
COVID-19 in Children: Expressions of Type I/II/III Interferons, TRIM28, SETDB1, and Endogenous Retroviruses in Mild and Severe Cases

 , , ,
, , ,
Abstract
1. Introduction
2. Results
2.1. Study Populations
2.2. Characteristics of Infected Children
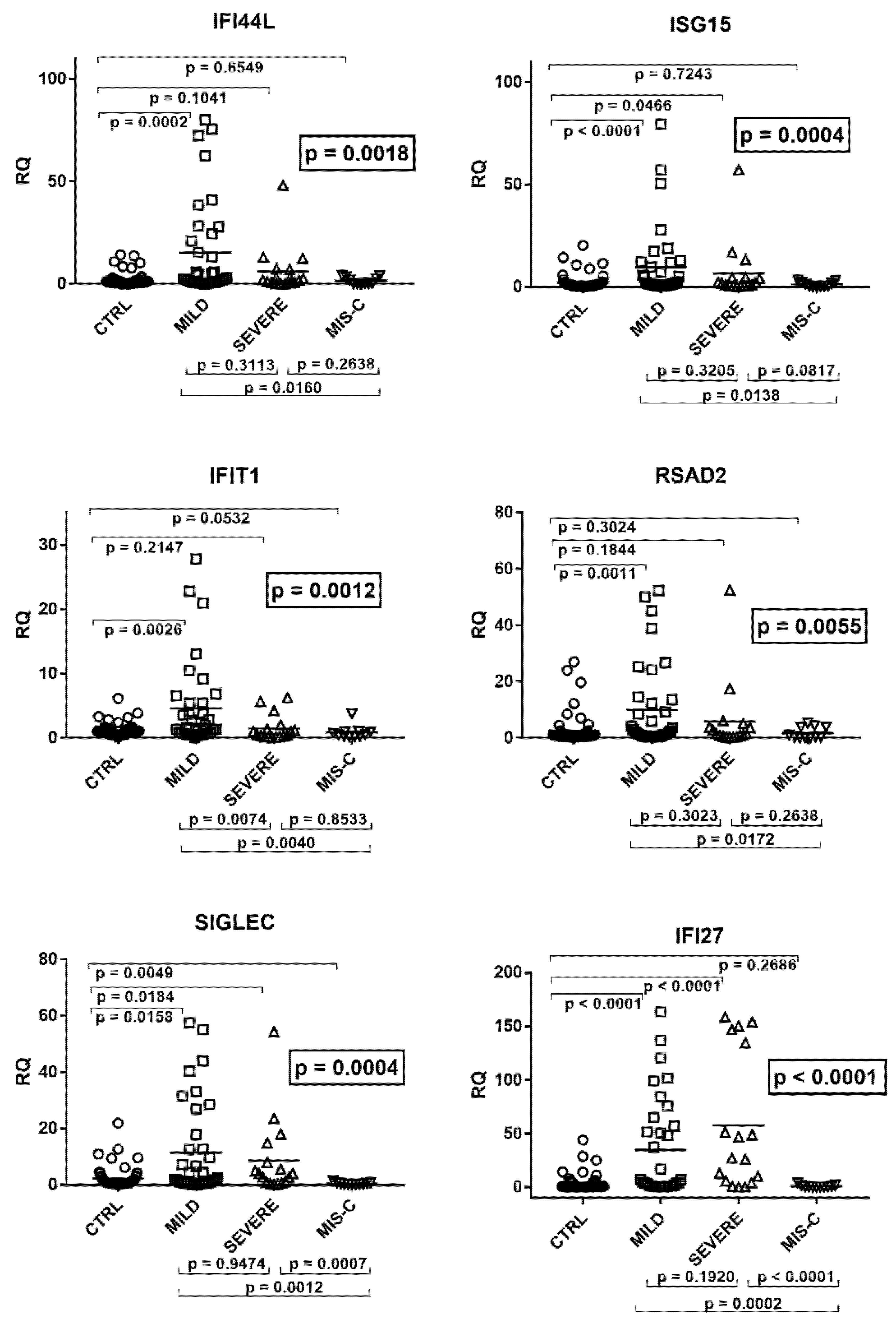
2.3. Type I IFN Signature
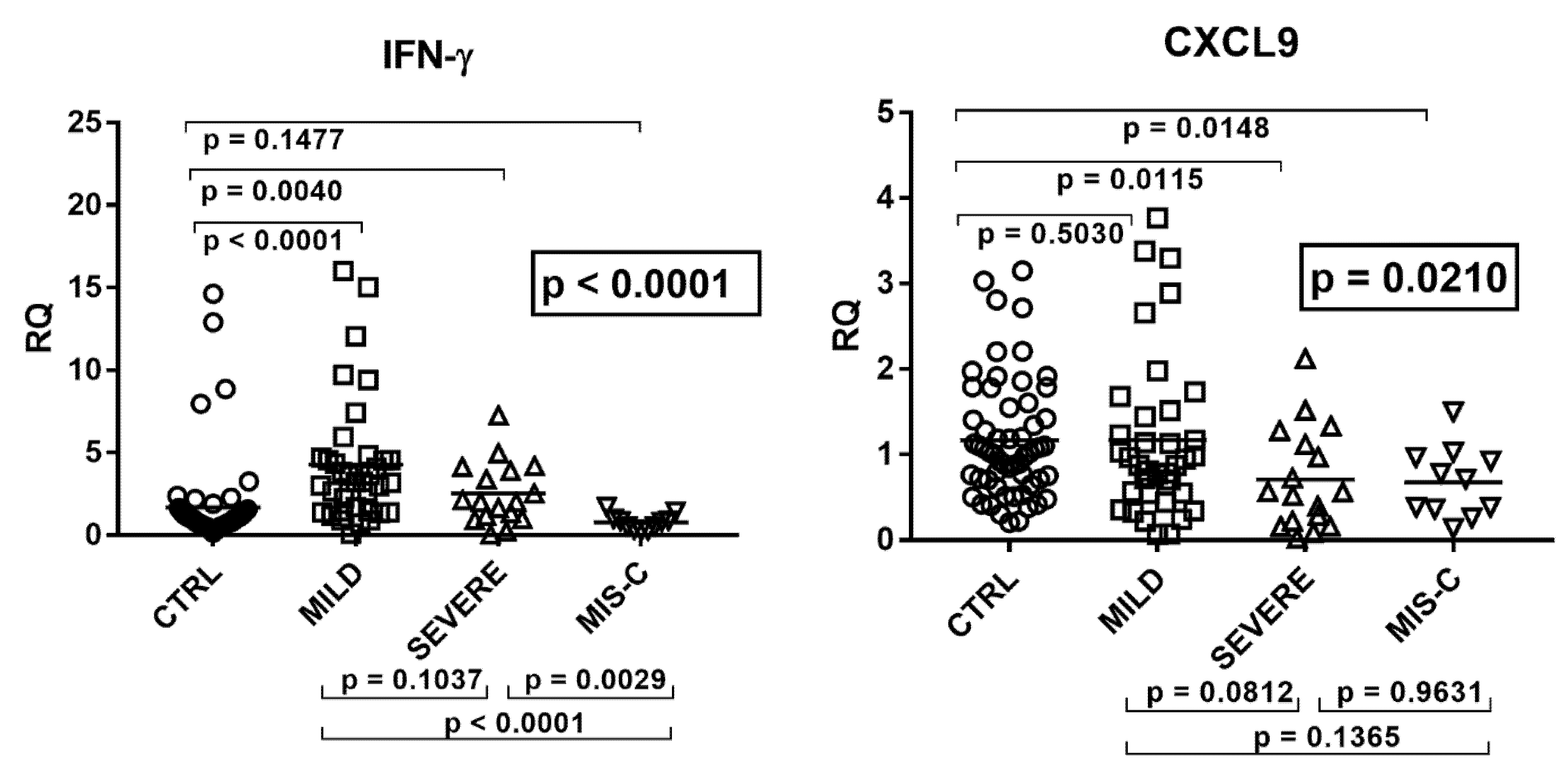
2.4. Type II IFN Signature
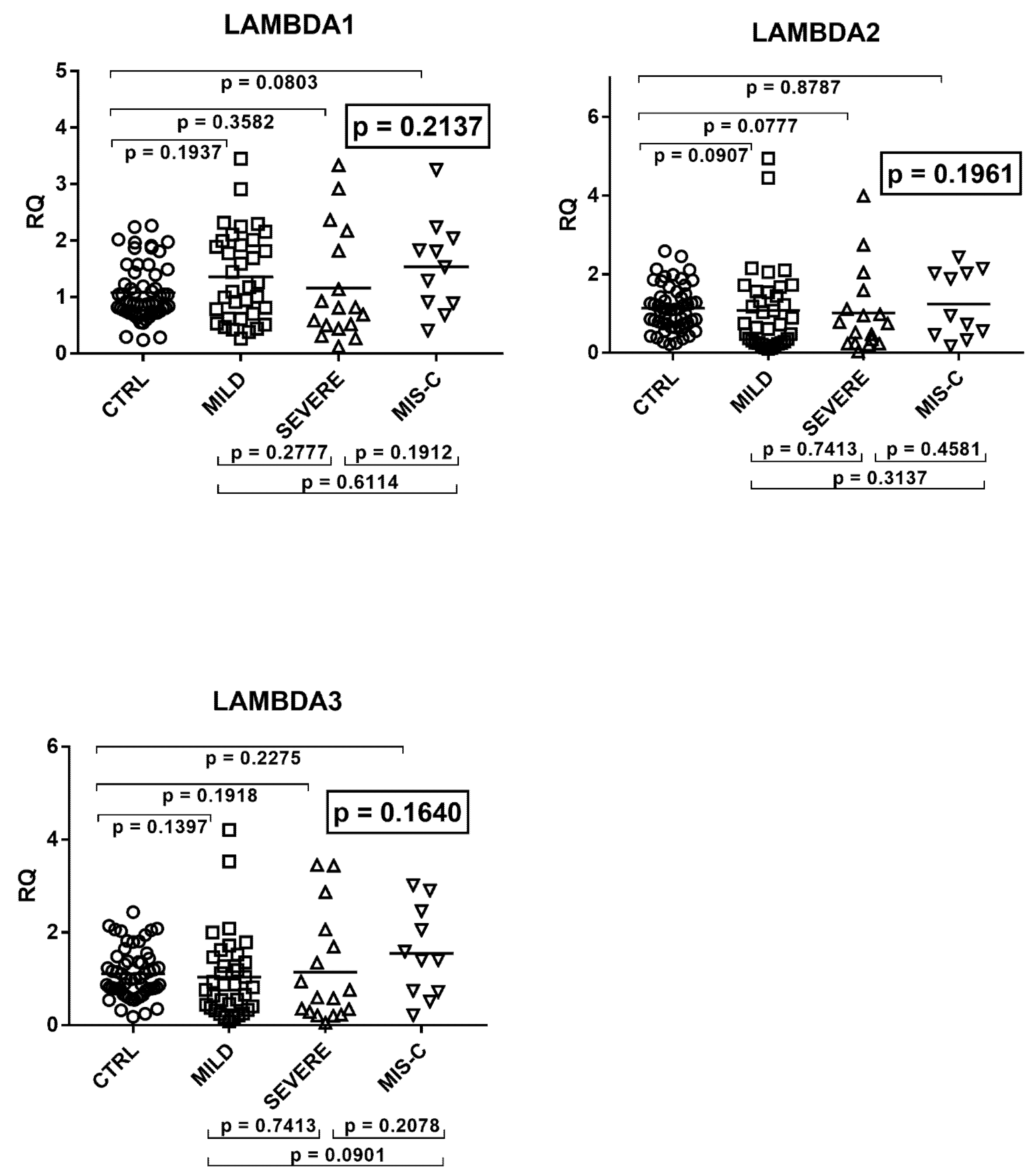
2.5. Type III IFNs
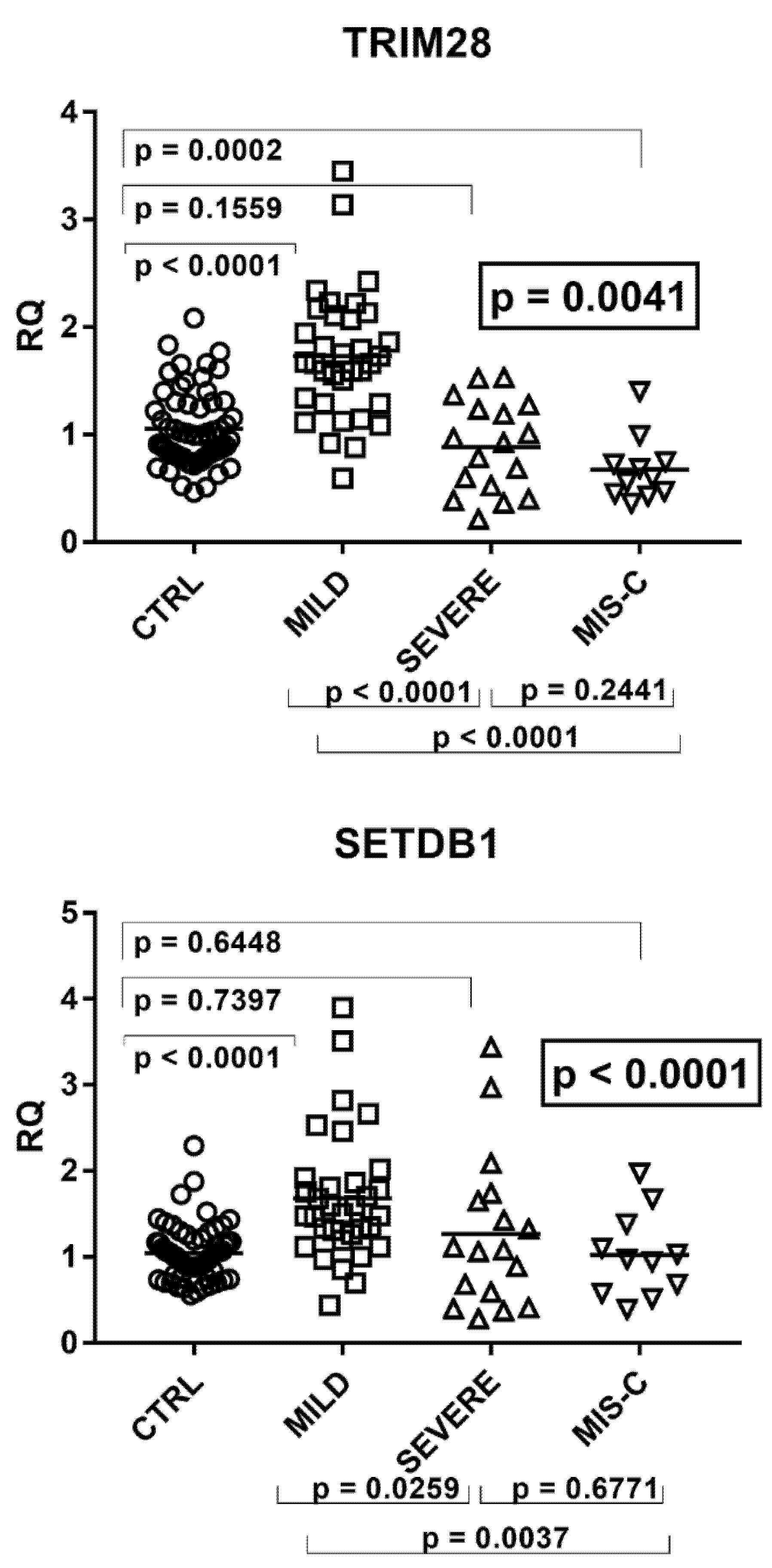
2.6. Expressions of TRIM28 and SETDB1
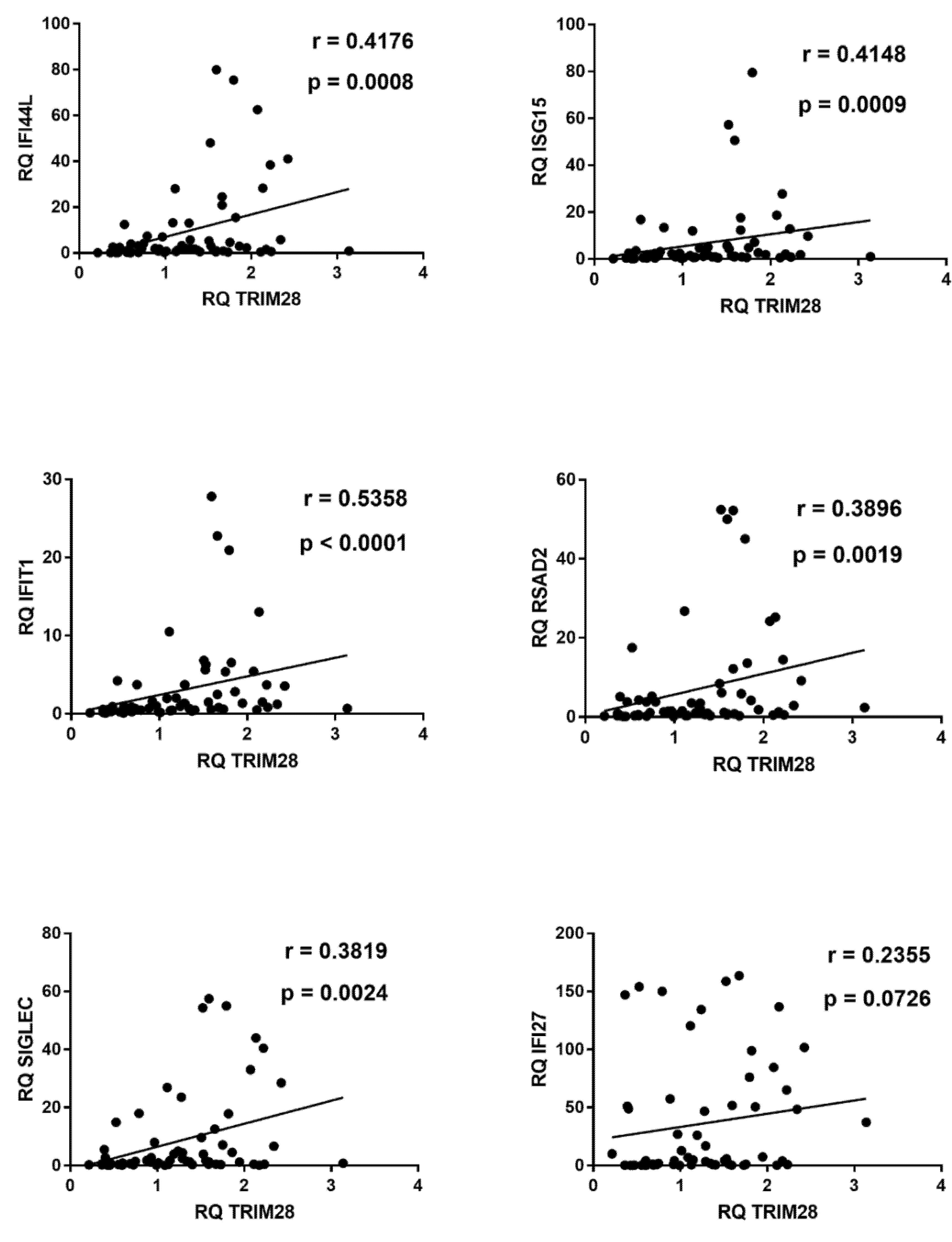
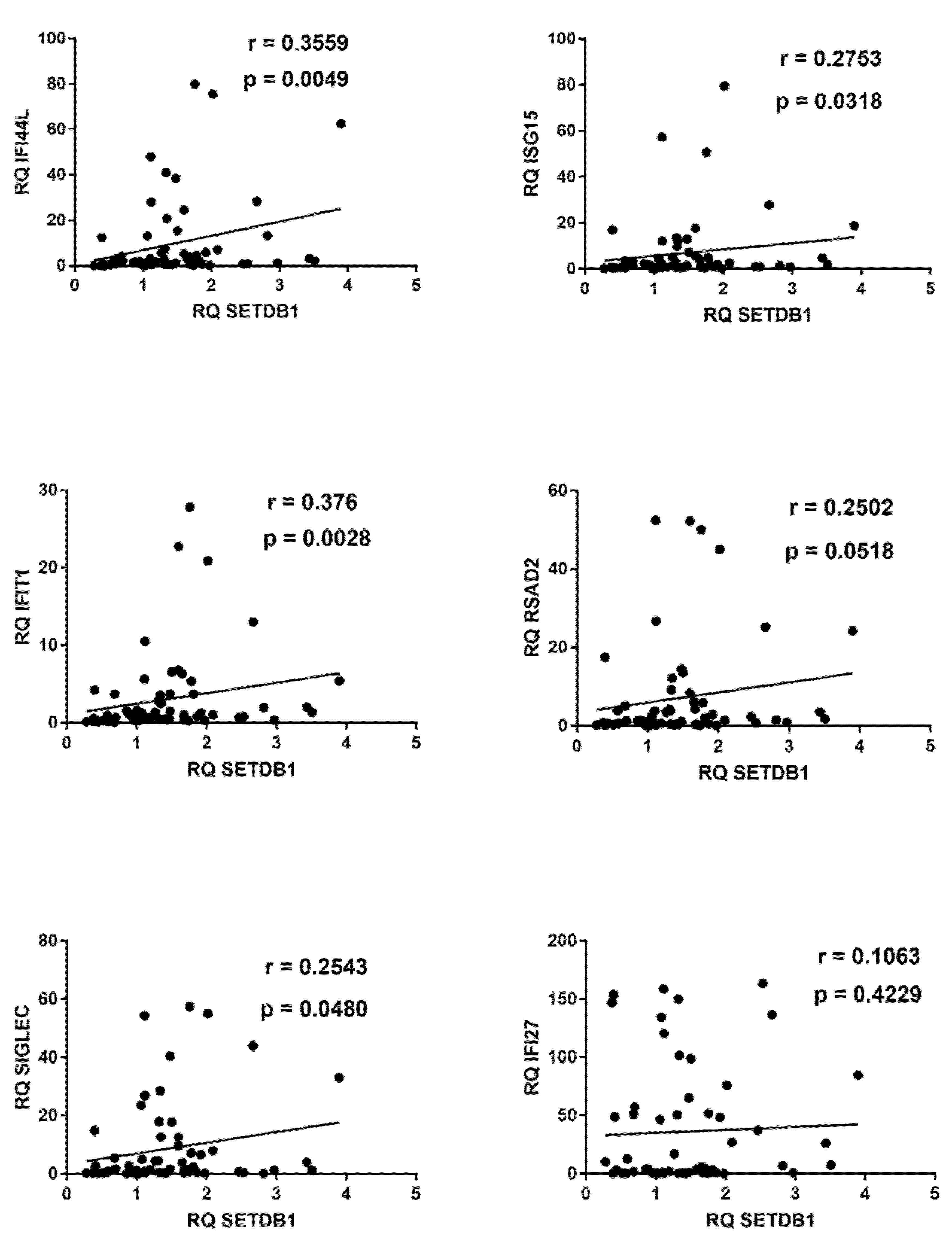
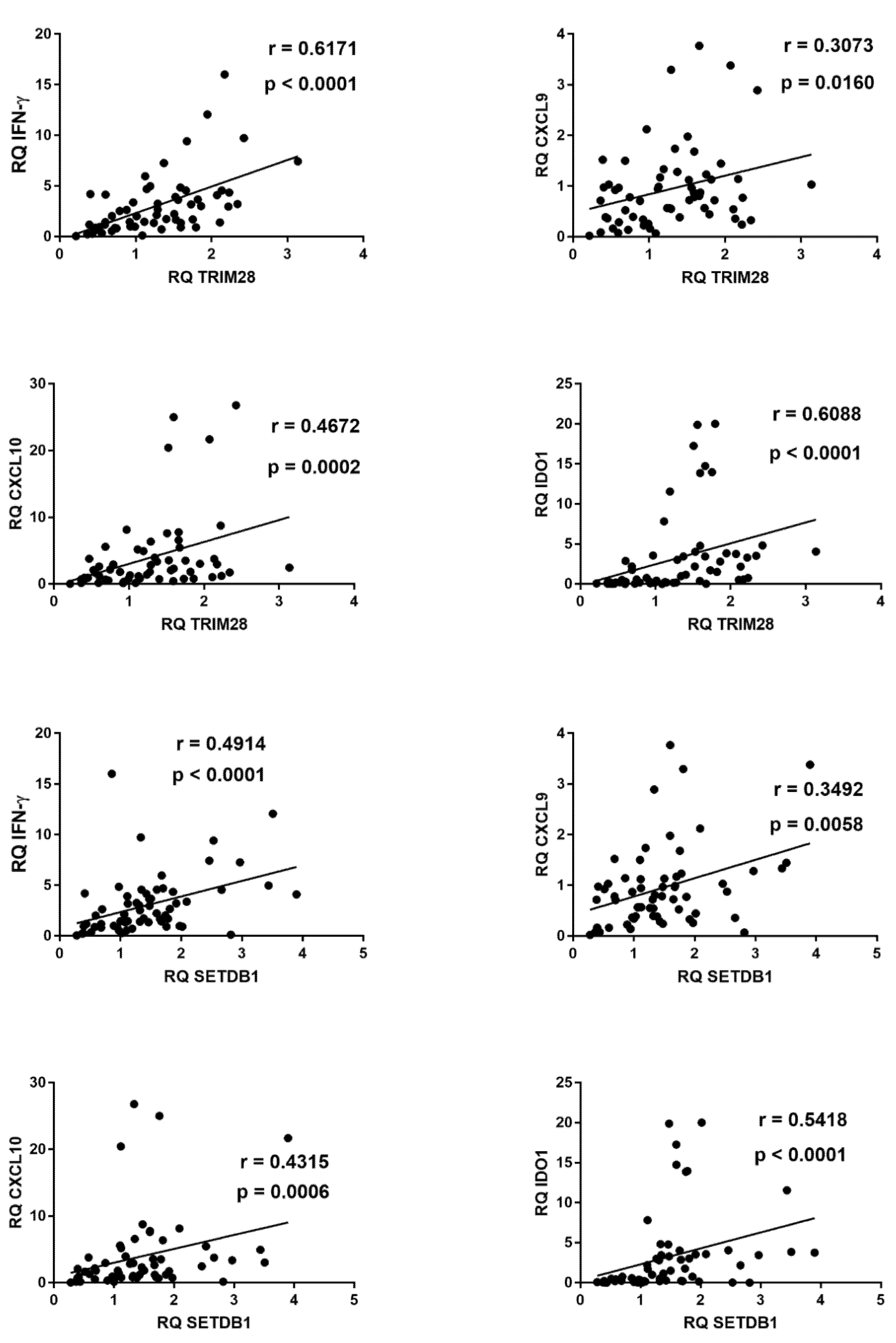
2.7. Correlations between Expressions of IFNs and ISGs and TRIM28 or SETDB1
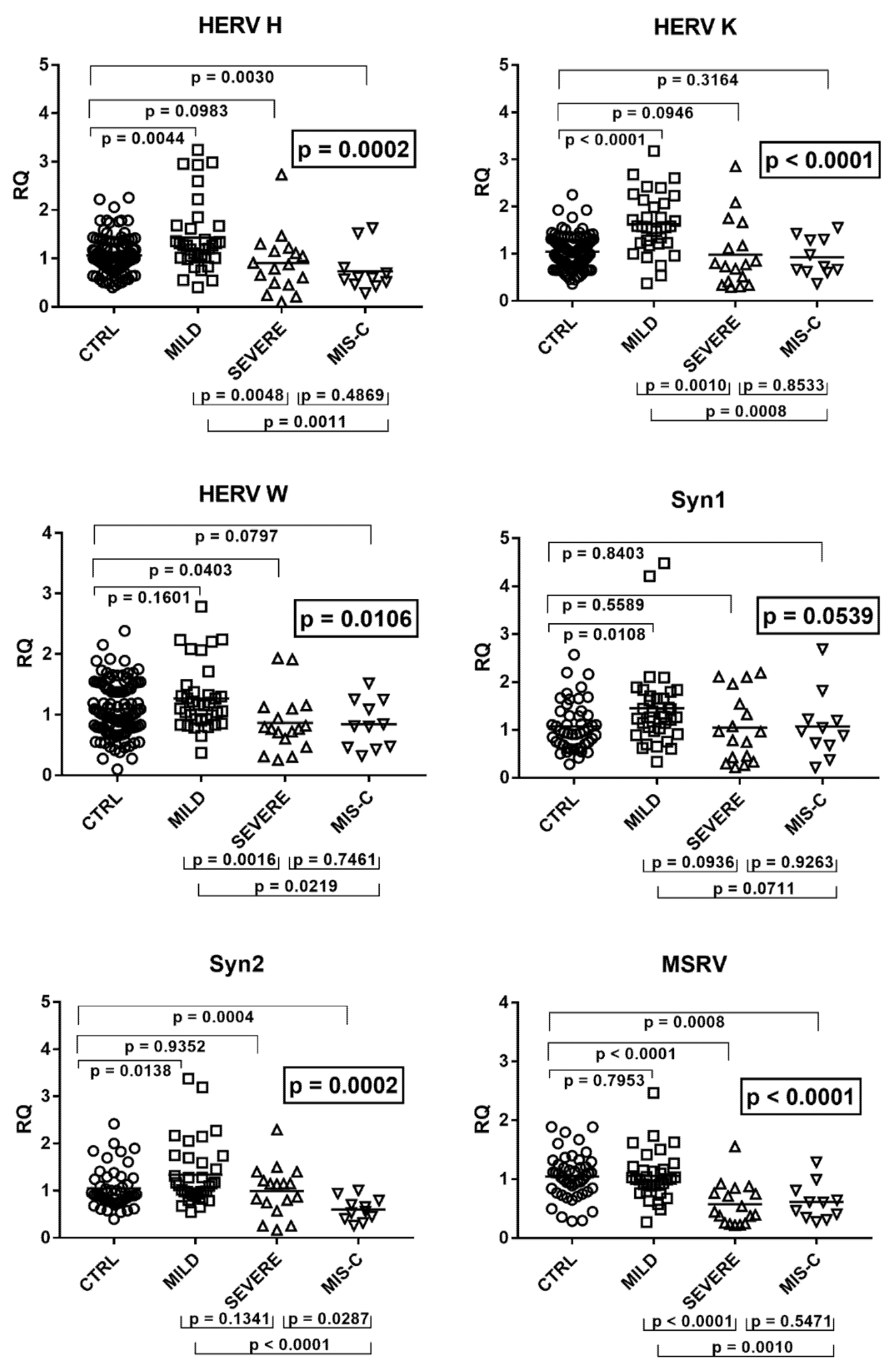
2.8. Expressions of HERV-H-pol, HERV-K-pol, HERV-W-pol, and of SYN1-env, SYN2-env, and MSRV-env
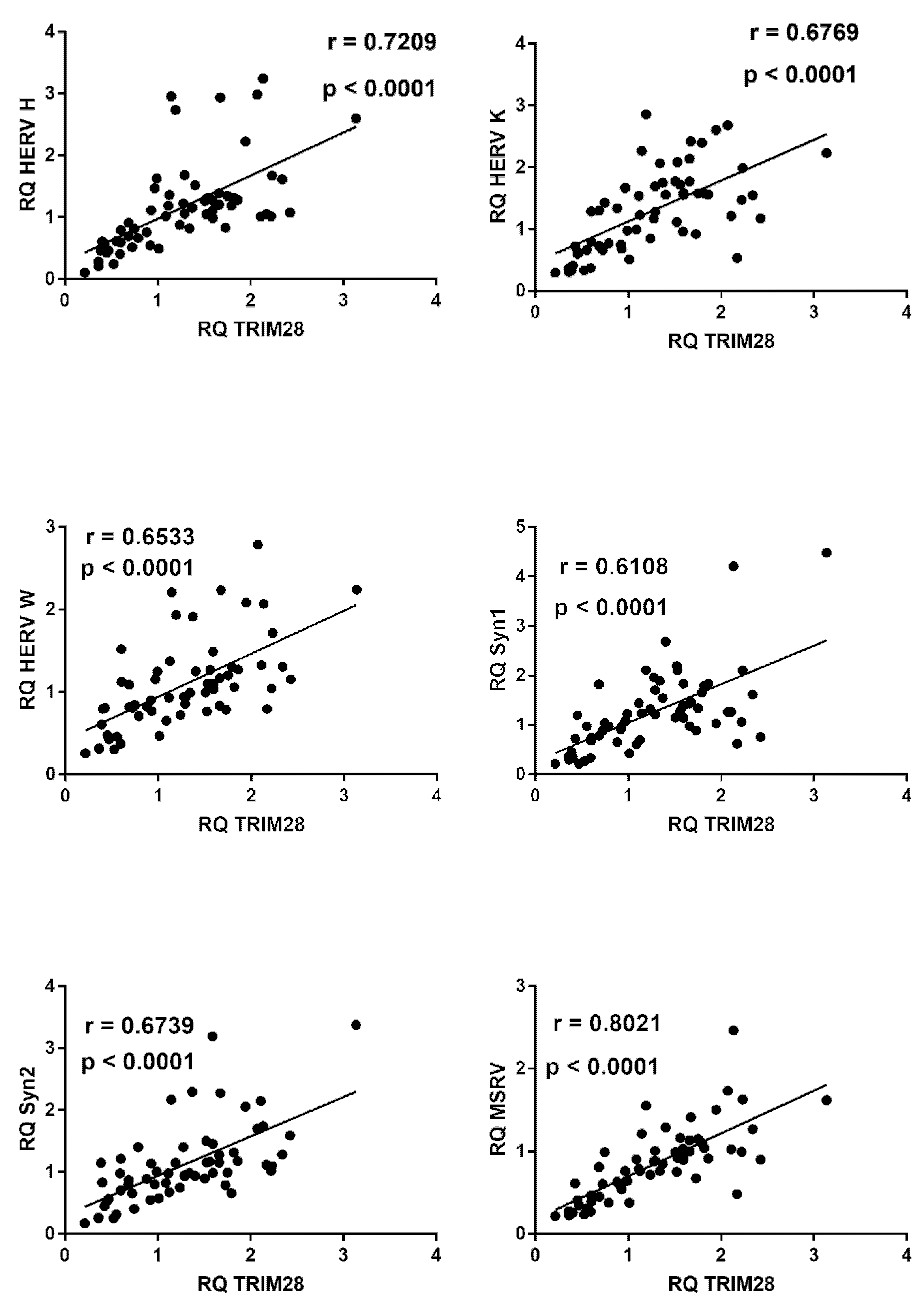
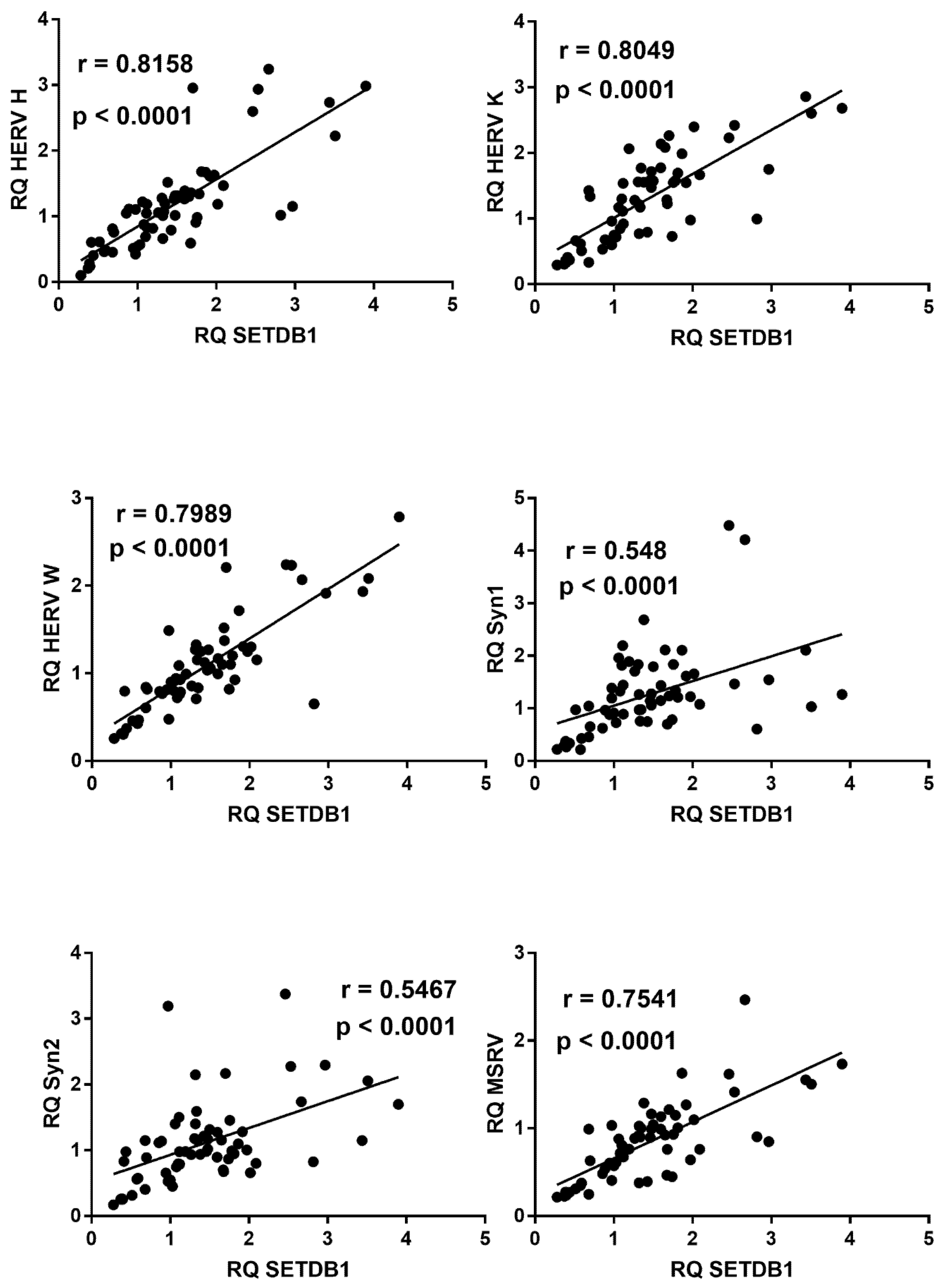
2.9. Correlations between Expressions of HERVs and TRIM28 or SETDB1
2.10. Correlations between Expressions of IFNs or ISGs and HERV Sequences
3. Discussion
4. Materials and Methods
4.1. Study Populations
4.2. Total RNA Extraction
4.3. Reverse Transcription
4.4. Transcription Levels of IFNs, ISGs, TRIM28, SETDB1, pol Genes of HERV-H, -K, and -W, and env Genes of SYN1, SYN2, and MSRV by Real-Time PCR Assays
4.5. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| COVID-19 | Coronavirus disease 2019 |
| HERVs | Human endogenous retroviruses |
| KRAB-ZFPs | Krüppel-associated box domain zinc finger proteins |
| IFN | Interferon |
| IFN-I | Type I interferon |
| IFN-II | Type II interferon |
| IFN-III | Type III interferon |
| MIS-C | Multisystem inflammatory syndrome in children |
| MSRV | Multiple sclerosis-associated retrovirus |
| PRR | Pattern recognition receptor |
| SAMHD1 | Sterile alpha motif and HD-domain–containing protein 1 |
| SARS-CoV-2 | Severe acute respiratory syndrome coronavirus 2 |
| SETDB1 | SET domain bifurcated histone lysine methyltrasferase 1 |
| SYN1 | Syncytin 1 |
| SYN2 | Syncytin 2 |
| TRIM28 | Tripartite motif containing 28 |
References
- Tsabouri, S.; Makis, A.; Kosmeri, C.; Siomou, E. Risk factors for severity in children with coronavirus disease 2019: A comprehensive literature review. Pediatr. Clin. N. Am. 2021, 68, 321–338. [Google Scholar] [CrossRef]
- Schreiber, G. The molecular basis for differential type I interferon signaling. J. Biol. Chem. 2017, 292, 7285–7294. [Google Scholar] [CrossRef]
- Park, A.; Iwasaki, A. Type I and type III interferons-induction, signaling, evasion, and application to combat COVID-19. Cell Host Microbe 2020, 27, 870–878. [Google Scholar] [CrossRef]
- Nile, S.H.; Nile, A.; Qiu, J.; Li, L.; Jia, X.; Kai, G. COVID-19: Pathogenesis, cytokine storm and therapeutic potential of interferons. Cytokine Growth Factor Rev. 2020, 53, 66–70. [Google Scholar] [CrossRef]
- Chen, L.; Ozato, K. Innate immune memory in hematopoietic stem/progenitor cells: Myeloid-biased differentiation and the role of interferon. Front. Immunol. 2021, 12, 621333. [Google Scholar] [CrossRef]
- Ayers, M.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, S.P.; Shankaran, V.; et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Investig. 2017, 127, 2930–2940. [Google Scholar] [CrossRef] [PubMed]
- Andreakos, E.; Zanoni, I.; Galani, I.E. Lambda interferons come to light: Dual function cytokines mediating antiviral immunity and damage control. Curr. Opin. Immunol. 2019, 56, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Kotenko, S.V.; Rivera, A.; Parker, D.; Durbin, J.E. Type III IFNs: Beyond antiviral protection. Semin. Immunol. 2019, 43, 101303. [Google Scholar] [CrossRef] [PubMed]
- Koltsida, O.; Hausding, M.; Stavropoulos, A.; Koch, S.; Tzelepis, G.; Ubel, C.; Kotenko, S.V.; Sideras, P.; Lehr, H.A.; Tepe, M.; et al. IL-28A (IFN-λ2) modulates lung DC function to promote Th1 immune skewing and suppress allergic airway disease. EMBO Mol. Med. 2011, 3, 348–361. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Schnepf, D.; Becker, J.; Ebert, K.; Tanriver, Y.; Bernasconi, V.; Gad, H.H.; Hartmann, R.; Lycke, N.; Staeheli, P. Interferon-λ enhances adaptive mucosal immunity by boosting release of thymic stromal lymphopoietin. Nat. Immunol. 2019, 20, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Busnadiego, I.; Fernbach, S.; Pohl, M.O.; Karakus, U.; Huber, M.; Trkola, A.; Stertz, S.; Hale, B.G. Antiviral activity of type I, II, and III interferons counterbalances ACE2 inducibility and restricts SARS-CoV-2. MBio 2020, 11, e01928–e02020. [Google Scholar] [CrossRef]
