
Critical Role of LSEC in Post-Hepatectomy Liver Regeneration and Failure

{kind=link}
{kind=link}
{kind=link}
Abstract
1. LSEC Are “Liver-Specialized” Endothelial Cells
2. Regulation of the Liver Blood Tone
3. Liver Endothelial Cells Origins and Subtypes
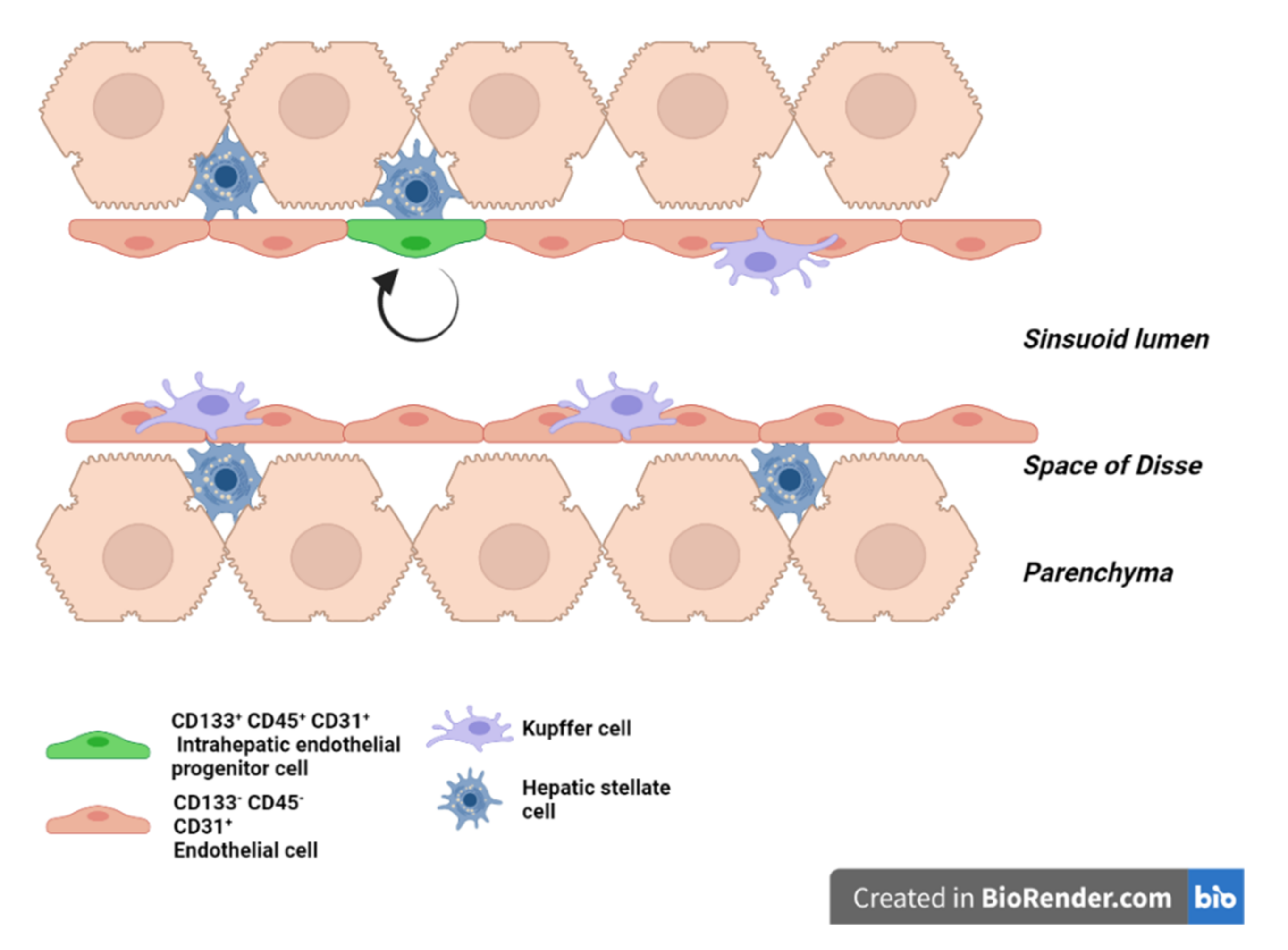
4. LSEC Renewal during Homeostasis
5. LSEC during Regeneration after Partial Hepatectomy
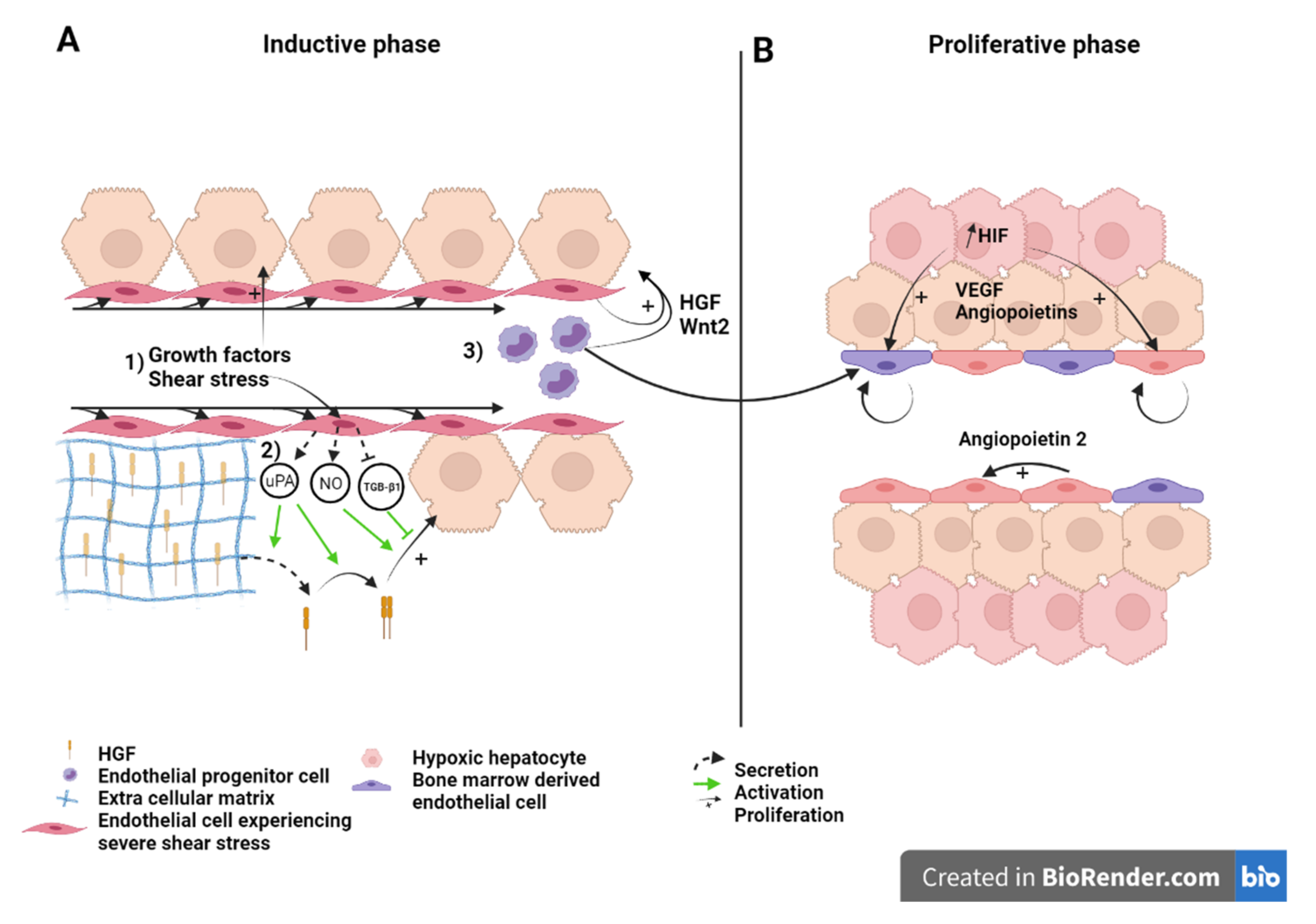
5.1. LSEC during the Inductive Phase of Liver Regeneration
5.1.1. How Do LSEC Modulate Hepatocyte Regeneration
5.1.2. How Do LSEC Interact with Hepatocytes, NPC’s and Circulating Progenitors
5.2. LSEC during the Angiogenic Phase of Liver Regeneration
5.3. LSEC during the Termination Phase of Liver Regeneration
6. Role of LSEC in Extended Hepatectomy
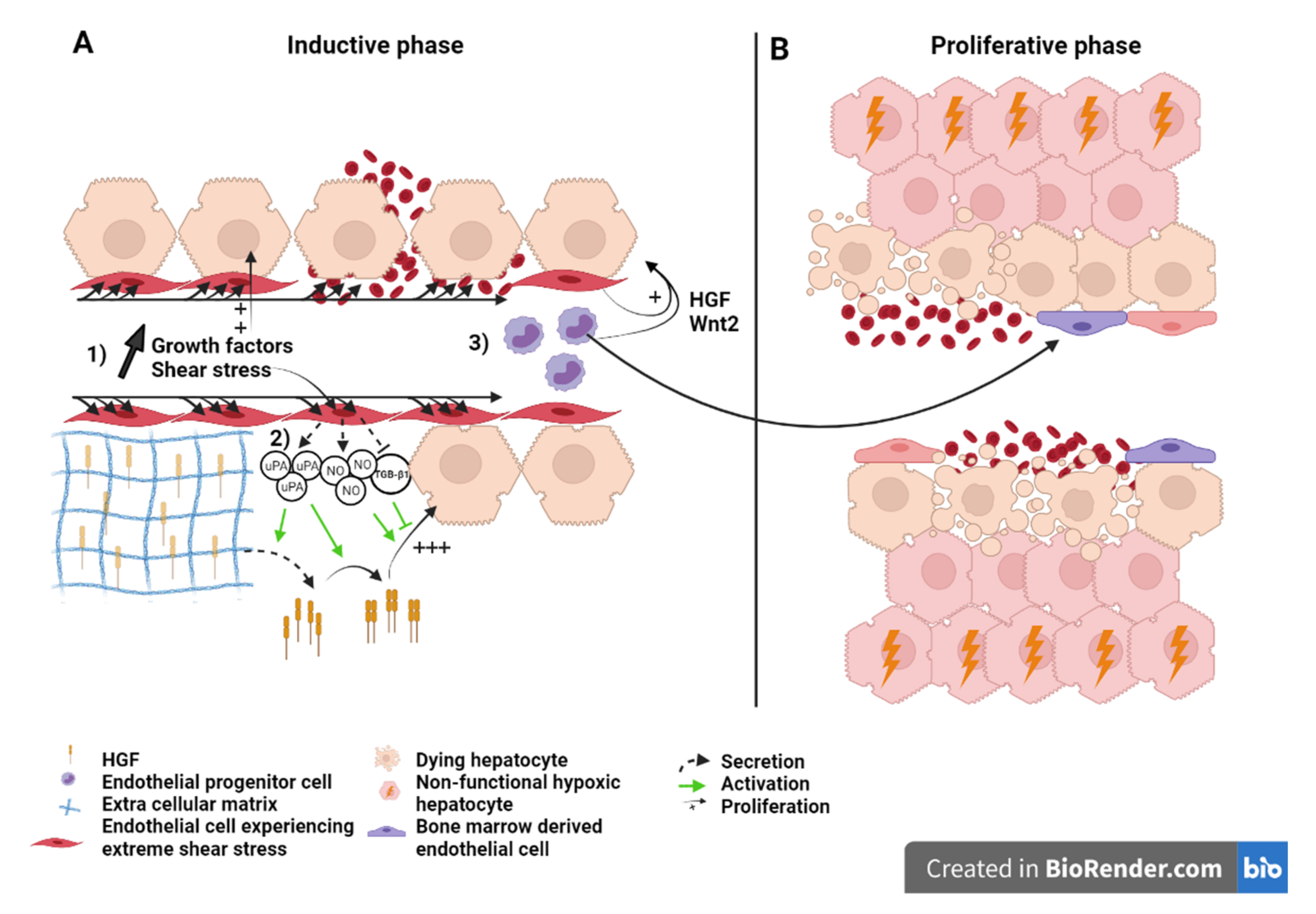
6.1. What Causes Mortality after Extended Hepatectomy?
6.2. Liver Failure Because of Sinusoid Insufficiency?
6.3. Effect of the Modulation of Portal Hyperflow and Shear Stress after Extended Hepatectomy
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Stanger, B.Z. Cellular Homeostasis and Repair in the Mammalian Liver. Annu. Rev. Physiol. 2015, 77, 179–200. [Google Scholar] [CrossRef]
- Wei, Y.; Wang, Y.G.; Jia, Y.; Li, L.; Yoon, J.; Zhang, S.; Wang, Z.; Zhang, Y.; Zhu, M.; Sharma, T.; et al. Liver homeostasis is maintained by midlobular zone 2 hepatocytes. Science 2021, 371, eabb1625. [Google Scholar] [CrossRef] [PubMed]
- Michalopoulos, G.K. Liver regeneration. J. Cell. Physiol. 2007, 213, 286–300. [Google Scholar] [CrossRef] [PubMed]
- Preziosi, M.E.; Monga, S.P. Update on the Mechanisms of Liver Regeneration. Semin. Liver Dis. 2017, 37, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Michalopoulos, G.K.; DeFrances, M.C.; Cressman, D.E.; Greenbaum, L.E.; DeAngelis, R.A.; Ciliberto, G.; Furth, E.E.; Poli, V.; Taub, R. Liver Regeneration. Science 1997, 276, 60–66. [Google Scholar] [CrossRef]
- Fausto, N. Liver regeneration. J. Hepatol. 2000, 32, 19–31. [Google Scholar] [CrossRef]
- Fausto, N.; Campbell, J.S.; Riehle, K.J. Liver regeneration. Hepatology 2006, 43, S45–S53. [Google Scholar] [CrossRef]
- Chi, J.-T.; Chang, H.Y.; Haraldsen, G.; Jahnsen, F.L.; Troyanskaya, O.G.; Chang, D.S.; Wang, Z.; Rockson, S.G.; van de Rijn, M.; Botstein, D.; et al. Endothelial cell diversity revealed by global expression profiling. Proc. Natl. Acad. Sci. USA 2003, 100, 10623–10628. [Google Scholar] [CrossRef]
- Marcu, R.; Choi, Y.J.; Xue, J.; Fortin, C.L.; Wang, Y.; Nagao, R.J.; Xu, J.; MacDonald, J.W.; Bammler, T.K.; Murry, C.E.; et al. Human Organ-Specific Endothelial Cell Heterogeneity. iScience 2018, 4, 20–35. [Google Scholar] [CrossRef]
- Shetty, S.; Lalor, P.F.; Adams, D.H. Liver sinusoidal endothelial cells—Gatekeepers of hepatic immunity. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 555–567. [Google Scholar] [CrossRef]
- Poisson, J.; Lemoinne, S.; Boulanger, C.M.; Durand, F.; Moreau, R.; Valla, D.; Rautou, P.-E. Liver sinusoidal endothelial cells: Physiology and role in liver diseases. J. Hepatol. 2017, 66, 212–227. [Google Scholar] [CrossRef]
- Deleve, L.D. Liver sinusoidal endothelial cells in hepatic fibrosis. Hepatology 2015, 61, 1740–1746. [Google Scholar] [CrossRef]
- Braet, F.; Wisse, E. Comparative Hepatology Structural and functional aspects of liver sinusoidal endothelial cell fenestrae: A review. Comp. Hepatol. 2002, 1, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Lautt, W.W. Regulatory processes interacting to maintain hepatic blood flow constancy: Vascular compliance, hepatic arterial buffer response, hepatorenal reflex, liver regeneration, escape from vasoconstriction. Hepatol. Res. 2007, 37, 891–903. [Google Scholar] [CrossRef]
- Eipel, C.; Abshagen, K.; Vollmar, B. Regulation of hepatic blood flow: The hepatic arterial buffer response revisited. World J. Gastroenterol. 2010, 16, 6046–6057. [Google Scholar] [CrossRef]
