



Mechanisms of Central Hypogonadism
 ,
,  ,
, 
Abstract
1. Introduction
2. Methods
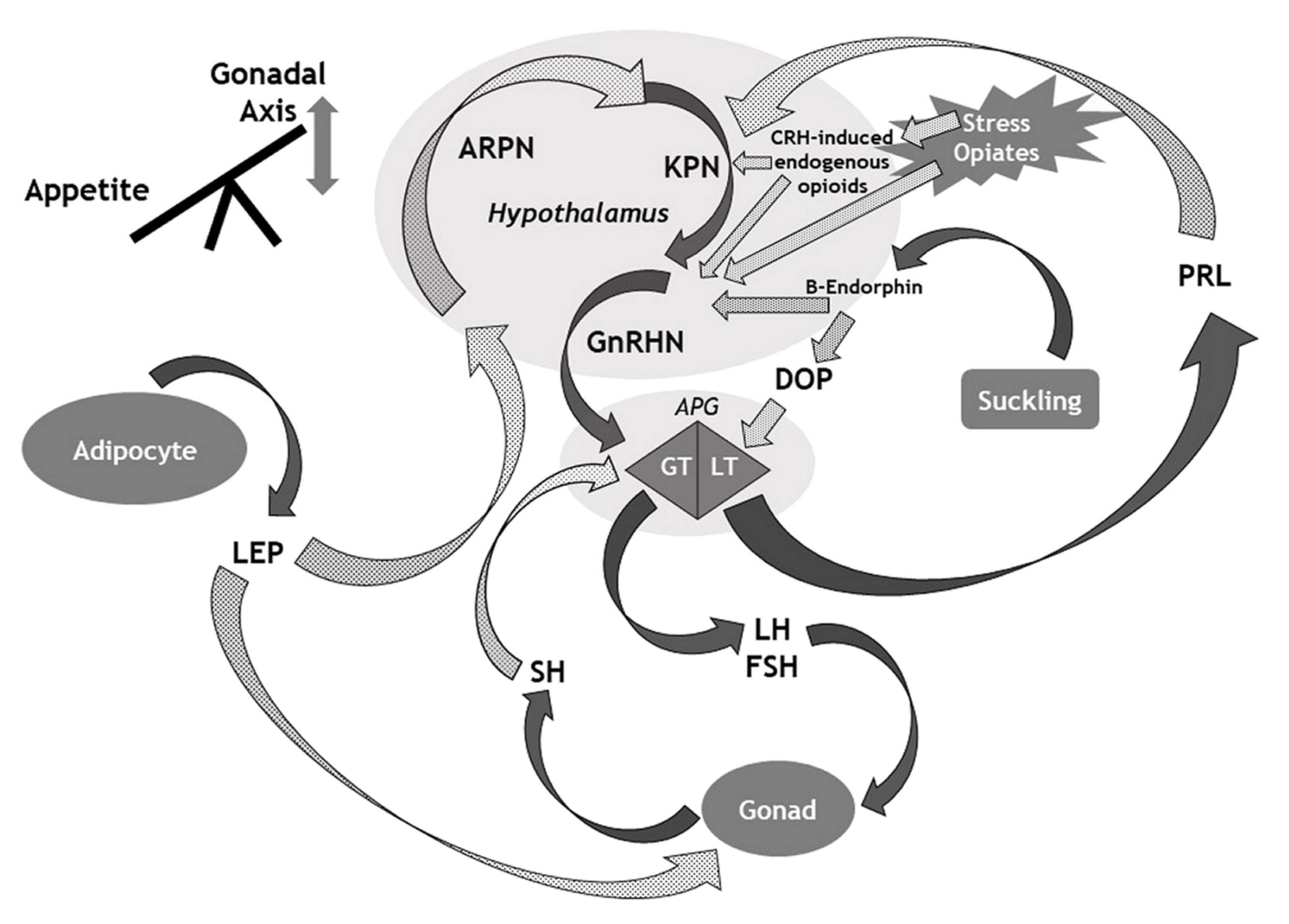
3. The HPG Axis: Lessons from the Genetics of Idiopathic Central Hypogonadism (ICH)
4. Prolactin as a Natural Contraceptive
5. Leptin as a Gatekeeper between Survival and Reproduction
5.1. Euleptinaemia and Hypoleptinaemia
5.2. Hyperleptinaemia
6. Signals from the Outside: Morphine, Anabolic-Androgenic Steroids, Physical Trauma and Stress
6.1. Morphine
6.2. Anabolic-Androgenic Steroids (AAS)
6.3. Physical Trauma
6.4. Stress
7. To Treat or Not to Treat?
8. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gallo-Payet, N. 60 YEARS OF POMC: Adrenal and extra-adrenal functions of ACTH. J. Mol. Endocrinol. 2016, 56, T135–T156. [Google Scholar] [CrossRef] [PubMed]
- Fliers, E.; Boelen, A.; van Trotsenburg, A.S. Central regulation of the hypothalamo-pituitary-thyroid (HPT) axis: Focus on clinical aspects. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2014; Volume 124, pp. 127–138. [Google Scholar]
- Stalder, T.; Steudte-Schmiedgen, S.; Alexander, N.; Klucken, T.; Vater, A.; Wichmann, S.; Kirschbaum, C.; Miller, R. Stress-related and basic determinants of hair cortisol in humans: A meta-analysis. Psychoneuroendocrinology 2017, 77, 261–274. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yao, Z.; Lin, L.; Sun, X.; Shi, X.; Zhang, L. Early life stress predicts cortisol response to psychosocial stress in healthy young adults. Psych J. 2019, 8, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Farwell, A.P. Euthyroid Sick Syndrome. Compr. Physiol. 2016, 6, 1071–1080. [Google Scholar]
- Honour, J.W. Biochemistry of the menopause. Ann. Clin. Biochem. 2018, 55, 18–33. [Google Scholar] [CrossRef]
- Tilbrook, A.J.; Turner, A.I.; Clarke, I.J. Stress and reproduction: Central mechanisms and sex differences in non-rodent species. Stress 2002, 5, 83–100. [Google Scholar] [CrossRef]
- Richard-Eaglin, A. Male and female hypogonadism. Nurs. Clin. N. Am. 2018, 53, 395–405. [Google Scholar] [CrossRef]
- Rey, R.A.; Grinspon, R.P.; Gottlieb, S.; Pasqualini, T.; Knoblovits, P.; Aszpis, S.; Pacenza, N.; Stewart Usher, J.; Bergada, I.; Campo, S.M. Male hypogonadism: An extended classification based on a developmental, endocrine physiology-based approach. Andrology 2013, 1, 3–16. [Google Scholar] [CrossRef]
- Esteves, S.C. Clinical management of infertile men with nonobstructive azoospermia. Asian J. Androl. 2015, 17, 459–470. [Google Scholar] [CrossRef]
- Irwig, M.S. Male hypogonadism and skeletal health. Curr. Opin. Endocrinol. Diabetes Obes. 2013, 20, 517–522. [Google Scholar] [CrossRef]
- Arver, S.; Lehtihet, M. Current guidelines for the diagnosis of testosterone deficiency. Adv. Manag. Testosterone Defic. 2009, 37, 5–20. [Google Scholar]
- Molina-Vega, M.; Munoz-Garach, A.; Damas-Fuentes, M.; Fernandez-Garcia, J.C.; Tinahones, F.J. Secondary male hypogonadism: A prevalent but overlooked comorbidity of obesity. Asian J. Androl. 2018, 20, 531–538. [Google Scholar] [PubMed]
- Salenave, S.; Trabado, S.; Maione, L.; Brailly-Tabard, S.; Young, J. Male acquired hypogonadotropic hypogonadism: Diagnosis and treatment. Ann. Endocrinol. 2012, 73, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Practice Committee of the American Society for Reproductive Medicine. Electronic address, a.a.o. Management of nonobstructive azoospermia: A committee opinion. Fertil. Steril. 2018, 110, 1239–1245. [Google Scholar] [CrossRef] [PubMed]
- Vargas, G.; Gonzalez, B.; Ramirez, C.; Ferreira, A.; Espinosa, E.; Mendoza, V.; Guinto, G.; Lopez-Felix, B.; Zepeda, E.; Mercado, M. Clinical characteristics and treatment outcome of 485 patients with nonfunctioning pituitary macroadenomas. Int. J. Endocrinol. 2015, 2015, 756069. [Google Scholar] [CrossRef]
