C. Butyrylcholinesterase is associated with β-amyloid plaques in the transgenic APPSWE/PSEN1dE9 mouse model of Alzheimer disease. J. Neuropathol. Exp. Neurol. 2012, 71, 2–14. [Google Scholar] [CrossRef]
- Sridhar, G.R.; Nirmala, G.; Apparao, A.; Madhavi, A.S.; Sreelatha, S.; Rani, J.S.; Vijayalakshmi, P. Serum butyrylcholinesterase in type 2 diabetes mellitus: A biochemical and bioinformatics approach. Lipids Health Dis. 2005, 4, 18. [Google Scholar] [CrossRef]
- Verdile, G.; Fuller, S.J.; Martins, R.N. The role of type 2 diabetes in neurodegeneration. Neurobiol. Dis. 2015, 84, 22–38. [Google Scholar] [CrossRef]
- Padnya, P.L.; Bayarashov, E.E.; Zueva, I.V.; Lushchekina, S.V.; Lenina, O.A.; Evtugyn, G.E.; Osin, Y.N.; Petrov, K.A.; Stoikov, I.I. Water-soluble betaines and amines based on thiacalix[4]arene scaffold as new cholinesterase inhibitors. Bioorg. Chem. 2020, 94, 103455. [Google Scholar] [CrossRef]
- Makhaeva, G.F.; Kovaleva, N.V.; Rudakova, E.V.; Boltneva, N.P.; Lushchekina, S.V.; Faingolg, I.I.; Poletaeva, D.A.; Soldanova, Y.V.; Kotelnikova, R.A.; Serkov, I.V.; et al. New Multifunctional Agents Based on Conjugates of 4-Amino-2,3-polymethylenequinoline and Butylated Hydroxytoluene for Alzheimer’s Disease Treatment. Molecules 2020, 25, 5891. [Google Scholar] [CrossRef]
- Kos, J.; Kozik, V.; Pindjakova, D.; Jankech, T.; Smolinski, A.; Stepankova, S.; Hosek, J.; Oravec, M.; Jampilek, J.; Bak, A. Synthesis and Hybrid SAR Property Modeling of Novel Cholinesterase Inhibitors. Int. J. Mol. Sci. 2021, 22, 3444. [Google Scholar] [CrossRef]
- Singh, Y.P.; Tej, G.N.V.C.; Pandey, A.; Priya, K.; Pandey, P.; Shankar, G.; Nayak, P.K.; Rai, G.; Chittiboyina, A.G.; Doerksen, R.J.; et al. Design, synthesis and biological evaluation of novel naturally-inspired multifunctional molecules for the management of Alzheimer’s disease. Eur. J. Med. Chem. 2020, 198, 112257. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef]
- Wang, S.; Dong, G.; Sheng, C. Structural Simplification of Natural Products. Chem. Rev. 2019, 119, 4180–4220. [Google Scholar] [CrossRef]
- Guo, Z. The modification of natural products for medical use. Acta Pharm. Sin. B 2017, 7, 119–136. [Google Scholar] [CrossRef]
- . Wu, X.; Cai, H.; Pan, L.; Cui, G.; Qin, F.; Li, Y.C.; Cai, Z. Small Molecule Natural Products and Alzheimer’s Disease. Curr. Top. Med. Chem. 2019, 19, 187–204. [Google Scholar] [CrossRef]
- Masondo, N.A.; Stafford, G.I.; Aremu, A.O.; Makunga, N.P. Acetylcholinesterase inhibitors from southern African plants: An overview of ethnobotanical, pharmacological potential and phytochemical research including and beyond Alzheimer’s disease treatment. S. Afr. J. Bot. 2019, 120, 39–64. [Google Scholar] [CrossRef]