- Vanderheiden, A.; Ralfs, P.; Chirkova, T.; Upadhyay, A.A.; Zimmerman, M.G.; Bedoya, S.; Aoued, H.; Tharp, G.M.; Pellegrini, K.L.; Manfredi, C.; et al. Type I and Type III Interferons restrict SARS-CoV-2 infection of human airway epithelial cultures. J. Virol. 2020, 94, e00985–e01020. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Bastard, P.; Liu, Z.; Le Pen, J.; Moncada-Velez, M.; Chen, J.; Ogishi, M.; Sabli, I.K.D.; Hodeib, S.; Korol, C.; et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science 2020, 370, eabd4570. [Google Scholar] [CrossRef] [PubMed]
- Bastard, P.; Rosen, L.B.; Zhang, Q.; Michailidis, E.; Hoffmann, H.-H.; Zhang, Y.; Dorgham, K.; Philippot, Q.; Rosain, J.; Béziat, V.; et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science 2020, 370, eabd4585. [Google Scholar] [CrossRef] [PubMed]
- Lopez, L.; Sang, P.C.; Tian, Y.; Sang, Y. Dysregulated interferon response underlying severe COVID-19. Viruses 2020, 12, 1433. [Google Scholar] [CrossRef] [PubMed]
- Galani, I.E.; Rovina, N.; Lampropoulou, V.; Triantafyllia, V.; Manioudaki, M.; Pavlos, E.; Koukaki, E.; Fragkou, P.C.; Panou, V.; Rapti, V.; et al. Untuned antiviral immunity in COVID-19 revealed by temporal type I/III interferon patterns and flu comparison. Nat. Immunol. 2021, 22, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Weber, F.; Kochs, G.; Haller, O. Inverse interference: How viruses fight the interferon system. Viral Immunol. 2004, 17, 498–515. [Google Scholar] [CrossRef] [PubMed]
- Murira, A.; Lamarre, A. Type-I interferon responses: From friend to foe in the battle against chronic viral infection. Front. Immunol. 2016, 7, 609. [Google Scholar] [CrossRef] [PubMed]
- Kopecky-Bromberg, S.A.; Martínez-Sobrido, L.; Frieman, M.; Baric, R.A.; Palese, P. Severe acute respiratory syndrome coronavirus open reading frame (ORF) 3b, ORF 6, and nucleocapsid proteins function as interferon antagonists. J. Virol. 2007, 81, 548–557. [Google Scholar] [CrossRef]
- Konno, Y.; Kimura, I.; Uriu, K.; Fukushi, M.; Irie, T.; Koyanagi, Y.; Sauter, D.; Gifford, R.J. USFQ-COVID19 Consortium, Nakagawa, S.; Sato, K. SARS-CoV-2 ORF3b is a potent interferon antagonist whose activity is increased by a naturally occurring elongation variant. Cell Rep. 2020, 32, 108185. [Google Scholar] [CrossRef]
- Sobocińska, J.; Molenda, S.; Machnik, M.; Oleksiewicz, U. KRAB-ZFP transcriptional regulators acting as oncogenes and tumor suppressors: An Overview. Int. J. Mol. Sci. 2021, 23, 2212. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.R.; Fredericks, W.J.; Jensen, D.E.; Speicher, D.W.; Huang, X.P.; Neilson, E.G.; Rauscher, F.J., III. KAP-1, a novel corepressor for the highly conserved KRAB repression domain. Genes Dev. 1996, 10, 2067–2078. [Google Scholar] [CrossRef] [PubMed]
- Schultz, D.C.; Ayyanathan, K.; Negorev, D.; Maul, G.G.; Rauscher, F.J., III. SETDB1: A novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev. 2002, 16, 919–932. [Google Scholar] [CrossRef]
- Jiang, Y.; Liu, Y.; Lu, H.; Sun, S.C.; Jin, W.; Wang, X.; Dong, C. Epigenetic activation during T helper 17 cell differentiation is mediated by Tripartite motif containing 28. Nat. Commun. 2018, 12, 1424. [Google Scholar] [CrossRef]
- Gehrmann, U.; Burbage, M.; Zueva, E.; Goudot, C.; Esnault, C.; Ye, M.; Carpier, J.M.; Burgdorf, N.; Hoyler, T.; Suarez, G.; et al. Critical role for TRIM28 and HP1β/γ in the epigenetic control of T cell metabolic reprograming and effector differentiation. Proc. Natl. Acad. Sci. USA 2019, 116, 25839–25849. [Google Scholar] [CrossRef]
- Czerwinska, P.; Jaworska, A.M.; Wlodarczyk, N.A.; Mackiewicz, A.A. Melanoma stem cell-like phenotype and significant suppression of immune response within a tumor are regulated by TRIM28 protein. Cancers 2020, 12, 2998. [Google Scholar] [CrossRef]