- Dalvi, M.; Walker, B.R.; Strachan, M.W.; Zammitt, N.N.; Gibb, F.W. The prevalence of structural pituitary abnormalities by MRI scanning in men presenting with isolated hypogonadotrophic hypogonadism. Clin. Endocrinol. 2016, 84, 858–861. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Qiu, M.; Qi, S.; Li, D.; Peng, Y. Hypopituitarism patterns among adult males with prolactinomas. Clin. Neurol. Neurosurg. 2016, 144, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Kashyap, A.S. Young male with headache, blindness, and hypogonadism. Diagnosis: Craniopharyngioma presenting with hypogonadotrophic hypogonadism. Postgrad. Med. J. 2000, 76, 513–519. [Google Scholar] [CrossRef][Green Version]
- Monteiro, D.M.; Freitas, P.; Vieira, R.; Carvalho, D. Hypogonadotropic hypogonadism in non-functioning pituitary adenomas: Impact of intervention. Exp. Clin. Endocrinol. Diabetes 2017, 125, 368–376. [Google Scholar] [CrossRef]
- Thakker, S.; Persily, J.; Najari, B.B. Kallman syndrome and central non-obstructive azoospermia. Best Pract. Res. Clin. Endocrinol. Metab. 2020, 34, 101475. [Google Scholar] [CrossRef]
- El Osta, R.; Grandpre, N.; Monnin, N.; Hubert, J.; Koscinski, I. Hypogonadotropic hypogonadism in men with hereditary hemochromatosis. Basic Clin. Androl. 2017, 27, 13. [Google Scholar] [CrossRef] [PubMed]
- Jarvi, K.; Lo, K.; Fischer, A.; Grantmyre, J.; Zini, A.; Chow, V.; Mak, V. CUA Guideline: The workup of azoospermic males. Can. Urol. Assoc. J. 2010, 4, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Beate, K.; Joseph, N.; Nicolas de, R.; Wolfram, K. Genetics of isolated hypogonadotropic hypogonadism: Role of GnRH receptor and other genes. Int. J. Endocrinol. 2012, 2012, 147893. [Google Scholar] [CrossRef]
- Cangiano, B.; Swee, D.S.; Quinton, R.; Bonomi, M. Genetics of congenital hypogonadotropic hypogonadism: Peculiarities and phenotype of an oligogenic disease. Hum. Genet. 2021, 140, 77–111. [Google Scholar] [CrossRef]
- Dode, C.; Hardelin, J.P. Kallmann syndrome. Eur. J. Hum. Genet. 2009, 17, 139–146. [Google Scholar] [CrossRef]
- Fraietta, R.; Zylberstejn, D.S.; Esteves, S.C. Hypogonadotropic hypogonadism revisited. Clinics 2013, 68 (Suppl. 1), 81–88. [Google Scholar] [CrossRef]
- Casoni, F.; Malone, S.A.; Belle, M.; Luzzati, F.; Collier, F.; Allet, C.; Hrabovszky, E.; Rasika, S.; Prevot, V.; Chedotal, A. Development of the neurons controlling fertility in humans: New insights from 3D imaging and transparent fetal brains. Development 2016, 143, 3969–3981. [Google Scholar] [CrossRef]
- Young, J.; Xu, C.; Papadakis, G.E.; Acierno, J.S.; Maione, L.; Hietamaki, J.; Raivio, T.; Pitteloud, N. Clinical management of congenital hypogonadotropic hypogonadism. Endocr. Rev. 2019, 40, 669–710. [Google Scholar] [CrossRef] [PubMed]
- Lewkowitz-Shpuntoff, H.M.; Hughes, V.A.; Plummer, L.; Au, M.G.; Doty, R.L.; Seminara, S.B.; Chan, Y.M.; Pitteloud, N.; Crowley, W.F., Jr.; Balasubramanian, R. Olfactory phenotypic spectrum in idiopathic hypogonadotropic hypogonadism: Pathophysiological and genetic implications. J. Clin. Endocrinol. Metab. 2012, 97, E136–E144. [Google Scholar] [CrossRef]
- Fechner, A.; Fong, S.; McGovern, P. A review of Kallmann syndrome: Genetics, pathophysiology, and clinical management. Obstet. Gynecol. Surv. 2008, 63, 189–194. [Google Scholar] [CrossRef]
- Topaloglu, A.K. Update on the genetics of idiopathic hypogonadotropic hypogonadism. J. Clin. Res. Pediatr. Endocrinol. 2017, 9 (Suppl. 2), 113–122. [Google Scholar] [CrossRef]