- Dey, A.; Bhattacharya, R.; Mukherjee, A.; Pandey, D.K. Natural products against Alzheimer’s disease: Pharmaco-therapeutics and biotechnological interventions. Biotechnol. Adv. 2017, 35, 178–216. [Google Scholar] [CrossRef]
- Houghton, P.J.; Ren, Y.; Howes, M. Acetylcholinesterase inhibitors from plants and fungi. Nat. Prod. Rep. 2006, 23, 181–199. [Google Scholar] [CrossRef]
- Mamun, A.A.; Maříková, J.; Hulcová, D.; Janoušek, J.; Šafratová, M.; Nováková, L.; Kučera, T.; Hrabinová, M.; Kuneš, J.; Korábečný, J.; et al. Amaryllidaceae Alkaloids of Belladine-Type from Narcissus pseudonarcissus cv. Carlton as New Selective Inhibitors of Butyrylcholinesterase. Biomolecules 2020, 10, 800. [Google Scholar] [CrossRef] [PubMed]
- Schulz, I.; Gerhartz, B.; Neubauer, A.; Holloschi, A.; Heiser, U.; Hafner, M.; Demuth, H. Modulation of inositol 1,4,5-triphosphate concentration by prolyl endopeptidase inhibition. Eur. J. Biochem. 2002, 269, 5813–5820. [Google Scholar] [CrossRef] [PubMed]
- Cahlíková, L.; Kawano, I.; Řezáčová, M.; Blunden, G.; Hulcová, D.; Havelek, R. The Amaryllidaceae alkaloids haemanthamine, haemanthidine and their semisynthetic derivatives as potential drugs. Phytochem. Rev. 2020, 20, 303–323. [Google Scholar] [CrossRef]
- He, M.; Qu, C.; Gao, O.; Hua, X.; Hong, X. Biological and pharmacological activities of amaryllidaceae alkaloids. RSC Adv. 2015, 5, 16562–16574. [Google Scholar] [CrossRef]
- Kornienko, A.; Evidente, A. Chemistry, biology, and medicinal potential of narciclasine and its congeners. Chem. Rev. 2008, 108, 1982–2014. [Google Scholar] [CrossRef]
- Koutová, D.; Maafi, N.; Havelek, R.; Opletal, L.; Blunden, G.; Řezáčová, M.; Cahlíková, L. Chemical and Biological Aspects of Montanine-Type Alkaloids Isolated from Plants of the Amaryllidaceae Family. Molecules 2020, 25, 2337. [Google Scholar] [CrossRef]
- Panek, D.; Wieckowska, A.; Pasieka, A.; Godyn, J.; Jonczyk, J.; Bajda, M.; Knez, D.; Gobec, S.; Malawska, B. Design, Synthesis, and Biological Evaluation of 2-(Benzylamino-2-Hydroxyalkyl)Isoindoline-1,3-Diones Derivatives as Potential Disease-Modifying Multifunctional Anti-Alzheimer Agents. Molecules 2018, 23, 347. [Google Scholar] [CrossRef]
- Panek, D.; Więckowska, A.; Wichur, T.; Bajda, M.; Godyń, J.; Jończyk, J.; Mika, K.; Janockova, J.; Soukup, O.; Knez, D.; et al. Design, synthesis and biological evaluation of new phthalimide and saccharin derivatives with alicyclic amines targeting cholinesterases, beta-secretase and amyloid beta aggregation. Eur. J. Med. Chem. 2017, 125, 676–695. [Google Scholar] [CrossRef]
- Panek, D.; Więckowska, A.; Jonczyk, J.; Godyn, J.; Bajda, M.; Wichur, T.; Pasieka, A.; Knez, D.; Pislar, A.; Korabecny, J.; et al. Design, Synthesis, and Biological Evaluation of 1-Benzylamino-2-hydroxyalkyl Derivatives as New Potential Disease-Modifying Multifunctional Anti-Alzheimer’s Agents. ACS Chem. Neurosci. 2018, 9, 1074–1094. [Google Scholar] [CrossRef]