- Kamitani, S.; Ohbayashi, N.; Ikeda, O.; Togi, S.; Muromoto, R.; Sekine, Y.; Ohta, K.; Ishiyama, H.; Matsuda, T. KAP1 regulates type I interferon/STAT1-mediated IRF-1 gene expression. Biochem. Biophys. Res. Commun. 2008, 370, 366–370. [Google Scholar] [CrossRef]
- Krischuns, T.; Günl, F.; Henschel, L.; Binder, M.; Willemsen, J.; Schloer, S.; Rescher, U.; Gerlt, V.; Zimmer, G.; Nordhoff, C.; et al. Phosphorylation of TRIM28 enhances the expression of IFN-β and proinflammatory cytokines during HPAIV infection of human lung epithelial cells. Front. Immunol. 2018, 9, 2229. [Google Scholar] [CrossRef]
- Nisole, S.; Stoye, J.P.; Saïb, A. TRIM family proteins: Retroviral restriction and antiviral defence. Nat. Rev. Microbiol. 2005, 3, 799–808. [Google Scholar] [CrossRef] [PubMed]
- Johnson, W.E. Origins and evolutionary consequences of ancient endogenous retroviruses. Nat. Rev. Microbiol. 2019, 17, 355–370. [Google Scholar] [CrossRef]
- Blond, J.L.; Lavillette, D.; Cheynet, V.; Bouton, O.; Oriol, G.; Chapel-Fernandes, S.; Mandrand, B.; Mallet, F.; Cosset, F.L. An envelope glycoprotein of the human endogenous retrovirus herv-w is expressed in the human placenta and fuses cells expressing the type d mammalian retrovirus receptor. J. Virol. 2000, 74, 3321–3329. [Google Scholar] [CrossRef]
- Blaise, S.; de Parseval, N.; Benit, L.; Heidmann, T. Genomewide screening for fusogenic human endogenous retrovirus envelopes identifies syncytin 2, a gene conserved on primate evolution. Proc. Natl. Acad. Sci. USA 2003, 100, 13013–13018. [Google Scholar] [CrossRef]
- Lokossou, A.G.; Toudic, C.; Barbeau, B. Implication of human endogenous retrovirus envelope proteins in placental functions. Viruses 2014, 6, 4609–4627. [Google Scholar] [CrossRef] [PubMed]
- Isbel, L.; Whitelaw, E. Endogenous retroviruses in mammals: An emerging picture of how ERVs modify expression of adjacent genes. BioEssays 2012, 34, 734–738. [Google Scholar] [CrossRef]
- Rolland, A.; Jouvin-Marche, E.; Viret, C.; Faure, M.; Perron, H.; Marche, P.N. The envelope protein of a human endogenous retrovirus-W family activates innate immunity through CD14/TLR4 and promotes Th1-like responses. J. Immunol. 2006, 76, 7636–7644. [Google Scholar] [CrossRef] [PubMed]
- Chuong, E.B.; Elde, N.C.; Feschotte, C. Regulatory evolution of innate immunity through co-option of endogenous retroviruses. Science 2016, 351, 1083–1087. [Google Scholar] [CrossRef] [PubMed]
- Mu, X.; Ahmad, S.; Hur, S. Endogenous retroelements and the host innate immune sensors. Adv. Immunol. 2016, 132, 47–69. [Google Scholar]
- Perron, H.; Garson, J.A.; Bedin, F.; Beseme, F.; Paranhos-Baccala, G.; Komurian-Pradel, F.; Mallet, F.; Tuke, P.W.; Voisset, C.; Blond, J.L.; et al. Molecular identification of a novel retrovirus repeatedly isolated from patients with multiple sclerosis. Proc. Natl. Acad. Sci. USA 1997, 94, 7583–7588. [Google Scholar] [CrossRef]
- Madeira, A.; Burgelin, I.; Perron, H.; Curtin, F.; Lang, A.B.; Faucard, R. MSRV envelope protein is a potent, endogenous and pathogenic agonist of human toll-like receptor 4: Relevance of GNbAC1 in multiple sclerosis treatment. J. Neuroimmunol. 2016, 291, 29–38. [Google Scholar] [CrossRef]
- Hummel, J.; Kämmerer, U.; Müller, N.; Avota, E.; Schneider-Schaulies, S. Human endogenous retrovirus envelope proteins target dendritic cells to suppress T-cell activation. Eur. J. Immunol. 2015, 45, 1748–1759. [Google Scholar] [CrossRef]
- Lokossou, A.G.; Toudic, C.; Nguyen, P.T.; Elisseeff, X.; Vargas, A.; Rassart, É.; Lafond, J.; LeDuc, L.; Bourgault, S.; Gilbert, C.; et al. Endogenous retrovirus-encoded Syncytin-2 contributes to exosome-mediated immunosuppression of T cells. Biol. Reprod. 2020, 102, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Montojo, M.; Rodriguez-Martin, E.; Ramos-Mozo, P.; Ortega-Madueño, I.; Dominguez-Mozo, M.I.; Arias-Leal, A.; García-Martínez, M.Á.; Casanova, I.; Galan, V.; Arroyo, R.; et al. Syncytin-1/HERV-W envelope is an early activation marker of leukocytes and is upregulated in multiple sclerosis patients. Eur. J. Immunol. 2020, 50, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Yu, P. The potential role of retroviruses in autoimmunity. Immunol. Rev. 2016, 269, 85–99. [Google Scholar] [CrossRef]