- Lautt, W.; Legare, D.J.; Ezzat, W. Quantitation of the hepatic arterial buffer response to graded changes in portal blood flow. Gastroenterology 1990, 98, 1024–1028. [Google Scholar] [CrossRef]
- Jakab, F.; Ráth, Z.; Schmal, F.; Nagy, P.; Faller, J. The interaction between hepatic arterial and portal venous blood flows; simultaneous measurement by transit time ultrasonic volume flowmetry. Hepatogastroenterology 1995, 42, 18–21. [Google Scholar]
- Lautt, W.W.; Legare, D.J.; D’Almeida, M.S. Adenosine as putative regulator of hepatic arterial flow (the buffer re-sponse). Am. J. Physiol. Heart Circ. Physiol. 1985, 248, H331–H338. [Google Scholar] [CrossRef]
- Ezzat, W.; Lautt, W.W. Hepatic arterial pressure-flow autoregulation is adenosine mediated. Am. J. Physiol. Heart Circ. Physiol. 1987, 252, H836–H845. [Google Scholar] [CrossRef]
- Benias, P.C.; Wells, R.G.; Sackey-Aboagye, B.; Klavan, H.; Reidy, J.; Buonocore, D.; Miranda, M.; Kornacki, S.; Wayne, M.; Carr-Locke, D.L.; et al. Structure and Distribution of an Unrecognized Interstitium in Human Tissues. Sci. Rep. 2018, 8, 4947. [Google Scholar] [CrossRef]
- Lautt, W. The hepatic artery: Subservient to hepatic metabolism or guardian of normal hepatic clearance rates of humoral substances. Gen. Pharmacol. Vasc. Syst. 1977, 8, 73–78. [Google Scholar] [CrossRef]
- Lautt, W.W. Control of hepatic arterial blood flow: Independence from liver metabolic activity. Am. J. Physiol. Heart Circ. Physiol. 1980, 239, H559. [Google Scholar] [CrossRef]
- Shah, V.; Haddad, F.G.; Garcia-Cardena, G.; Frangos, J.A.; Mennone, A.; Groszmann, R.J.; Sessa, W.C. Liver sinusoidal endothelial cells are responsible for nitric oxide modulation of resistance in the hepatic sinusoids. J. Clin. Investig. 1997, 100, 2923–2930. [Google Scholar] [CrossRef]
- Vollmar, B.; Menger, M.D. The Hepatic Microcirculation: Mechanistic Contributions and Therapeutic Targets in Liver Injury and Repair. Physiol. Rev. 2009, 89, 1269–1339. [Google Scholar] [CrossRef]
- Soydemir, S.; Comella, O.; Abdelmottaleb, D.; Pritchett, J. Does Mechanocrine Signaling by Liver Sinusoidal Endothelial Cells Offer New Opportunities for the Development of Anti-fibrotics? Front. Med. 2020, 6, 312. [Google Scholar] [CrossRef]
- Gracia-Sancho, J.; Russo, L.; García-Calderó, H.; Garcia-Pagan, J.C.; García-Cardeña, G.; Bosch, J. Endothelial expression of transcription factor Kruppel-like factor 2 and its vasoprotective target genes in the normal and cirrhotic rat liver. Gut 2010, 60, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Parmar, K.M.; Larman, H.B.; Dai, G.; Zhang, Y.; Wang, E.T.; Moorthy, S.N.; Kratz, J.R.; Lin, Z.; Jain, M.K.; Gimbrone, M.A.G., Jr.; et al. Integration of flow-dependent endothelial phenotypes by Kruppel-like factor 2. J. Clin. Investig. 2005, 116, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Maretti-Mira, A.C.; Deleve, L.D. Liver Sinusoidal Endothelial Cell: An Update. Semin. Liver Dis. 2017, 37, 377–387. [Google Scholar] [CrossRef]
- Strauss, O.; Phillips, A.; Ruggiero, K.; Bartlett, A.; Dunbar, R. Immunofluorescence identifies distinct subsets of endothelial cells in the human liver. Sci. Rep. 2017, 7, 44356. [Google Scholar] [CrossRef] [PubMed]
- Dingle, A.M.; Yap, K.K.; Gerrand, Y.-W.; Taylor, C.J.; Keramidaris, E.; Lokmic, Z.; Kong, A.M.; Peters, H.L.; Morrison, W.A.; Mitchell, G.M. Characterization of isolated liver sinusoidal endothelial cells for liver bioengineering. Angiogenesis 2018, 21, 581–597. [Google Scholar] [CrossRef]
- Zhang, H.; Pu, W.; Tian, X.; Huang, X.; He, L.; Liu, Q.; Lingjuan, H.; Zhang, L.; He, L.; Liu, K.; et al. Genetic lineage tracing identifies endocardial origin of liver vasculature. Nat. Genet. 2016, 48, 537–543. [Google Scholar] [CrossRef]
- Oberlin, E.; El Hafny, B.; Petit, L.; Souyri, M. Definitive human and mouse hematopoiesis originates from the embryonic endothelium: A new class of HSCs based on VE-cadherin expression. Int. J. Dev. Biol. 2010, 54, 1165–1173. [Google Scholar] [CrossRef]