- Kodama, S.; Takita, H.; Kajimoto, T.; Nishide, K.; Node, M. Synthesis of Amaryllidaceae alkaloids, siculine, oxocrinine, epicrinine, and buflavine. Tetrahedron 2004, 60, 4901–4907. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.D.; Andresjr, V.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Xing, S.; Li, Q.; Xiong, B.; Chen, Y.; Feng, F.; Liu, W.; Sun, H. Structure and therapeutic uses of butyrylcholinesterase: Application in detoxification, Alzheimer’s disease, and fat metabolism. Med. Res. Rev. 2021, 41, 858–901. [Google Scholar] [CrossRef]
- Carmona-Viglianco, F.; Zaragoza-Puchol, D.; Parravicini, O.; Garro, A.; Enriz, R.D.; Feresin, G.E.; Kurina-Sanz, M.; Orden, A.A. Synthesis, biological evaluation and molecular modeling studies of substituted N-benzyl-2-phenylethanamines as cholinesterase inhibitors. New J. Chem. 2020, 44, 9466–9476. [Google Scholar] [CrossRef]
- Tarrago, T.; Kichik, N.; Segui, J.; Giralt, E. The natural product berberine is a human prolyl oligopeptidase inhibitor. Chem. Med. Chem. 2007, 2, 354–359. [Google Scholar] [CrossRef]
- Gupta, M.; Lee, H.J.; Barden, C.J.; Weaver, D.F. The Blood–Brain Barrier (BBB) Score. J. Med. Chem. 2019, 62, 9824–9836. [Google Scholar] [CrossRef]
- Meden, A.; Knez, D.; Jukič, M.; Brazzolotto, X.; Gršič, M.; Pišlar, A.; Zahirović, A.; Kos, J.; Nachon, F.; Svete, J.; et al. Tryptophan-derived butyrylcholinesterase inhibitors as promising leads against Alzheimer’s disease. Chem. Commun. 2019, 55, 3765–3768. [Google Scholar] [CrossRef]
- Brus, B.; Košak, U.; Turk, S.; Pišlar, A.; Coquelle, N.; Kos, J.; Stojan, J.; Colletier, J.; Gobec, S. Discovery, Biological Evaluation, and Crystal Structure of a Novel Nanomolar Selective Butyrylcholinesterase Inhibitor. J. Med. Chem. 2014, 57, 8167–8179. [Google Scholar] [CrossRef]
- Hartmann, J.; Kiewert, C.; Duysen, E.G.; Lockridge, O.; Greig, N.H.; Klein, J. Excessive hippocampal acetylcholine levels in acetylcholinesterase-deficient mice are moderated by butyrylcholinesterase activity. J. Neurochem. 2007, 100, 1421–1429. [Google Scholar] [CrossRef]
- Greig, N.H.; Utsuki, T.; Ingram, D.K.; Wang, Y.; Pepeu, G.; Scali, C.; Yu, Q.-S.; Mamczarz, J.; Giordano, T.; Chen, D.; et al. Selective butyrylcholinesterase inhibition elevates brain acetylcholine, augments learning and lowers Alzheimer beta-amyloid peptide in rodent. Proc. Natl. Acad. Sci. USA 2005, 102, 17213–17218. [Google Scholar] [CrossRef]
- Meden, A.; Knez, D.; Malikowska-Racia, N.; Brazzolotto, X.; Nachon, F.; Svete, J.; Sałat, K.; Grošelj, U.; Gobec, S. Structure-activity relationship study of tryptophan-based butyrylcholinesterase inhibitors. Eur. J. Med. Chem. 2020, 208, 112766. [Google Scholar] [CrossRef]