- Grandi, N.; Tramontano, E. Human endogenous retroviruses are ancient acquired elements still shaping innate immune responses. Front. Immunol. 2018, 9, 2039. [Google Scholar] [CrossRef] [PubMed]
- Tovo, P.A.; Rabbone, I.; Tinti, D.; Galliano, I.; Trada, M.; Daprà, V.; Cerutti, F.; Bergallo, M. Enhanced expression of human endogenous retroviruses in new-onset type 1 diabetes: Pathogenetic and therapeutic implications. Autoimmunity 2020, 53, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Ruprecht, K.; Obojes, K.; Wengel, V.; Gronen, F.; Kim, K.S.; Perron, H.; Schneider-Schaulies, J.; Rieckmann, P. Regulation of human endogenous retrovirus W protein expression by herpes simplex virus type 1: Implications for multiple sclerosis. J. Neurovirol. 2006, 12, 65–71. [Google Scholar] [CrossRef]
- Mameli, G.; Poddighe, L.; Mei, A.; Uleri, E.; Sotgiu, S.; Serra, C.; Manetti, R.; Dolei, A. Expression and activation by Epstein Barr virus of human endogenous retroviruses-W in blood cells and astrocytes: Inference for multiple sclerosis. PLoS ONE 2012, 7, e44991. [Google Scholar] [CrossRef]
- Van der Kuyl, A.C. HIV infection and HERV expression: A review. Retrovirology 2012, 9, 6. [Google Scholar] [CrossRef]
- Assinger, A.; Yaiw, K.C.; Göttesdorfer, I.; Leib-Mösch, C.; Söderberg-Nauclér, C. Human cytomegalovirus (HCMV) induces human endogenous retrovirus (HERV) transcription. Retrovirology 2013, 10, 132. [Google Scholar] [CrossRef]
- Schmidt, N.; Domingues, P.; Golebiowski, F.; Patzina, C.; Tatham, M.H.; Hay, R.T.; Hale, B.G. An influenza virus-triggered SUMO switch orchestrates co-opted endogenous retroviruses to stimulate host antiviral immunity. Proc. Natl. Acad. Sci. USA 2019, 116, 17399–17408. [Google Scholar] [CrossRef]
- Tovo, P.A.; Garazzino, S.; Daprà, V.; Alliaudi, C.; Silvestro, E.; Calvi, C.; Montanari, P.; Galliano, I.; Bergallo, M. Chronic HCV infection is associated with overexpression of human endogenous retroviruses that persists after drug-induced viral clearance. Int. J. Mol. Sci. 2020, 21, 3980. [Google Scholar] [CrossRef]
- Manghera, M.; Ferguson-Parry, J.; Lin, R.; Douville, R.N. NF-κB and IRF1 induce endogenous retrovirus expression via interferon-stimulated response elements in its 5’ long terminal repeat. J. Virol. 2016, 90, 9338–9349. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Richards, A.; Khalil, A.; Wogram, E.; Ma, H.; Richard, R.A.; Jaenish, R. SARS-CoV-2 RNA reverse-transcribed and integrated into the human genome. BioRxiv 2020. [Google Scholar] [CrossRef]
- Yin, Y.; Liu, X.-Z.; He, X.; Zhou, L.-Q. Exogenous coronavirus interacts with endogenous retrotransposon in human cells. Front. Cell Infect. Microbiol. 2021, 11, 609160. [Google Scholar] [CrossRef] [PubMed]
- Balestrieri, E.; Minutolo, A.; Petrone, V.; Fanelli, M.; Iannetta, M.; Malagnino, V.; Zordan, M.; Vitale, P.; Charvet, B.; Horvat, B.; et al. Evidence of the pathogenic HERV-W envelope expression in T lymphocytes in association with the respiratory outcome of COVID-19 patients. EBioMedicine 2021, 66, 103341. [Google Scholar] [CrossRef] [PubMed]
- Bergallo, M.; Marozio, L.; Botta, G.; Tancredi, A.; Daprà, V.; Galliano, I.; Montanari, P.; Coscia, A.; Benedetto, C.; Tovo, P.A. Human endogenous retroviruses are preferentially expressed in mononuclear cells from cord blood than from maternal blood and in the fetal part of placenta. Front. Pediatr. 2020, 8, 244. [Google Scholar] [CrossRef] [PubMed]
- Hadjadj, J.; Yatim, N.; Barnabei, L.; Corneau, A.; Boussier, J.; Smith, N.; Péré, H.; Charbit, B.; Bondet, V.; Chenevier-Gobeaux, C.; et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 2020, 369, 718–724. [Google Scholar] [CrossRef] [PubMed]
- Guan, W.-J.; Ni, Z.-Y.; Hu, Y.; Liang, W.-H.; Ou, C.-Q.; He, J.-X.; Liu, L.; Shan, H.; Lei, C.-L.; Hui, D.S.C.; et al. Clinical characteristics of coronavirus disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.G.; Lee, K.M.; Hsiao, M.J.; Yang, S.L.; Huang, P.N.; Gong, Y.N. Culture-based virus isolation to evaluate potential infectivity of clinical specimens tested for COVID-19. J. Clin. Microbiol. 2020, 58, e01068-20. [Google Scholar] [CrossRef]