- Franco, B.; Guioli, S.; Pragliola, A.; Incerti, B.; Bardoni, B.; Tonlorenzi, R.; Carrozzo, R.; Maestrini, E.; Pieretti, M.; Taillon-Miller, P. A gene deleted in Kallmann’s syndrome shares homology with neural cell adhesion and axonal path-finding molecules. Nature 1991, 353, 529–536. [Google Scholar] [CrossRef]
- Pedersen-White, J.R.; Chorich, L.P.; Bick, D.P.; Sherins, R.J.; Layman, L.C. The prevalence of intragenic deletions in patients with idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Mol. Hum. Reprod. 2008, 14, 367–370. [Google Scholar] [CrossRef][Green Version]
- Massin, N.; Pecheux, C.; Eloit, C.; Bensimon, J.L.; Galey, J.; Kuttenn, F.; Hardelin, J.P.; Dode, C.; Touraine, P. X chromosome-linked Kallmann syndrome: Clinical heterogeneity in three siblings carrying an intragenic deletion of the KAL-1 gene. J. Clin. Endocrinol. Metab. 2003, 88, 2003–2008. [Google Scholar] [CrossRef]
- Goncalves, C.; Bastos, M.; Pignatelli, D.; Borges, T.; Aragues, J.M.; Fonseca, F.; Pereira, B.D.; Socorro, S.; Lemos, M.C. Novel FGFR1 mutations in Kallmann syndrome and normosmic idiopathic hypogonadotropic hypogonadism: Evidence for the involvement of an alternatively spliced isoform. Fertil. Steril. 2015, 104, 1261–1267. [Google Scholar] [CrossRef]
- Hebert, J.M.; Lin, M.; Partanen, J.; Rossant, J.; McConnell, S.K. FGF signaling through FGFR1 is required for olfactory bulb morphogenesis. Development 2003, 130, 1101–1111. [Google Scholar] [CrossRef] [PubMed]
- Dode, C.; Rondard, P. PROK2/PROKR2 signaling and Kallmann syndrome. Front. Endocrinol. 2013, 4, 19. [Google Scholar] [CrossRef]
- Beranova, M.; Oliveira, L.M.; Bedecarrats, G.Y.; Schipani, E.; Vallejo, M.; Ammini, A.C.; Quintos, J.B.; Hall, J.E.; Martin, K.A.; Hayes, F.J. Prevalence, phenotypic spectrum, and modes of inheritance of gonadotropin-releasing hormone receptor mutations in idiopathic hypogonadotropic hypogonadism. J. Clin. Endocrinol. Metab. 2001, 86, 1580–1588. [Google Scholar] [CrossRef] [PubMed]
- Jongmans, M.C.; van Ravenswaaij-Arts, C.M.; Pitteloud, N.; Ogata, T.; Sato, N.; Claahsen-van der Grinten, H.L.; van der Donk, K.; Seminara, S.; Bergman, J.E.; Brunner, H.G. CHD7 mutations in patients initially diagnosed with Kallmann syndrome--the clinical overlap with CHARGE syndrome. Clin. Genet. 2009, 75, 65–71. [Google Scholar] [CrossRef]
- Trarbach, E.B.; Abreu, A.P.; Silveira, L.F.; Garmes, H.M.; Baptista, M.T.; Teles, M.G.; Costa, E.M.; Mohammadi, M.; Pitteloud, N.; Mendonca, B.B. Nonsense mutations in FGF8 gene causing different degrees of human gonadotropin-releasing deficiency. J. Clin. Endocrinol. Metab. 2010, 95, 3491–3496. [Google Scholar] [CrossRef] [PubMed]
- Costa-Barbosa, F.A.; Balasubramanian, R.; Keefe, K.W.; Shaw, N.D.; Al-Tassan, N.; Plummer, L.; Dwyer, A.A.; Buck, C.L.; Choi, J.H.; Seminara, S.B. Prioritizing genetic testing in patients with Kallmann syndrome using clinical phenotypes. J. Clin. Endocrinol. Metab. 2013, 98, E943–E953. [Google Scholar] [CrossRef]
- Bonomi, M.; Vezzoli, V.; Krausz, C.; Guizzardi, F.; Vezzani, S.; Simoni, M.; Bassi, I.; Duminuco, P.; Di Iorgi, N.; Giavoli, C. Characteristics of a nationwide cohort of patients presenting with isolated hypogonadotropic hypogonadism (IHH). Eur. J. Endocrinol. 2018, 178, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zhao, S.; Xie, Y.; Yang, W.; Mo, Z. De novo SOX10 nonsense mutation in a patient with Kallmann syndrome, deafness, iris hypopigmentation, and hyperthyroidism. Ann. Clin. Lab. Sci. 2018, 48, 248–252. [Google Scholar]
- Stamou, M.I.; Georgopoulos, N.A. Kallmann syndrome: Phenotype and genotype of hypogonadotropic hypogonadism. Metabolism 2018, 86, 124–134. [Google Scholar] [CrossRef]
- Pingault, V.; Bodereau, V.; Baral, V.; Marcos, S.; Watanabe, Y.; Chaoui, A.; Fouveaut, C.; Leroy, C.; Verier-Mine, O.; Francannet, C. Loss-of-function mutations in SOX10 cause Kallmann syndrome with deafness. Am. J. Hum. Genet. 2013, 92, 707–724. [Google Scholar] [CrossRef] [PubMed]