- Knez, D.; Coquelle, N.; Pišlar, A.; Žakelj, S.; Jukič, M.; Sova, M.; Mravljak, J.; Nachon, F.; Brazzolotto, X.; Kos, J.; et al. Multi-target-directed ligands for treating Alzheimer’s disease: Butyrylcholinesterase inhibitors displaying antioxidant and neuroprotective activities. Eur. J. Med. Chem. 2018, 156, 598–617. [Google Scholar] [CrossRef]
- Henry, S.; Kidner, R.; Reisenauer, M.R.; Magedov, I.V.; Kiss, R.; Mathieu, V.; Lefranc, F.; Dasari, R.; Evidente, A.; Yu, X.; et al. 5,10b-Ethanophenanthridine amaryllidaceae alkaloids inspire the discovery of novel bicyclic ring systems with activity against drug resistant cancer cells. Eur. J. Med. Chem. 2016, 120, 313–328. [Google Scholar] [CrossRef]
- Hostalkova, A.; Marikova, J.; Opletal, L.; Korabecny, J.; Hulcova, D.; Kunes, J.; Novakova, L.; Perez, D.I.; Jun, D.; Kucera, T.; et al. Isoquinoline Alkaloids from Berberis vulgaris as Potential Lead Compounds for the Treatment of Alzheimer’s Disease. J. Nat. Prod. 2019, 82, 239–248. [Google Scholar] [CrossRef]
- Mezeiova, E.; Janockova, J.; Andrys, R.; Soukup, O.; Kobrlova, T.; Muckova, L.; Pejchal, J.; Simunkova, M.; Hand, J.; Micankova, P.; et al. 2-Propargylamino-naphthoquinone derivatives as multipotent agents for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2021, 211, 113112. [Google Scholar] [CrossRef] [PubMed]
- Fotakis, G.; Timbrell, J.A. In vitro cytotoxicity assays: Comparison of LDH, neutral red, MTT and protein assay in hepatoma cell lines following exposure to cadmium chloride. Toxicol. Lett. 2006, 160, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Pall, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Mark, P.; Nilsson, L. Structure and Dynamics of the TIP3P, SPC, and SPC/E Water Models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | %Inhibition hAChE ± SEM a | IC50, hAChE ± SEM (µM) b | % Inhibition hBuChE ± SEM a | IC50, hBuChE ± SEM (µM) b | SI for hBuChE c | IC50, POP ± SEM (µM) b | BBB Score d |
|---|---|---|---|---|---|---|---|
| 1 | 30.4 ± 2.1 | >100 | 98.7 ± 0.3 | 0.36 ± 0.03 | >277 | 186 ± 14 | 4.53 |
| 2 | 35.8 ± 1.2 | >100 | 97.7 ± 0.5 | 0.29 ± 0.02 | >348 | >79 f | 4.53 |
| 3 | 20.8 ± 0.9 | >100 | 96.8 ± 1.1 | 0.61 ± 0.04 | >163 | >200 f | 4.79 |
| 4 | 45.2 ± 2.4 | >100 | 97.9 ± 0.6 | 0.25 ± 0.01 | >394 | >79 f | 4.79 |
| 5 | 3.4 ± 0.5 | >100 | 38.9 ± 0.9 | >100 | - | n.s. | 4.87 |
| 6 | 10.1 ± 0.6 | >100 | 98.6 ± 0.9 | 0.07 ± 0.01 | >1,389 | >79 f | 4.87 |
| 7 | 23.4 ± 2.5 | >100 | 94.5 ± 0.9 | 1.28 ± 0.05 | >78 | n.s. | 5.15 |
| 8 | 12.6 ± 0.5 | >100 | 96.6 ± 0.4 | 1.10 ± 0.05 | >90 | n.s. | 5.15 |
| 9 | 18.8 ± 1.9 | >100 | 74.9 ± 2.4 | 5.19 ± 0.28 | >19 | n.s. | 5.04 |
| 10 | 72.4 ± 1.1 | 21.5 ± 0.6 | 92.0 ± 2.4 | 1.17 ± 0.04 | 18 | n.s. | 5.04 |
| 11 | 27.9 ± 0.7 | >100 | 93.5 ± 0.3 | 2.39 ± 0.27 | >41 | n.s. | 4.87 |
| 12 | 0.0 ± 0.0 | >100 | 94.6 ± 0.6 | 1.12 ± 0.11 | >89 | n.s. | 4.87 |