- Zovein, A.; Hofmann, J.J.; Lynch, M.; French, W.J.; Turlo, K.A.; Yang, Y.; Becker, M.S.; Zanetta, L.; Dejana, E.; Gasson, J.C.; et al. Fate Tracing Reveals the Endothelial Origin of Hematopoietic Stem Cells. Cell Stem Cell 2008, 3, 625–636. [Google Scholar] [CrossRef]
- Lacaud, G.; Kouskoff, V. Hemangioblast, hemogenic endothelium, and primitive versus definitive hematopoiesis. Exp. Hematol. 2017, 49, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Halpern, K.B.; Shenhav, R.; Massalha, H.; Toth, B.; Egozi, A.; Massasa, E.E.; Medgalia, C.; David, E.; Giladi, A.; Moor, A.E.; et al. Paired-cell sequencing enables spatial gene expression mapping of liver endothelial cells. Nat. Biotechnol. 2018, 36, 962–970. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Zhao, L.; Fish, M.; Logan, C.Y.; Nusse, R. Self-renewing diploid Axin2+ cells fuel homeostatic renewal of the liver. Nat. Cell Biol. 2015, 524, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Planas-Paz, L.; Orsini, V.; Boulter, L.; Calabrese, D.; Pikiolek, M.; Nigsch, F.; Xie, Y.; Roma, G.; Donovan, A.; Marti, P.; et al. The RSPO–LGR4/5–ZNRF3/RNF43 module controls liver zonation and size. Nat. Cell Biol. 2016, 18, 467–479. [Google Scholar] [CrossRef] [PubMed]
- Rocha, A.S.; Vidal, V.; Mertz, M.; Kendall, T.; Charlet, A.; Okamoto, H.; Schedl, A. The Angiocrine Factor Rspondin3 Is a Key Determinant of Liver Zonation. Cell Rep. 2015, 13, 1757–1764. [Google Scholar] [CrossRef]
- Canali, S.; Zumbrennen-Bullough, K.B.; Core, A.B.; Wang, C.-Y.; Nairz, M.; Bouley, R.; Swirski, F.; Babitt, J.L. Endothelial cells produce bone morphogenetic protein 6 required for iron homeostasis in mice. Blood 2017, 129, 405–414. [Google Scholar] [CrossRef]
- Koch, P.-S.; Olsavszky, V.; Ulbrich, F.; Sticht, C.; Demory, A.; Leibing, T.; Henzler, T.; Meyer, M.; Zierow, J.; Schneider, S.; et al. Angiocrine Bmp2 signaling in murine liver controls normal iron homeostasis. Blood 2017, 129, 415–419. [Google Scholar] [CrossRef]
- Wang, L.; Wang, X.; Xie, G.; Wang, L.; Hill, C.K.; Deleve, L.D. Liver sinusoidal endothelial cell progenitor cells promote liver regeneration in rats. J. Clin. Investig. 2012, 122, 1567–1573. [Google Scholar] [CrossRef] [PubMed]
- Deleve, L.D. Liver sinusoidal endothelial cells and liver regeneration. J. Clin. Investig. 2013, 123, 1861–1866. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, T.; Liu, Z.; Murase, N.; Ezure, T.; Yokomuro, S.; Poli, V.; Demetris, A.J. Mitosis and apoptosis in the liver of interleukin-6-deficient mice after partial hepatectomy. Hepatology 1999, 29, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Koyama, S.; Tsukada, K.; Hatakeyama, K. Acute portal hypertension reflecting shear stress as a trigger of liver regeneration following partial hepatectomy. Surg. Today 1997, 27, 518–526. [Google Scholar] [CrossRef] [PubMed]
- Schoen, J.M.; Wangb, H.H.; Minukac, G.Y.; Lautt, W. Shear Stress-Induced Nitric Oxide Release Triggers the Liver Regeneration Cascade. Nitric Oxide 2001, 5, 453–464. [Google Scholar] [CrossRef]
- Albinsson, S.; Hellstrand, P. Integration of signal pathways for stretch-dependent growth and differentiation in vascular smooth muscle. Am. J. Physiol. Cell Physiol. 2007, 293, C772–C782. [Google Scholar] [CrossRef][Green Version]
- Weinbaum, S.; Zhang, X.; Han, Y.; Vink, H.; Cowin, S.C. Mechanotransduction and flow across the endothelial glycocalyx. Proc. Natl. Acad. Sci. USA 2003, 100, 7988–7995. [Google Scholar] [CrossRef]
- Díaz-Juárez, J.A.; Hernández-Muñoz, R. Rat Liver Enzyme Release Depends on Blood Flow-Bearing Physical Forces Acting in Endothelium Glycocalyx rather than on Liver Damage. Oxidative Med. Cell. Longev. 2017, 2017, 1360565. [Google Scholar] [CrossRef]
- Schoen, J.M.; Lautt, W.W. Nitric Oxide Potentiates C-Fos mRNA Expression after 2/3 Partial Hepatectomy. Proc. West. Pharmacol. Soc. 2002, 45, 47–48. [Google Scholar]
- Isomura, H.; Sawada, N.; Nakajima, Y.; Sakamoto, H.; Ikeda, T.; Kojima, T.; Enomoto, K.; Mori, M. Increase in portal flow induces c-myc expression in isolated perfused rat liver. J. Cell. Physiol. 1993, 154, 329–332. [Google Scholar] [CrossRef]