- Laing, A.G.; Lorenc, A.; del Barrio, I.D.; Das, A.; Fish, M.; Monin, L.; Muñoz-Ruiz, M.; McKenzie, D.R.; Hayday, T.S.; Francos-Quijorna, I.; et al. A dynamic COVID-19 immune signature includes associations with poor prognosis. Nat. Med. 2020, 26, 1623–1635. [Google Scholar] [CrossRef]
- Arunachalam, P.S.; Wimmers, F.; Mok, C.K.P.; Perera, R.; Scott, M.; Hagan, T.; Sigal, N.; Feng, Y.; Bristow, L.; Tak, Y.; et al. Systems biological assessment of immunity to mild versus severe COVID-19 infection in humans. Science 2020, 369, 1210–1220. [Google Scholar] [CrossRef] [PubMed]
- Trouillet-Assant, S.; Viel, S.; Gaymard, A. Type I IFN immunoprofiling in COVID-19 patients. J. Allergy Clin. Immunol. 2020, 146, 206–208. [Google Scholar] [CrossRef] [PubMed]
- Scagnolari, C.; Pierangeli, A.; Frasca, F.; Bitossi, C.; Viscido, A.; Oliveto, G.; Scordio, M.; Mazzuti, L.; Di Carlo, D.; Gentile, M.; et al. Differential induction of type I and III interferon genes in the upper respiratory tract of patients with coronavirus disease 2019 (COVID-19). Virus Res. 2021, 295, 198283. [Google Scholar] [CrossRef] [PubMed]
- Lucas, C.; Wong, P.; Klein, J.; Castro, T.B.R.; Silva, J.; Sundaram, M.; Ellingson, M.K.; Mao, T.; Oh, J.E.; Israelow, B.; et al. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature 2020, 584, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Abers, M.S.; Delmonte, O.M.; Ricotta, E.E.; Fintzi, J.; Fink, D.L.; de Jesus, A.A.A.; Zarember, K.A.; Alehashemi, S.; Oikonomou, V.; Desai, J.V.; et al. An immune-based biomarker signature is associated with mortality in COVID-19 patients. JCI Insight. 2021, 6, 144455. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.C. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell 2020, 18, 1036–1045. [Google Scholar] [CrossRef]
- Galani, I.E.; Triantafyllia, V.; Eleminiadou, E.E.; Koltsida, O.; Stavropoulos, A.; Manioudaki, M.; Thanos, D.; Doyle, S.E.; Kotenko, S.V.; Thanopoulou, K.; et al. Interferon-λ mediates non-redundant font-line antiviral protection against influenza virus infection without compromising Host itness. Immunity 2017, 46, 875–890.e6. [Google Scholar] [CrossRef]
- Andreakos, E.; Tsiodras, S. COVID19: Lambda interferon against viral load and hyperinflammation. EMBO Mol. Med. 2020, 12, e12465. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Zhan, Y.; Zhu, L.; Hou, Z.; Liu, F.; Song, P.; Qiu, F.; Wang, X.; Zou, X.; Wan, D.; et al. Retrospective multicenter cohort study shows early interferon therapy is associated with favorable clinical responses in COVID-19 patients. Cell Host Microbe 2020, 28, 455–464.e2. [Google Scholar] [CrossRef]
- Karki, R.; Sharma, B.R.; Tuladhar, S.; Williams, E.P.; Zalduondo, L.; Samir, P.; Zheng, M.; Sundaram, B.; Banoth, B.; Malireddi, R.K.S.; et al. Synergism of TNF-α and IFN-γ triggers inflammatory cell death, tissue damage, and mortality in SARS-CoV-2 infection and cytokine shock syndromes. Cell 2021, 184, 149–168.e17. [Google Scholar] [CrossRef] [PubMed]
- Sims, J.T.; Krishnan, V.; Chang, C.Y.; Engle, S.M.; Casalini, G.; Rodgers, G.H.; Bivi, N.; Nickoloff, B.J.; Konrad, R.J.; de Bono, S.; et al. Characterization of the cytokine storm reflects hyperinflammatory endothelial dysfunction in COVID-19. J. Allergy Clin. Immunol. 2021, 147, 107–111. [Google Scholar] [CrossRef]
- Hu, Z.-J.; Xu, J.; Yin, J.-M.; Li, L.; Hou, W.; Zhang, L.-L.; Zhou, Z.; Yu, Y.Z.; Li, H.-J.; Feng, Y.-M.; et al. Lower circulating interferon-gamma is a risk factor for lung fibrosis in COVID-19 patients. Front. Immunol. 2020, 11, 585647. [Google Scholar] [CrossRef]
- Schäfer, A.; Baric, R.S. Epigenetic landscape during coronavirus infection. Pathogens 2017, 6, 8. [Google Scholar] [CrossRef] [PubMed]
- Di Pietro, A.; Kajaste-Rudnitski, A.; Oteiza, A.; Nicora, L.; Towers, G.J.; Mechti, N.; Vicenzi, E. TRIM22 inhibits influenza A virus infection by targeting the viral nucleoprotein for degradation. J. Virol. 2013, 87, 4523–4533. [Google Scholar] [CrossRef]
- Liu, B.; Li, N.-L.; Shen, Y.; Bao, X.; Fabrizio, T.; Elbahesh, H.; Webby, R.J.; Li, K. The C-terminal tail of TRIM56 dictates antiviral restriction of influenza A and B viruses by impeding viral RNA synthesis. J. Virol. 2016, 90, 4369–4382. [Google Scholar] [CrossRef]