- Newbern, K.; Natrajan, N.; Kim, H.G.; Chorich, L.P.; Halvorson, L.M.; Cameron, R.S.; Layman, L.C. Identification of HESX1 mutations in Kallmann syndrome. Fertil. Steril. 2013, 99, 1831–1837. [Google Scholar] [CrossRef][Green Version]
- Short, R.V. Lactation--the central control of reproduction. In Ciba Foundation Symposium; Ciba Foundation: London, UK, 1976; pp. 73–86. [Google Scholar]
- Short, R.V. Lactational infertility in family planning. Ann. Med. 1993, 25, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Grattan, D.R.; Szawka, R.E. Kisspeptin and Prolactin. Semin. Reprod. Med. 2019, 37, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Grossman, A.; Moult, P.J.; McIntyre, H.; Evans, J.; Silverstone, T.; Rees, L.H.; Besser, G.M. Opiate mediation of amenorrhoea in hyperprolactinaemia and in weight-loss related amenorrhoea. Clin. Endocrinol. 1982, 17, 379–388. [Google Scholar] [CrossRef]
- Sobrinho, L.G. Prolactin, psychological stress and environment in humans: Adaptation and maladaptation. Pituitary 2003, 6, 35–39. [Google Scholar] [CrossRef]
- Barber, T.M.; Kenkre, J.; Garnett, C.; Scott, R.V.; Byrne, J.V.; Wass, J.A. Recurrence of hyperprolactinaemia following discontinuation of dopamine agonist therapy in patients with prolactinoma occurs commonly especially in macroprolactinoma. Clin. Endocrinol. 2011, 75, 819–824. [Google Scholar] [CrossRef] [PubMed]
- Duskin-Bitan, H.; Shimon, I. Prolactinomas in males: Any differences? Pituitary 2020, 23, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Grant, P.; Jackson, G.; Baig, I.; Quin, J. Erectile dysfunction in general medicine. Clin. Med. 2013, 13, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Lopes, M.B.S. Pathology of prolactinomas: Any predictive value? Pituitary 2020, 23, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Kumran, T.; Haspani, S.; Malin Abdullah, J.; Alias, A.; Ven, F.R. Factors influencing disconnection hyperprolactinemia and reversal of serum prolactin after pituitary surgery in a non-functioning pituitary macroadenoma. Malays. J. Med. Sci. 2016, 23, 72–76. [Google Scholar] [PubMed]
- Ishibashi, M.; Yamaji, T. Mechanism of the inhibitory action of dopamine and somatostatin on prolactin secretion from human lactotrophs in culture. J. Clin. Endocrinol. Metab. 1985, 60, 599–606. [Google Scholar] [CrossRef]
- Korevaar, T.; Wass, J.A.; Grossman, A.B.; Karavitaki, N. Disconnection hyperprolactinaemia in nonadenomatous sellar/parasellar lesions practically never exceeds 2000 mU/l. Clin. Endocrinol. 2012, 76, 602–603. [Google Scholar] [CrossRef]
- Bostwick, J.R.; Guthrie, S.K.; Ellingrod, V.L. Antipsychotic-induced hyperprolactinemia. Pharmacotherapy 2009, 29, 64–73. [Google Scholar] [CrossRef]
- Vilar, L.; Vilar, C.F.; Lyra, R.; Freitas, M.D.C. Pitfalls in the diagnostic evaluation of hyperprolactinemia. Neuroendocrinology 2019, 109, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Ajmal, A.; Joffe, H.; Nachtigall, L.B. Psychotropic-induced hyperprolactinemia: A clinical review. Psychosomatics 2014, 55, 29–36. [Google Scholar] [CrossRef]
- Reeves, K.W.; Okereke, O.I.; Qian, J.; Tworoger, S.S.; Rice, M.S.; Hankinson, S.E. Antidepressant use and circulating prolactin levels. Cancer Causes Control 2016, 27, 853–861. [Google Scholar] [CrossRef]
- Coker, F.; Taylor, D. Antidepressant-induced hyperprolactinaemia: Incidence, mechanisms and management. CNS Drugs 2010, 24, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Srikantha, M.; Butterworth, R. Pharmacological hyperprolactinaemia. BMJ Case Rep. 2009. [Google Scholar] [CrossRef]