- Gan, L.-M.; Doroudi, R.; Hägg, U.; Johansson, A.-M.; Selin-Sjögren, L.; Jern, S. Differential immediate-early gene responses to shear stress and intraluminal pressure in intact human conduit vessels. FEBS Lett. 2000, 477, 89–94. [Google Scholar] [CrossRef]
- Lauber, D.T.; Tihanyi, D.K.; Czigány, Z.; Kovács, T.; Budai, A.; Drozgyik, D.; Fülöp, A.; Szijártó, A. Liver regeneration after different degrees of portal vein ligation. J. Surg. Res. 2016, 203, 451–458. [Google Scholar] [CrossRef]
- Meier, M.; Andersen, K.J.; Knudsen, A.R.; Nyengaard, J.R.; Hamilton-Dutoit, S.; Mortensen, F.V. Liver regeneration is dependent on the extent of hepatectomy. J. Surg. Res. 2016, 205, 76–84. [Google Scholar] [CrossRef]
- Shimazu, M.; Kato, Y.; Kawachi, S.; Tanabe, M.; Hoshino, K.; Wakabayashi, G.; Kitagawa, Y.; Kitajima, M. Impact of Portal Hemodynamic Changes in Partial Liver Grafts on Short-Term Graft Regeneration in Living Donor Liver Transplantation. Transplant. Proc. 2016, 48, 2747–2755. [Google Scholar] [CrossRef]
- Eguchi, S.; Yanaga, K.; Sugiyama, N.; Okudaira, S.; Furui, J.; Kanematsu, T. Relationship between portal venous flow and liver regeneration in patients after living donor right-lobe liver transplantation. Liver Transplant. 2003, 9, 547–551. [Google Scholar] [CrossRef]
- Oyama, T.; Sadamori, H.; Matsukawa, H.; Murata, H.; Umeda, Y.; Watanabe, Y.; Ozaki, M.; Iwagaki, H.; Tanaka, N.; Yagi, T. Small liver graft regenerates through immediate increase of HGF and IL-6--possible involvement of sinusoidal tensile/shear stress in small liver graft. Hepatogastroenterology 2008, 54, 2078–2083. [Google Scholar]
- Jones, D.E.; Tran-Patterson, R.; Cui, D.M.; Davin, D.; Estell, K.P.; Miller, D.M. Epidermal growth factor secreted from the salivary gland is necessary for liver regeneration. Am. J. Physiol. Gastrointest. Liver Physiol. 1995, 268, G872–G878. [Google Scholar] [CrossRef] [PubMed]
- Rubin, R.A.; O’Keefe, E.J.; Earp, H.S. Alteration of epidermal growth factor-dependent phosphorylation during rat liver regeneration. Proc. Natl. Acad. Sci. USA 1982, 79, 776–780. [Google Scholar] [CrossRef]
- Olsen, P.S.; Boesby, S.; Kirkegaard, P.; Therkelsen, K.; Almdal, T.; Poulsen, S.S.; Nexø, E. Influence of epidermal growth factor on liver regeneration after partial hepatectomy in rats. Hepatology 1988, 8, 992–996. [Google Scholar] [CrossRef] [PubMed]
- Shavandi, A.; Saeedi, P.; Gérard, P.; Jalalvandi, E.; Cannella, D.; Bekhit, A.E. The role of microbiota in tissue repair and regeneration. J. Tissue Eng. Regen. Med. 2020, 14, 539–555. [Google Scholar] [CrossRef] [PubMed]
- Patijn, G.A.; Lieber, A.; Schowalter, D.B.; Schwall, R.; Kay, M.A. Hepatocyte growth factor induces hepatocyte proliferationin vivo and allows for efficient retroviral-mediated gene transfer in mice. Hepatology 1998, 28, 707–716. [Google Scholar] [CrossRef]
- Sokabe, T.; Yamamoto, K.; Ohura, N.; Nakatsuka, H.; Qin, K.; Obi, S.; Kamiya, A.; Ando, J. Differential regulation of urokinase-type plasminogen activator expression by fluid shear stress in human coronary artery endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H2027–H2034. [Google Scholar] [CrossRef][Green Version]
- Dolan, J.M.; Sim, F.J.; Meng, H.; Kolega, J. Endothelial cells express a unique transcriptional profile under very high wall shear stress known to induce expansive arterial remodeling. Am. J. Physiol. Cell Physiol. 2012, 302, C1109–C1118. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Mars, W.M.; Stolz, D.B.; Petersen, B.E.; Michalopoulos, G.K. Extracellular matrix remodeling at the early stages of liver regeneration in the rat. Hepatology 1997, 26, 896–904. [Google Scholar] [CrossRef] [PubMed]
- Nejak-Bowen, K.; Orr, A.; Bowen, W.C., Jr.; Michalopoulos, G.K. Conditional Genetic Elimination of Hepatocyte Growth Factor in Mice Compromises Liver Regeneration after Partial Hepatectomy. PLoS ONE 2013, 8, e59836. [Google Scholar] [CrossRef] [PubMed]