- Van Tol, S.; Hage, A.; Giraldo, M.I.; Bharaj, P.; Rajsbaum, R. The TRIMendous role of TRIMs in virus-host interactions. Vaccines 2017, 5, 23. [Google Scholar] [CrossRef]
- Ivanov, A.V.; Peng, H.; Yurchenko, V.; Yap, K.L.; Negorev, D.G.; Schultz, D.C.; Psulkowski, E.; Fredericks, W.J.; White, D.E.; Maul, G.G.; et al. PHD domain-mediated E3 ligase activity directs intramolecular sumoylation of an adjacent bromodomain required for gene silencing. Mol. Cell 2007, 28, 823–837. [Google Scholar] [CrossRef]
- Liang, Q.; Deng, H.; Li, X.; Wu, X.; Tang, Q.; Chang, T.H.; Peng, H.; Rauscher, F.J.; Ozato, K.; Zhu, F. Tripartite motif-containing protein 28 is a small ubiquitin-related modifier E3 ligase and negative regulator of IFN regulatory factor 7. J. Immunol. 2011, 187, 4754–4763. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zheng, H.; Zhu, J.; Dong, Q.; Wang, J.; Fan, H.; Chen, Y.; Zhang, X.; Han, X.; Li, Q.; et al. Ubiquitin-modified proteome of SARS-CoV-2-infected host cells reveals insights into virus—Host interaction and pathogenesis. J. Proteome Res. 2021, 20, 2224–2239. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Zhang, J.; Wang, C.; Jain, P.P.; Xiong, M.; Shi, X.; Lei, Y.; Chen, S.; Yin, Q.; Thistlethwaite, P.A.; et al. MDM2-Mediated ubiquitination of angiotensin-converting enzyme 2 contributes to the development of pulmonary arterial hypertension. Circulation 2020, 142, 1190–1204. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhao, Q.; Zhang, H.; Liang, F.; Zhang, C.; Wang, J.; Chen, Z.; Wu, R.; Yu, H.; Sun, B.; et al. Degradation of SARS-CoV-2 receptor ACE2 by the E3 ubiquitin ligase Skp2 in lung epithelial cells. Front. Med. 2021, 15, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Cuellar, T.L.; Herzner, A.M.; Zhang, X.; Goyal, Y.; Watanabe, C.; Friedman, B.A.; Janakiraman, V.; Durinck, S.; Stinson, J.; Arnott, D. Silencing of retrotransposons by SETDB1 inhibits the interferon response in acute myeloid leukemia. J. Cell. Biol. 2017, 216, 3535–3549. [Google Scholar] [CrossRef]
- Zhou, X.-F.; Yu, J.; Chang, M.; Zhang, M.; Zhou, D.; Cammas, F.; Sun, S.C. TRIM28 mediates chromatin modifications at the TCRα enhancer and regulates the development of T and natural killer T cells. Proc. Natl. Acad. Sci. USA 2012, 109, 20083–20088. [Google Scholar] [CrossRef] [PubMed]
- Chikuma, S.; Yamanaka, S.; Nakagawa, S.; Ueda, M.T.; Hayabuchi, H.; Tokifuji, Y.; Kanayama, M.; Okamura, T.; Arase, H.; Yoshimura, A. TRIM28 expression on dendritic cells prevents excessive T cell priming by silencing endogenous retrovirus. J. Immunol. 2021, ji2001003. [Google Scholar]
- Ehrenfeld, M.; Tincani, A.; Andreoli, M.; Greenbaum, A.; Kanduc, D.; Alijotas-Reig, J.; Zinserling, V.; Semenova, N.; Amital, H.; Shoenfeld, Y. Covid-19 and autoimmunity. Autoimmun. Rev. 2020, 19, 102597. [Google Scholar] [CrossRef] [PubMed]
- Klimczak, M.; Czerwińska, P.; Mazurek, S.; Sozańska, B.; Biecek, P.; Mackiewicz, A.; Wiznerowicz, M. TRIM28 epigenetic corepressor is indispensable for stable induced pluripotent stem cell formation. Stem Cell Res. 2017, 23, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Debuc, B.; Smadja, D.M. Is COVID-19 a new hematologic disease? Stem Cell Rev. Rep. 2021, 17, 4–8. [Google Scholar] [CrossRef] [PubMed]
- Adoue, V.; Binet, B.; Malbec, A.; Fourquet, J.; Romagnoli, P.; van Meerwijk, J.P.M.; Amigorena, S.; Joffre, O.P. The histone methyltransferase SETDB1 controls T helper cell lineage integrity by repressing endogenous retroviruses. Immunity 2019, 50, 629–644.e8. [Google Scholar] [CrossRef]
- Južnić, L.; Peuker, K.; Strigli, A.; Brosch, M.; Herrmann, A.; Häsler, R.; Koch, M.; Matthiesen, L.; Zeissig, Y.; Löscher, B.S.; et al. SETDB1 is required for intestinal epithelial differentiation and the prevention of intestinal inflammation. Gut 2021, 70, 485–498. [Google Scholar] [CrossRef]
- Wang, R.; Li, H.; Wu, J.; Cai, Z.-Y.; Li, B.; Ni, H.; Qiu, X.; Chen, H.; Liu, W.; Yang, Z.H.; et al. Gut stem cell necroptosis by genome instability triggers bowel inflammation. Nature 2020, 580, 386–390. [Google Scholar] [CrossRef]