- Molitch, M.E. Drugs and prolactin. Pituitary 2008, 11, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Arze, R.S.; Ramos, J.M.; Rashid, H.U.; Kerr, D.N. Amenorrhoea, galactorrhoea, and hyperprolactinaemia induced by methyldopa. Br. Med. J. (Clin. Res. Ed.) 1981, 283, 194. [Google Scholar] [CrossRef] [PubMed]
- Kelley, S.R.; Kamal, T.J.; Molitch, M.E. Mechanism of verapamil calcium channel blockade-induced hyperprolactinemia. Am. J. Physiol. 1996, 270 Pt 1, E96–E100. [Google Scholar] [CrossRef]
- Fountas, A.; Chai, S.T.; Kourkouti, C.; Karavitaki, N. Mechanisms of Endocrinology: Endocrinology of opioids. Eur. J. Endocrinol. 2018, 179, R183–R196. [Google Scholar] [CrossRef]
- Melmed, S.; Casanueva, F.F.; Hoffman, A.R.; Kleinberg, D.L.; Montori, V.M.; Schlechte, J.A.; Wass, J.A.; Endocrine, Society. Diagnosis and treatment of hyperprolactinemia: An Endocrine Society clinical practice guideline. J. Clin. Endocrinol. Metab. 2011, 96, 273–288. [Google Scholar] [CrossRef]
- Childs, G.V.; Odle, A.K.; MacNicol, M.C.; MacNicol, A.M. The importance of leptin to reproduction. Endocrinology 2021, 162, bqaa204. [Google Scholar] [CrossRef]
- Caron, A.; Lee, S.; Elmquist, J.K.; Gautron, L. Leptin and brain-adipose crosstalks. Nat. Rev. Neurosci. 2018, 19, 153–165. [Google Scholar] [CrossRef]
- Tsatsanis, C.; Dermitzaki, E.; Avgoustinaki, P.; Malliaraki, N.; Mytaras, V.; Margioris, A.N. The impact of adipose tissue-derived factors on the hypothalamic-pituitary-gonadal (HPG) axis. Hormones 2015, 14, 549–562. [Google Scholar] [CrossRef]
- Garcia-Galiano, D.; Allen, S.J.; Elias, C.F. Role of the adipocyte-derived hormone leptin in reproductive control. Horm. Mol. Biol. Clin. Investig. 2014, 19, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Sarmento-Cabral, A.; Peinado, J.R.; Halliday, L.C.; Malagon, M.M.; Castano, J.P.; Kineman, R.D.; Luque, R.M. Adipokines (leptin, adiponectin, resistin) differentially regulate all hormonal cell types in primary anterior pituitary cell cultures from two primate species. Sci. Rep. 2017, 7, 43537. [Google Scholar] [CrossRef] [PubMed]
- Petrine, J.C.P.; Franci, C.R.; Del Bianco-Borges, B. Leptin actions through the nitrergic system to modulate the hypothalamic expression of the kiss1 mRNA in the female rat. Brain Res. 2020, 1728, 146574. [Google Scholar] [CrossRef] [PubMed]
- Donato, J., Jr.; Silva, R.J.; Sita, L.V.; Lee, S.; Lee, C.; Lacchini, S.; Bittencourt, J.C.; Franci, C.R.; Canteras, N.S.; Elias, C.F. The ventral premammillary nucleus links fasting-induced changes in leptin levels and coordinated luteinizing hormone secretion. J. Neurosci. 2009, 29, 5240–5250. [Google Scholar] [CrossRef]
- Grossman, A.B.; Rossmanith, W.G.; Kabigting, E.B.; Cadd, G.; Clifton, D.; Steiner, R.A. The distribution of hypothalamic nitric oxide synthase mRNA in relation to gonadotrophin-releasing hormone neurons. J. Endocrinol. 1994, 140, R5–R8. [Google Scholar] [CrossRef]
- Boutari, C.; Pappas, P.D.; Mintziori, G.; Nigdelis, M.P.; Athanasiadis, L.; Goulis, D.G.; Mantzoros, C.S. The effect of underweight on female and male reproduction. Metabolism 2020, 107, 154229. [Google Scholar] [CrossRef]
- Jenkins, P.J.; Ibanez-Santos, X.; Holly, J.; Cotterill, A.; Perry, L.; Wolman, R.; Harries, M.; Grossman, A. IGFBP-1: A metabolic signal associated with exercise-induced amenorrhoea. Neuroendocrinology 1993, 57, 600–604. [Google Scholar] [CrossRef]
- Isidori, A.M.; Caprio, M.; Strollo, F.; Moretti, C.; Frajese, G.; Isidori, A.; Fabbri, A. Leptin and androgens in male obesity: Evidence for leptin contribution to reduced androgen levels. J. Clin. Endocrinol. Metab. 1999, 84, 3673–3680. [Google Scholar]