- Mars, W.M.; Zarnegar, R.; Michalopoulos, G.K. Activation of Hepatocyte Growth Factor by the Plasminogen Activators uPA and tPA. Am. J. Pathol. 1993, 143, 949–958. [Google Scholar] [PubMed]
- Mars, W.M.; Kim, T.H.; Stolz, D.B.; Liu, M.L.; Michalopoulos, G.K. Presence of urokinase in serum-free primary rat hepatocyte cultures and its role in activating hepatocyte growth factor. Cancer Res. 1996, 56, 2837–2843. [Google Scholar]
- Shanmukhappa, K.; Sabla, G.E.; Degen, J.L.; Bezerra, J.A. Urokinase-type plasminogen activator supports liver repair independent of its cellular receptor. BMC Gastroenterol. 2006, 6, 40. [Google Scholar] [CrossRef]
- Ding, B.-S.; Nolan, D.J.; Butler, J.M.; James, D.; Babazadeh, A.O.; Rosenwaks, Z.; Mittal, V.; Kobayashi, H.; Shido, K.; Lyden, D.; et al. Inductive angiocrine signals from sinusoidal endothelium are required for liver regeneration. Nat. Cell Biol. 2010, 468, 310–315. [Google Scholar] [CrossRef]
- Döme, B.; Dobos, J.; Tóvári, J.; Paku, S.; Kovács, G.; Ostoros, G.; Tímár, J. Circulating bone marrow-derived endothelial progenitor cells: Characterization, mobilization, and therapeutic considerations in malignant disease. Cytom. Part A 2008, 73, 186–193. [Google Scholar] [CrossRef]
- Timmermans, F.; Plum, J.; Yoder, M.; Ingram, D.A.; Vandekerckhove, B.; Case, J. Endothelial progenitor cells: Identity defined? J. Cell. Mol. Med. 2008, 13, 87–102. [Google Scholar] [CrossRef] [PubMed]
- Hristov, M.; Erl, W.; Weber, P.C. Endothelial progenitor cells: Mobilization, differentiation, and homing. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1185–1189. [Google Scholar] [CrossRef]
- Fujii, H.; Hirose, T.; Oe, S.; Yasuchika, K.; Azuma, H.; Fujikawa, T.; Nagao, M.; Yamaoka, Y. Contribution of bone marrow cells to liver regeneration after partial hepatectomy in mice. J. Hepatol. 2002, 36, 653–659. [Google Scholar] [CrossRef]
- Li, N.; Hua, J. Immune cells in liver regeneration. Oncotarget 2016, 8, 3628–3639. [Google Scholar] [CrossRef]
- Harb, R.; Xie, G.; Lutzko, C.; Guo, Y.; Wang, X.; Hill, C.K.; Kanel, G.C.; DeLeve, L.D. Bone Marrow Progenitor Cells Repair Rat Hepatic Sinusoidal Endothelial Cells After Liver Injury. Gastroenterology 2009, 137, 704–712. [Google Scholar] [CrossRef]
- Deleve, L.D.; Wang, X.; Wang, L. VEGF-sdf1 recruitment of CXCR7 bone marrow progenitors of liver sinusoidal endothelial cells promotes rat liver regeneration. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 310, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Odent (Grigorescu), G.; Rosca, A.-M.; Preda, M.B.; Tutuianu, R.; Simionescu, M.; Burlacu, A. Synergic effects of VEGF-A and SDF-1 on the angiogenic properties of endothelial progenitor cells. J. Tissue Eng. Regen. Med. 2016, 11, 3241–3252. [Google Scholar] [CrossRef]
- Moore, M.A.; Hattori, K.; Heissig, B.; Shieh, J.-H.; Dias, S.; Crystal, R.G.; Rafii, S. Mobilization of Endothelial and Hematopoietic Stem and Progenitor Cells by Adenovector-Mediated Elevation of Serum Levels of SDF-1, VEGF, and Angiopoietin-1. Ann. N. Y. Acad. Sci. 2006, 938, 36–47. [Google Scholar] [CrossRef]
- Peled, A.; Grabovsky, V.; Habler, L.; Sandbank, J.; Arenzana-Seisdedos, F.; Petit, I.; Ben-Hur, H.; Lapidot, T.; Alon, R. The chemokine SDF-1 stimulates integrin-mediated arrest of CD34+ cells on vascular endothelium under shear flow. J. Clin. Investig. 1999, 104, 1199–1211. [Google Scholar] [CrossRef]
- Silverman, M.D.; Haas, C.S.; Rad, A.M.; Arbab, A.S.; Koch, A.E. The role of vascular cell adhesion molecule 1/very late activation antigen 4 in endothelial progenitor cell recruitment to rheumatoid arthritis synovium. Arthritis Rheum. 2007, 56, 1817–1826. [Google Scholar] [CrossRef]
- Yoon, C.-H.; Hur, J.; Oh, I.-Y.; Park, K.-W.; Kim, T.-Y.; Shin, J.-H.; Kim, J.-H.; Lee, C.-S.; Chung, J.-K.; Park, Y.-B.; et al. Intercellular Adhesion Molecule-1 is Upregulated in Ischemic Muscle, Which Mediates Trafficking of Endothelial Progenitor Cells. Arter. Thromb. Vasc. Biol. 2006, 26, 1066–1072. [Google Scholar] [CrossRef]