- Carthagena, L.; Bergamaschi, A.; Luna, J.M.; David, A.; Uchil, P.D.; Margottin-Goguet, F.; Mothes, W.; Hazan, U.; Transy, C.; Pancino, G.; et al. Human TRIM gene expression in response to interferons. PLoS ONE 2009, 4, e4894. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wang, L.; Liu, H.; Chen, J.; Liu, D. Transcriptome analyses implicate endogenous retroviruses involved in the host antiviral immune system through the interferon pathway. Virol. Sin. 2021, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Cañadas, I.; Thummalapalli, R.; Kim, J.W.; Kitajima, S.; Jenkins, R.W.; Christensen, C.L.; Campisi, M.; Kuang, Y.; Zhang, Y.; Gjini, E.; et al. Tumor innate immunity primed by specific interferon-stimulated endogenous retroviruses. Nat. Med. 2018, 24, 1143–1150. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Sheehan, M.M.; Reddy, A.J.; Rothberg, M.B. Reinfection rates among patients who previously tested positive for COVID-19: A retrospective cohort study. Clin Infect Dis. 2021. [Google Scholar] [CrossRef]
- Tolosa, J.M.; Parsons, K.S.; Hansbro, P.M.; Smith, R.; Wark, P.A. The placental protein syncytin-1 impairs antiviral responses and exaggerates inflammatory responses to influenza. PLoS ONE 2015, 10, e0118629. [Google Scholar]
- Holder, B.S.; Tower, C.L.; Forbes, K.; Mulla, M.J.; Aplin, J.D.; Abrahams, V.M. Immune cell activation by trophoblast-derived microvesicles is mediated by syncytin 1. Immunology 2012, 136, 184–191. [Google Scholar] [CrossRef]
- Wang, X.; Liu, Z.; Wang, P.; Li, S.; Zeng, J.; Tu, X.; Yan, Q.; Xiao, Z.; Pan, M.; Zhu, F. Syncytin-1, an endogenous retroviral protein, triggers the activation of CRP via TLR3 signal cascade in glial cells. Brain Behav. Immun. 2018, 67, 324–334. [Google Scholar] [CrossRef]
- Bodnar, B.; Patel, K.; Ho, W.; Luo, J.J.; Hu, W. Cellular mechanisms underlying neurological/neuropsychiatric manifestations of COVID-19. J. Med. Virol. 2021, 93, 1983–1998. [Google Scholar] [CrossRef]
- Shimba, A.; Ikuta, K. Control of immunity by glucocorticoids in health and disease. Semin. Immunopathol. 2020, 42, 669–680. [Google Scholar] [CrossRef]
- Finney, L.J.; Glanville, N.; Farne, H.; Aniscenko, J.; Fenwick, P.; Kemp, S.V.; Trujillo-Torralbo, M.B.; Loo, S.L.; Calderazzo, M.A.; Wedzicha, J.A.; et al. Inhaled corticosteroids downregulate the SARS-CoV-2 receptor ACE2 in COPD through suppression of type I interferon. J. Allergy Clin. Immunol. 2021, 147, 510–519.e5. [Google Scholar] [CrossRef]
- Fiegl, M.; Strasser-Wozak, E.; Geley, S.; Gsur, A.; Drach, J.; Kofler, R. Glucocorticoid-mediated immunomodulation: Hydrocortisone enhances immunosuppressive endogenous retroviral protein (p15E) expression in mouse immune cells. Clin. Exp. Immunol. 1995, 101, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Hsu, K.; Lee, Y.K.; Chew, A.; Chiu, S.; Lim, D.; Greenhalgh, D.G.; Cho, K. Inherently variable responses to glucocorticoid stress among endogenous retroviruses isolated from 23 mouse strains. Biochim. Biophys. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 2594–2600. [Google Scholar] [CrossRef] [PubMed]
- Moskophidis, D.; Lechner, F.; Pircher, H.; Zinkernagel, R.M. Virus persistence in acutely infected immunocompetent mice by exhaustion of antiviral cytotoxic effector T cells. Nature 1993, 362, 758–761. [Google Scholar] [CrossRef]
- Sen, D.R.; Kaminski, J.; Barnitz, R.A.; Kurachi, M.; Gerdemann, U.; Yates, K.B.; Tsao, H.W.; Godec, J.; LaFleur, M.W.; Brown, F.D.; et al. The epigenetic landscape of T cell exhaustion. Science 2016, 354, 1165–1169. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.C.; Sen, D.R.; Al Abosy, R.; Bi, K.; Virkud, Y.V.; LaFleur, M.W.; Yates, K.B.; Lako, A.; Felt, K.; Naik, G.S.; et al. Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint. Nat. Immunol. 2019, 20, 326–336. [Google Scholar] [CrossRef]
- Chen, S.; Bonifati, S.; Qin, Z.; St Gelais, C.; Kodigepalli, K.M.; Barrett, B.S.; Kim, S.H.; Antonucci, J.M.; Ladner, K.J.; Buzovetsky, O.; et al. SAMHD1 suppresses innate immune responses to viral infections and inflammatory stimuli by inhibiting the NF-κB and interferon pathways. Proc. Natl. Acad. Sci. USA 2018, 115, E3798–E3807. [Google Scholar] [CrossRef]