- Saboor Aftab, S.A.; Kumar, S.; Barber, T.M. The role of obesity and type 2 diabetes mellitus in the development of male obesity-associated secondary hypogonadism. Clin. Endocrinol. 2013, 78, 330–337. [Google Scholar] [CrossRef]
- Khodamoradi, K.; Parmar, M.; Khosravizadeh, Z.; Kuchakulla, M.; Manoharan, M.; Arora, H. The role of leptin and obesity on male infertility. Curr. Opin. Urol. 2020, 30, 334–339. [Google Scholar] [CrossRef] [PubMed]
- Costanzo, P.R.; Knoblovits, P. Male gonadal axis function in patients with type 2 diabetes. Horm. Mol. Biol. Clin. Investig. 2016, 26, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Mammi, C.; Calanchini, M.; Antelmi, A.; Cinti, F.; Rosano, G.M.; Lenzi, A.; Caprio, M.; Fabbri, A. Androgens and adipose tissue in males: A complex and reciprocal interplay. Int. J. Endocrinol. 2012, 2012, 789653. [Google Scholar] [CrossRef] [PubMed]
- Dandona, P.; Dhindsa, S. Update: Hypogonadotropic hypogonadism in type 2 diabetes and obesity. J. Clin. Endocrinol. Metab. 2011, 96, 2643–2651. [Google Scholar] [CrossRef] [PubMed]
- Herman, A.P.; Krawczynska, A.; Bochenek, J.; Dobek, E.; Herman, A.; Tomaszewska-Zaremba, D. LPS-induced inflammation potentiates the IL-1beta-mediated reduction of LH secretion from the anterior pituitary explants. Clin. Dev. Immunol. 2013, 2013, 926937. [Google Scholar] [CrossRef]
- Gautier, A.; Bonnet, F.; Dubois, S.; Massart, C.; Grosheny, C.; Bachelot, A.; Aube, C.; Balkau, B.; Ducluzeau, P.H. Associations between visceral adipose tissue, inflammation and sex steroid concentrations in men. Clin. Endocrinol. 2013, 78, 373–378. [Google Scholar] [CrossRef]
- Maggio, M.; Basaria, S.; Ceda, G.P.; Ble, A.; Ling, S.M.; Bandinelli, S.; Valenti, G.; Ferrucci, L. The relationship between testosterone and molecular markers of inflammation in older men. J. Endocrinol. Investig. 2005, 28 (Suppl. 11), 116–119. [Google Scholar]
- Burney, B.O.; Hayes, T.G.; Smiechowska, J.; Cardwell, G.; Papusha, V.; Bhargava, P.; Konda, B.; Auchus, R.J.; Garcia, J.M. Low testosterone levels and increased inflammatory markers in patients with cancer and relationship with cachexia. J. Clin. Endocrinol. Metab. 2012, 97, E700–E709. [Google Scholar] [CrossRef]
- Brook, K.; Bennett, J.; Desai, S.P. The Chemical History of Morphine: An 8000-year Journey, from Resin to de-novo Synthesis. J. Anesth. Hist. 2017, 3, 50–55. [Google Scholar] [CrossRef]
- Rose, M.E. Are prescription opioids driving the opioid crisis? Assumptions vs facts. Pain Med. 2018, 19, 793–807. [Google Scholar] [CrossRef]
- Drobnis, E.Z.; Nangia, A.K. Pain medications and male reproduction. Adv. Exp. Med. Biol. 2017, 1034, 39–57. [Google Scholar]
- De Vries, F.; Bruin, M.; Lobatto, D.J.; Dekkers, O.M.; Schoones, J.W.; van Furth, W.R.; Pereira, A.M.; Karavitaki, N.; Biermasz, N.R.; Zamanipoor Najafabadi, A.H. Opioids and their Endocrine effects: A systematic review and meta-analysis. J. Clin. Endocrinol. Metab. 2020, 105, 1020–1029. [Google Scholar] [CrossRef] [PubMed]
- Grossman, A.; Moult, P.J.; Gaillard, R.C.; Delitala, G.; Toff, W.D.; Rees, L.H.; Besser, G.M. The opioid control of LH and FSH release: Effects of a met-enkephalin analogue and naloxone. Clin. Endocrinol. 1981, 14, 41–47. [Google Scholar] [CrossRef]
- Moult, P.J.; Grossman, A.; Evans, J.M.; Rees, L.H.; Besser, G.M. The effect of naloxone on pulsatile gonadotrophin release in normal subjects. Clin. Endocrinol. 1981, 14, 321–324. [Google Scholar] [CrossRef]
- Christou, M.A.; Christou, P.A.; Markozannes, G.; Tsatsoulis, A.; Mastorakos, G.; Tigas, S. Effects of anabolic androgenic steroids on the reproductive system of athletes and recreational users: A systematic review and meta-analysis. Sports Med. 2017, 47, 1869–1883. [Google Scholar] [CrossRef] [PubMed]