- Jin, H.; Aiyer, A.; Su, J.; Borgström, P.; Stupack, D.; Friedlander, M.; Varner, J. A homing mechanism for bone marrow–derived progenitor cell recruitment to the neovasculature. J. Clin. Investig. 2006, 116, 652–662. [Google Scholar] [CrossRef]
- Chavakis, E.; Aicher, A.; Heeschen, C.; Sasaki, K.-I.; Kaiser, R.; El Makhfi, N.; Urbich, C.; Peters, T.; Scharffetter-Kochanek, K.; Zeiher, A.M.; et al. Role of β2-integrins for homing and neovascularization capacity of endothelial progenitor cells. J. Exp. Med. 2004, 201, 63–72. [Google Scholar] [CrossRef]
- Tilling, L.; Chowienczyk, P.; Clapp, B. Progenitors in motion: Mechanisms of mobilization of endothelial progenitor cells. Br. J. Clin. Pharmacol. 2009, 68, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Myronovych, A.; Murata, S.; Chiba, M.; Matsuo, R.; Ikeda, O.; Watanabe, M.; Hisakura, K.; Nakano, Y.; Kohno, K.; Kawasaki, T.; et al. Role of platelets on liver regeneration after 90% hepatectomy in mice. J. Hepatol. 2008, 49, 363–372. [Google Scholar] [CrossRef]
- Meyer, J.; Lejmi, E.; Fontana, P.; Morel, P.; Gonelle-Gispert, C.; Bühler, L. A focus on the role of platelets in liver regeneration: Do platelet-endothelial cell interactions initiate the regenerative process? J. Hepatol. 2015, 63, 1263–1271. [Google Scholar] [CrossRef] [PubMed]
- Melgar-Lesmes, P.; Edelman, E. Monocyte-endothelial cell interactions in the regulation of vascular sprouting and liver regeneration in mouse. J. Hepatol. 2015, 63, 917–925. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Srivastava, K.; Wieland, M.; Runge, A.; Mogler, C.; Besemfelder, E.; Terhardt, D.; Vogel, M.J.; Cao, L.; Korn, C.; et al. Endothelial Cell-Derived Angiopoietin-2 Controls Liver Regeneration as a Spatiotemporal Rheostat. Science 2014, 343, 416–419. [Google Scholar] [CrossRef]
- Taniguchi, E.; Sakisaka, S.; Matsuo, K.; Tanikawa, K.; Sata, M. Expression and role of vascular endothelial growth factor in liver regeneration after partial hepatectomy in rats. J. Histochem. Cytochem. 2001, 49, 121–129. [Google Scholar] [CrossRef]
- Shimizu, H.; Miyazaki, M.; Wakabayashi, Y.; Mitsuhashi, N.; Kato, A.; Ito, H.; Nakagawa, K.; Yoshidome, H.; Kataoka, M.; Nakajima, N. Vascular endothelial growth factor secreted by replicating hepatocytes induces sinusoidal endothelial cell proliferation during regeneration after partial hepatectomy in rats. J. Hepatol. 2001, 34, 683–689. [Google Scholar] [CrossRef]
- Furnus, C.; Inda, A.; Andrini, L.; García, M.; García, A.; Badrán, A.; Errecalde, A. Chronobiology of the proliferative events related to angiogenesis in mice liver regeneration after partial hepatectomy. Cell Biol. Int. 2003, 27, 383–386. [Google Scholar] [CrossRef]
- Kron, P.; Linecker, M.; Limani, P.; Schlegel, A.; Kambakamba, P.; Lehn, J.-M.; Nicolau, C.; Graf, R.; Humar, B.; Clavien, P.-A. Hypoxia-driven Hif2α coordinates mouse liver regeneration by coupling parenchymal growth to vascular expansion. Hepatology 2016, 64, 2198–2209. [Google Scholar] [CrossRef] [PubMed]
- Braun, L.; Mead, J.E.; Panzica, M.; Mikumo, R.; Bell, G.I.; Fausto, N. Transforming growth factor beta mRNA increases during liver regeneration: A possible paracrine mechanism of growth regulation. Proc. Natl. Acad. Sci. USA 1988, 85, 1539–1543. [Google Scholar] [CrossRef] [PubMed]
- Michalopoulos, G.K.; Bhushan, B. Liver regeneration: Biological and pathological mechanisms and implications. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 40–55. [Google Scholar] [CrossRef]
- Poon, R.T.; Fan, S.T.; Lo, C.M.; Liu, C.L.; Lam, C.M.; Yuen, W.K.; Yeung, C.; Wong, J.; Nagorney, D.M.; Henderson, J.M.; et al. Improving perioperative outcome expands the role of hepatectomy in management of benign and malignant hepatobiliary diseases: Analysis of 1222 consecutive patients from a prospective database. Ann. Surg. 2004, 240, 698–710. [Google Scholar] [CrossRef] [PubMed]