| 13 | 32.7 ± 1.6 | >100 | 90.9 ± 1.5 | 2.72 ± 0.50 | >37 | n.s. | 4.96 |
| 14 | 60.9 ± 0.4 | 37.7 ± 1.7 | 95.8 ± 0.9 | 0.38 ± 0.01 | 98 | >200 * | 4.96 |
| 15 | 25.8 ± 1.3 | >100 | 75.3 ± 0.6 | 15.06 ± 2.34 | >6 | n.s. | 4.80 |
| 16 | 28.3 ± 1.1 | >100 | 91.7 ± 0.4 | 1.21 ± 0.08 | >82 | n.s. | 5.21 |
| 17 | 29.3 ± 3.9 | >100 | 77.0 ± 1.0 | 9.89 ± 1.37 | 10 | n.s. | 5.39 |
| 18 | 0.0 ± 0.0 | >100 | 60.0 ± 1.6 | 41.1 ± 2.6 | >2 | n.s. | 5.53 |
| 19 | 5.9 ± 2.1 | >100 | 80.2 ± 0.2 | 4.63 ± 0.48 | >22 | n.s. | 5.60 |
| 20 | 49.5 ± 0.8 | >100 | 82.5 ± 1.0 | 0.69 ± 0.03 | >145 | n.s. | 5.13 |
| galantamine e | 98.8 ± 1.1 | 2.0 ± 0.1 | 68.2 ± 1.2 | 29.31 ± 3.49 | 0.07 | n.s. | 5.01 |
| eserine e | 99.8 ± 0.6 | 0.20 ± 0.01 | 99.9 ± 0.5 | 0.30 ± 0.01 | 0.67 | n.s. | 5.02 |
| berberine e | - | - | - | - | - | 142 ± 21 | n.s. |
| chlorothiazide e | - | - | - | - | - | - | 2.14 |
| promazine e | - | - | - | - | - | - | 5.64 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mamun, A.A.; Pidaný, F.; Hulcová, D.; Maříková, J.; Kučera, T.; Schmidt, M.; Catapano, M.C.; Hrabinová, M.; Jun, D.; Múčková, L.; et al. Amaryllidaceae Alkaloids of Norbelladine-Type as Inspiration for Development of Highly Selective Butyrylcholinesterase Inhibitors: Synthesis, Biological Activity Evaluation, and Docking Studies. Int. J. Mol. Sci. 2021, 22, 8308. https://doi.org/10.3390/ijms22158308
Mamun AA, Pidaný F, Hulcová D, Maříková J, Kučera T, Schmidt M, Catapano MC, Hrabinová M, Jun D, Múčková L, et al. Amaryllidaceae Alkaloids of Norbelladine-Type as Inspiration for Development of Highly Selective Butyrylcholinesterase Inhibitors: Synthesis, Biological Activity Evaluation, and Docking Studies. International Journal of Molecular Sciences. 2021; 22(15):8308. https://doi.org/10.3390/ijms22158308
Chicago/Turabian StyleMamun, Abdullah Al, Filip Pidaný, Daniela Hulcová, Jana Maříková, Tomáš Kučera, Monika Schmidt, Maria Carmen Catapano, Martina Hrabinová, Daniel Jun, Lubica Múčková, and et al. 2021. "Amaryllidaceae Alkaloids of Norbelladine-Type as Inspiration for Development of Highly Selective Butyrylcholinesterase Inhibitors: Synthesis, Biological Activity Evaluation, and Docking Studies" International Journal of Molecular Sciences 22, no. 15: 8308. https://doi.org/10.3390/ijms22158308
APA StyleMamun, A. A., Pidaný, F., Hulcová, D., Maříková, J., Kučera, T., Schmidt, M., Catapano, M. C., Hrabinová, M., Jun, D., Múčková, L., Kuneš, J., Janoušek, J., Andrýs, R., Nováková, L., Peřinová, R., Maafi, N., Soukup, O., Korábečný, J., & Cahlíková, L. (2021). Amaryllidaceae Alkaloids of Norbelladine-Type as Inspiration for Development of Highly Selective Butyrylcholinesterase Inhibitors: Synthesis, Biological Activity Evaluation, and Docking Studies. International Journal of Molecular Sciences, 22(15), 8308. https://doi.org/10.3390/ijms22158308