- Nieschlag, E.; Vorona, E. MECHANISMS IN ENDOCRINOLOGY: Medical consequences of doping with anabolic androgenic steroids: Effects on reproductive functions. Eur. J. Endocrinol. 2015, 173, R47–R58. [Google Scholar] [CrossRef]
- Kanayama, G.; Hudson, J.I.; DeLuca, J.; Isaacs, S.; Baggish, A.; Weiner, R.; Bhasin, S.; Pope, H.G., Jr. Prolonged hypogonadism in males following withdrawal from anabolic-androgenic steroids: An under-recognized problem. Addiction 2015, 110, 823–831. [Google Scholar] [CrossRef]
- Rahnema, C.D.; Lipshultz, L.I.; Crosnoe, L.E.; Kovac, J.R.; Kim, E.D. Anabolic steroid-induced hypogonadism: Diagnosis and treatment. Fertil. Steril. 2014, 101, 1271–1279. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, M.J.; Kanayama, G.; Hudson, J.I.; Pope, H.G., Jr. Supraphysiologic-dose anabolic-androgenic steroid use: A risk factor for dementia? Neurosci. Biobehav. Rev. 2019, 100, 180–207. [Google Scholar] [CrossRef] [PubMed]
- Sav, A.; Rotondo, F.; Syro, L.V.; Serna, C.A.; Kovacs, K. Pituitary pathology in traumatic brain injury: A review. Pituitary 2019, 22, 201–211. [Google Scholar] [CrossRef]
- Auer, M.; Stalla, G.K.; Athanasoulia, A.P. Isolated gonadotropic deficiency after multiple concussions in a professional soccer player. Dtsch. Med. Wochenschr. 2013, 138, 831–833. [Google Scholar]
- Gilis-Januszewska, A.; Kluczynski, L.; Hubalewska-Dydejczyk, A. Traumatic brain injuries induced pituitary dysfunction: A call for algorithms. Endocr. Connect. 2020, 9, R112–R123. [Google Scholar] [CrossRef]
- Tanriverdi, F.; Unluhizarci, K.; Kocyigit, I.; Tuna, I.S.; Karaca, Z.; Durak, A.C.; Selcuklu, A.; Casanueva, F.F.; Kelestimur, F. Brief communication: Pituitary volume and function in competing and retired male boxers. Ann. Intern. Med. 2008, 148, 827–831. [Google Scholar] [CrossRef] [PubMed]
- Kelly, D.F.; Chaloner, C.; Evans, D.; Mathews, A.; Cohan, P.; Wang, C.; Swerdloff, R.; Sim, M.S.; Lee, J.; Wright, M.J. Prevalence of pituitary hormone dysfunction, metabolic syndrome, and impaired quality of life in retired professional football players: A prospective study. J. Neurotrauma 2014, 31, 1161–1171. [Google Scholar] [CrossRef] [PubMed]
- McCrory, P.; Feddermann-Demont, N.; Dvorak, J.; Cassidy, J.D.; McIntosh, A.; Vos, P.E.; Echemendia, R.J.; Meeuwisse, W.; Tarnutzer, A.A. What is the definition of sports-related concussion: A systematic review. Br. J. Sports Med. 2017, 51, 877–887. [Google Scholar] [CrossRef]
- Hutmacher, F. Putting Stress in Historical Context: Why it is important that being stressed out was not a way to be a person 2,000 years ago. Front. Psychol. 2021, 12, 539799. [Google Scholar] [CrossRef] [PubMed]
- Lecic-Tosevski, D.; Vukovic, O.; Stepanovic, J. Stress and personality. Psychiatriki 2011, 22, 290–297. [Google Scholar] [PubMed]
- Siegrist, J.; Li, J. Work stress and the development of chronic diseases. Int. J. Environ. Res. Public Health 2018, 15, 536. [Google Scholar] [CrossRef] [PubMed]
- Sowinska-Przepiera, E.; Andrysiak-Mamos, E.; Jarzabek-Bielecka, G.; Walkowiak, A.; Osowicz-Korolonek, L.; Syrenicz, M.; Kędzia, W.; Syrenicz, A. Functional hypothalamic amenorrhoea—Diagnostic challenges, monitoring, and treatment. Endokrynol. Pol. 2015, 66, 252–260. [Google Scholar] [PubMed]
- Lania, A.; Gianotti, L.; Gagliardi, I.; Bondanelli, M.; Vena, W.; Ambrosio, M.R. Functional hypothalamic and drug-induced amenorrhea: An overview. J. Endocrinol. Investig. 2019, 42, 1001–1010. [Google Scholar] [CrossRef]
- Gordon, C.M.; Ackerman, K.E.; Berga, S.L.; Kaplan, J.R.; Mastorakos, G.; Misra, M.; Murad, M.H.; Santoro, N.F.; Warren, M.P. Functional hypothalamic amenorrhea: An Endocrine Society clinical practice guideline. J. Clin. Endocrinol. Metab. 2017, 102, 1413–1439. [Google Scholar] [CrossRef] [PubMed]