- Imamura, H.; Seyama, Y.; Kokudo, N.; Aoki, T.; Sano, K.; Minagawa, M.; Sugawara, Y.; Makuuchi, M. Single and multiple resections of multiple hepatic metastases of colorectal origin. Surgery 2004, 135, 508–517. [Google Scholar] [CrossRef] [PubMed]
- Dahm, F.; Georgiev, P.; Clavien, P.-A. Small-for-Size Syndrome after Partial Liver Transplantation: Definition, Mechanisms of Disease and Clinical Implications. Arab. Archaeol. Epigr. 2005, 5, 2605–2610. [Google Scholar] [CrossRef]
- Yagi, S.; Iida, T.; Hori, T.; Taniguchi, K.; Yamamoto, C.; Yamagiwa, K.; Uemoto, S. Optimal Portal Venous Circulation for Liver Graft Function after Living-Donor Liver Transplantation. Transplantation 2006, 81, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Guglielmi, A.; Ruzzenente, A.; Conci, S.; Valdegamberi, A.; Iacono, C. How Much Remnant Is Enough in Liver Resection? Dig. Surg. 2012, 29, 6–17. [Google Scholar] [CrossRef]
- Lehmann, K.; Tschuor, C.; Rickenbacher, A.; Jang, J.; Oberkofler, C.E.; Tschopp, O.; Schultze, S.M.; Raptis, D.A.; Weber, A.; Graf, R.; et al. Liver Failure After Extended Hepatectomy in Mice Is Mediated by a p21-Dependent Barrier to Liver Regeneration. Gastroenterology 2012, 143, 1609–1619.e4. [Google Scholar] [CrossRef]
- Zieve, L.; Anderson, W.R.; Lindblad, S. Course of hepatic regeneration after 80% to 90% resection of normal rat liver. Comparison with two-lobe and one-lobe hepatectomy. J. Lab. Clin. Med. 1985, 105, 331–336. [Google Scholar]
- Moser, M.J.; Gong, Y.; Zhang, M.N.; Johnston, J.; Lipschitz, J.; Minuk, G.Y. Immediate-Early Protooncogene Expression and Liver Function Following Various Extents of Partial Hepatectomy in the Rat. Dig. Dis. Sci. 2001, 46, 907–914. [Google Scholar] [CrossRef]
- Ninomiya, M.; Shirabe, K.; Terashi, T.; Ijichi, H.; Yonemura, Y.; Harada, N.; Soejima, Y.; Taketomi, A.; Shimada, M.; Maehara, Y. Deceleration of Regenerative Response Improves the Outcome of Rat with Massive Hepatectomy. Arab. Archaeol. Epigr. 2010, 10, 1580–1587. [Google Scholar] [CrossRef] [PubMed]
- Gridelli, B.G.; Gruttadauria, S.; Pagano, D.; Liotta, R.; Tropea, A.; Tuzzolino, F.; Marrone, G.; Mamone, G.; Marsh, J.W.; Miraglia, R.; et al. Liver Volume Restoration and Hepatic Microarchitecture in Small-for-Size Syndrome. Ann. Transplant. 2015, 20, 381–389. [Google Scholar] [CrossRef]
- Byun, S.H.; Yang, H.S.; Kim, J.H. Liver graft hyperperfusion in the early postoperative period promotes hepatic regeneration 2 weeks after living donor liver transplantation. Medicine 2016, 95, e5404. [Google Scholar] [CrossRef]
- Demetris, A.J.; Kelly, D.M.; Eghtesad, B.; Fontes, P.; Marsh, J.W.; Tom, K.; Tan, H.P.; Shaw-Stiffel, T.; Boig, L.; Novelli, P.; et al. Pathophysiologic Observations and Histopathologic Recognition of the Portal Hyperperfusion or Small-for-Size Syndrome. Am. J. Surg. Pathol. 2006, 30, 986–993. [Google Scholar] [CrossRef] [PubMed]
- Vasavada, B.; Chen, C.L.; Zakaria, M. Portal flow is the main predictor of early graft dysfunction regardless of the GRWR status in living donor liver transplantation—A retrospective analysis of 134 patients. Int. J. Surg. 2014, 12, 177–180. [Google Scholar] [CrossRef]
- Asencio, J.; Vaquero, J.; Olmedilla, L.; Sabrido, J.G. “Small-for-flow” syndrome: Shifting the “size” paradigm. Med. Hypotheses 2013, 80, 573–577. [Google Scholar] [CrossRef] [PubMed]
- Allard, M.-A.; Adam, R.; Bucur, P.-O.; Termos, S.; Cunha, A.S.; Bismuth, H.; Castaing, D.; Vibert, E. Posthepatectomy Portal Vein Pressure Predicts Liver Failure and Mortality after Major Liver Resection on Noncirrhotic Liver. Ann. Surg. 2013, 258, 822–830. [Google Scholar] [CrossRef]
- Brown, R.S. Live Donors in Liver Transplantation. Gastroenterology 2008, 134, 1802–1813. [Google Scholar] [CrossRef]
- Golse, N.; Bucur, P.O.; Adam, R.; Castaing, D.; Cunha, A.S.; Vibert, E. New Paradigms in Post-hepatectomy Liver Failure. J. Gastrointest. Surg. 2012, 17, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Umeda, Y.; Yagi, T.; Sadamori, H.; Fujiwara, T. Small-for-Size Syndrome after Living Donor Liver Transplantation. In Liver Transplantation: Technical Issues and Complications; Abdeldayem, H., Ed.; IntechOpen: London, UK, 2012; ISBN 978-953-51-0015-7. [Google Scholar]
- Troisi, R.; Ricciardi, S.; Smeets, P.; Petrovic, M.; Van Maele, G.; Colle, I.; Van Vlierberghe, H.; De Hemptinne, B. Effects of Hemi-Portocaval Shunts for Inflow Modulation on the Outcome of Small-for-Size Grafts in Living Donor Liver Transplantation. Arab. Archaeol. Epigr. 2005, 5, 1397–1404. [Google Scholar] [CrossRef]
- Sato, Y.; Yamamoto, S.; Oya, H.; Nakatsuka, H.; Tsukahara, A.; Kobayashi, T.; Watanabe, T.; Hatakeyama, K. Sple-nectomy for reduction of excessive portal hypertension after adult living-related donor liver transplantation. Hepatogastroenterology 2002, 49, 1652–1655. [Google Scholar]
- Yamada, T.; Tanaka, K.; Uryuhara, K.; Ito, K.; Takada, Y.; Uemoto, S. Selective Hemi-Portocaval Shunt Based on Portal Vein Pressure for Small-for-Size Graft in Adult Living Donor Liver Transplantation. Arab. Archaeol. Epigr. 2008, 8, 847–853. [Google Scholar] [CrossRef] [PubMed]
- Boillot, O. Portomesenteric disconnection for small-for-size grafts in liver transplantation: Preclinical studies in pigs. Liver Transplant. 2003, 9, S42–S46. [Google Scholar] [CrossRef]
- Yamanaka, K.; Hatano, E.; Narita, M.; Kitamura, K.; Yanagida, A.; Asechi, H.; Nagata, H.; Taura, K.; Nitta, T.; Uemoto, S. Olprinone attenuates excessive shear stress through up-regulation of endothelial nitric oxide synthase in a rat excessive hepatectomy model. Liver Transplant. 2010, 17, 60–69. [Google Scholar] [CrossRef]
- Morioka, D.; Matsuo, K.; Endo, I.; Togo, S.; Kubota, T.; Sekido, H.; Saito, S.; Ichikawa, Y.; Shimada, H. Prostaglandin E1 improved the function of transplanted fatty liver in a rat reduced-size-liver transplantation model under conditions of permissible cold preservation. Liver Transplant. 2003, 9, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Man, K.; Lee, K.W.; Liang, T.B.; Lo, C.M.; Fung, P.C.-W.; Tsui, S.H.; Li, X.L.; Ng, K.T.-P.; Fan, S.T. FK 409 Ameliorates Small-for-Size Liver Graft Injury by Attenuation of Portal Hypertension and Down-Regulation of Egr-1 Pathway. Ann. Surg. 2004, 240, 159–168. [Google Scholar] [CrossRef]
- Xu, X.; Man, K.; Zheng, S.S.; Liang, T.B.; Lee, K.W.; Ng, K.T.-P.; Fan, S.T.; Lo, C.M. Attenuation of acute phase shear stress by somatostatin improves small-for-size liver graft survival. Liver Transplant. 2006, 12, 621–627. [Google Scholar] [CrossRef]
- Mohkam, K.; Darnis, B.; Schmitt, Z.; Duperret, S.; Ducerf, C.; Mabrut, J.-Y. Successful modulation of portal inflow by somatostatin in a porcine model of small-for-size syndrome. Am. J. Surg. 2016, 212, 321–326. [Google Scholar] [CrossRef]
- Dili, A.; Bertrand, C.; Lebrun, V.; Pirlot, B.; Leclercq, I.A. Hypoxia protects the liver from Small for Size Syndrome: A lesson learned from the associated liver partition and portal vein ligation for staged hepatectomy (ALPPS) procedure in rats. Arab. Archaeol. Epigr. 2019, 19, 2979–2990. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Rudder, M.; Dili, A.; Stärkel, P.; Leclercq, I.A. Critical Role of LSEC in Post-Hepatectomy Liver Regeneration and Failure. Int. J. Mol. Sci. 2021, 22, 8053. https://doi.org/10.3390/ijms22158053
De Rudder M, Dili A, Stärkel P, Leclercq IA. Critical Role of LSEC in Post-Hepatectomy Liver Regeneration and Failure. International Journal of Molecular Sciences. 2021; 22(15):8053. https://doi.org/10.3390/ijms22158053
Chicago/Turabian StyleDe Rudder, Maxime, Alexandra Dili, Peter Stärkel, and Isabelle A. Leclercq. 2021. "Critical Role of LSEC in Post-Hepatectomy Liver Regeneration and Failure" International Journal of Molecular Sciences 22, no. 15: 8053. https://doi.org/10.3390/ijms22158053
APA StyleDe Rudder, M., Dili, A., Stärkel, P., & Leclercq, I. A. (2021). Critical Role of LSEC in Post-Hepatectomy Liver Regeneration and Failure. International Journal of Molecular Sciences, 22(15), 8053. https://doi.org/10.3390/ijms22158053