- Schneiderman, N.; Ironson, G.; Siegel, S.D. Stress and health: Psychological, behavioral, and biological determinants. Annu. Rev. Clin. Psychol. 2005, 1, 607–628. [Google Scholar] [CrossRef] [PubMed]
- Meczekalski, B.; Katulski, K.; Czyzyk, A.; Podfigurna-Stopa, A.; Maciejewska-Jeske, M. Functional hypothalamic amenorrhea and its influence on women’s health. J. Endocrinol. Investig. 2014, 37, 1049–1056. [Google Scholar] [CrossRef] [PubMed]
- Genazzani, A.D.; Chierchia, E.; Santagni, S.; Rattighieri, E.; Farinetti, A.; Lanzoni, C. Hypothalamic amenorrhea: From diagnosis to therapeutical approach. Ann. Endocrinol. 2010, 71, 163–169. [Google Scholar] [CrossRef]
- Bhasin, S.; Brito, J.P.; Cunningham, G.R.; Hayes, F.J.; Hodis, H.N.; Matsumoto, A.M.; Snyder, P.J.; Swerdloff, R.S.; Wu, F.C.; Yialamas, M.A. Testosterone therapy in men with hypogonadism: An Endocrine Society clinical practice guideline. J. Clin. Endocrinol. Metab. 2018, 103, 1715–1744. [Google Scholar] [CrossRef]
- Elagizi, A.; Kohler, T.S.; Lavie, C.J. Testosterone and cardiovascular health. Mayo Clin. Proc. 2018, 93, 83–100. [Google Scholar] [CrossRef]
- Yang, J.; Wu, G.; Feng, Y.; Lv, Q.; Lin, S.; Hu, J. Effects of taurine on male reproduction in rats of different ages. J. Biomed. Sci. 2010, 17 (Suppl. 1), S9. [Google Scholar] [CrossRef]
- McKee, A.; Morley, J.E.; Matsumoto, A.M.; Vinik, A. Sarcopenia: An Endocrine disorder? Endocr. Pract. 2017, 23, 1140–1149. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, A.A.; Quinton, R. The metabolic syndrome in central hypogonadotrophic hypogonadism. Front. Horm. Res. 2018, 49, 156–169. [Google Scholar]
- Penn, D.J.; Smith, K.R. Differential fitness costs of reproduction between the sexes. Proc. Natl. Acad. Sci. USA 2007, 104, 553–558. [Google Scholar] [CrossRef]


{kind=link}
| Drug Group | Drug Class | Drug Name |
|---|---|---|
| Neuroleptic | Classical anti-psychotic | Phenothiazines |
| Butyrophenones | ||
| Thioxanthenes | ||
| Atypical anti-psychotic | Risperidone | |
| Anti-depressant | Selective Serotonin Reuptake Inhibitor | Sertraline, Citalopram, Fluoxetine, Paroxetine |
| Serotonin and Noradrenaline Reuptake Inhibitor | Duloxetine, Venlafaxine | |
| Tricyclic antidepressant | Amitriptyline, Nortriptyline | |
| Anti-emetic | Prokinetic | Metoclopramide |
| Domperidone | ||
| Anti-hypertensive | Centrally-acting alpha-2 adrenergic agonist | Methyldopa |
| Anti-arrhythmic and Anti-hypertensive | Calcium-Channel Blocker | Verapamil |
| Depressant | Analgesic | Opiates |
| Recreational and “social” drug | Alcohol | |
| Stimulant | Illicit drug | Cocaine |
| Depressant, Stimulant and Hallucinogen | Recreational drug | Marijuana |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barber, T.M.; Kyrou, I.; Kaltsas, G.; Grossman, A.B.; Randeva, H.S.; Weickert, M.O. Mechanisms of Central Hypogonadism. Int. J. Mol. Sci. 2021, 22, 8217. https://doi.org/10.3390/ijms22158217
Barber TM, Kyrou I, Kaltsas G, Grossman AB, Randeva HS, Weickert MO. Mechanisms of Central Hypogonadism. International Journal of Molecular Sciences. 2021; 22(15):8217. https://doi.org/10.3390/ijms22158217
Chicago/Turabian StyleBarber, Thomas M., Ioannis Kyrou, Gregory Kaltsas, Ashley B. Grossman, Harpal S Randeva, and Martin O. Weickert. 2021. "Mechanisms of Central Hypogonadism" International Journal of Molecular Sciences 22, no. 15: 8217. https://doi.org/10.3390/ijms22158217
APA StyleBarber, T. M., Kyrou, I., Kaltsas, G., Grossman, A. B., Randeva, H. S., & Weickert, M. O. (2021). Mechanisms of Central Hypogonadism. International Journal of Molecular Sciences, 22(15), 8217. https://doi.org/10.3390/ijms22158217