- Wilmes, S.; Beutel, O.; Li, Z.; Francois-Newton, V.; Richter, C.P.; Janning, D.; Kroll, C.; Hanhart, P.; Hötte, K.; You, C.; et al. Receptor dimerization dynamics as a regulatory valve for plasticity of type I interferon signaling. J. Cell Biol. 2015, 209, 579–593. [Google Scholar] [CrossRef]
- Kamada, R.; Yang, W.; Zhang, Y.; Patel, M.C.; Yang, Y.; Ouda, R.; Dey, A.; Wakabayashi, Y.; Sakaguchi, K.; Fujita, T.; et al. Interferon stimulation creates chromatin marks and establishes transcriptional memory. Proc. Natl. Acad. Sci. USA 2018, 115, E9162–E9171. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Onai, N.; Yoshihara, H.; Arai, F.; Suda, T.; Ohteki, T. Interferon regulatory factor-2 protects quiescent hematopoietic stem cells from type I interferon-dependent exhaustion. Nat. Med. 2009, 15, 696–700. [Google Scholar] [CrossRef]
- De Biasi, S.; Meschiari, M.; Gibellini, L.; Bellinazzi, C.; Borella, R.; Fidanza, L.; Gozzi, L.; Iannone, A.; Lo Tartaro, D.; Mattioli, M.; et al. Marked T cell activation, senescence, exhaustion and skewing towards TH17 in patients with COVID-19 pneumonia. Nat. Commun. 2020, 11, 3434. [Google Scholar] [CrossRef]
- Mukhopadhyay, D.; Dasso, M. Modification in reverse: The SUMO proteases. Trends Biochem. Sci. 2007, 32, 286–295. [Google Scholar] [CrossRef]
- Zheng, Q.; Cao, Y.; Chen, Y.; Wang, J.; Fan, Q.; Huang, X.; Wang, Y.; Wang, T.; Wang, X.; Ma, J.; et al. Senp2 regulates adipose lipid storage by de-SUMOylation of Setdb1. J. Mol. Cell Biol. 2018, 10, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Nalbandian, A.; Sehgal, K.; Gupta, A.; Madhavan, M.V.; McGroder, C.; Stevens, J.S.; Cook, J.R.; Nordvig, A.S.; Shalev, D.; Sehrawat, T.S.; et al. Post-acute COVID-19 syndrome. Nat. Med. 2021, 27, 6010–6015. [Google Scholar] [CrossRef] [PubMed]
- Al-Aly, Z.; Xie, Y.; Bowe, B. High-dimensional characterization of post-acute sequalae of COVID-19. Nature 2021, 27, 601–615. [Google Scholar] [CrossRef]
- Dong, Y.; Mo, X.; Hu, Y.; Qi, X.; Jiang, F.; Jiang, Z.; Tong, S. Epidemiology of COVID-19 among children in China. Pediatrics 2020, 145, e20200702. [Google Scholar] [CrossRef]
- Pokorska-Śpiewak, M.; Talarek, E.; Popielska, J.; Nowicka, K.; Ołdakowska, A.; Zawadka, K.; Kowalik-Mikołajewska, B.; Tomasik, A.; Dobrzeniecka, A.; Lipińska, M.; et al. Comparison of clinical severity and epidemiological spectrum between coronavirus disease 2019 and influenza in children. Sci. Rep. 2021, 11, 5760. [Google Scholar] [CrossRef]
- Bergallo, M.; Ferrari, L.; Faolotto, G.; Balbo, P.E.; Montanari, P.; Patrucco, F.; Gavelli, F.; Daverio, M.; Bellan, M.; Salmi, L.; et al. Interferon signature in immunosuppressed patients with lower respiratory tract infections: Dosage on bronchoalveolar lavage. Minerva Med. 2020, 111, 245–253. [Google Scholar] [CrossRef]
- Fang, L.; Choudhary, S.; Tian, B.; Boldogh, I.; Yang, C.; Ivanciuc, T.; Ma, Y.; Garofalo, R.P.; Brasier, A.R. Ataxia telangiectasia mutated kinase mediates NF-κB serine 276 phosphorylation and interferon expression via the IRF7-RIG-I amplification loop in paramyxovirus infection. J. Virol. 2015, 89, 2628–2642. [Google Scholar] [CrossRef][Green Version]
- Schanab, O.; Humer, J.; Gleiss, A.; Mikula, M.; Sturlan, S.; Grunt, S.; Okamoto, I.; Muster, T.; Pehamberger, H.; Waltenberger, A. Expression of human endogenous retrovirus K is stimulated by ultraviolet radiation in melanoma. Pigment. Cell Melanoma Res. 2011, 24, 656–665. [Google Scholar] [CrossRef]
- Mameli, G.; Poddighe, L.; Astone, V.; Delogu, G.; Arru, G.; Sotgiu, S.; Serra, C.; Dolei, A. Novel reliable real-time PCR for differential detection of MSRVenv and syncytin-1 in RNA and DNA from patients with multiple sclerosis. J. Virol. Methods 2009, 161, 98–106. [Google Scholar] [CrossRef]
- NIH. NIH Guidelines for Research Involving Recombinant or Synthetic Nucleic Acid Molecules. 2019. Available online: https://osp.od.nih.gov/wp-content/uploads/2019_NIH_Guidelines.htm (accessed on 25 April 2019).
- WHO. Laboratory Biosafety Guidance Related to Coronavirus Disease (COVID-19): Interim Guidance. 2020. Available online: https://www.who.int/publications/i/item/laboratory-biosafety-guidance-related-to-coronavirus-disease-(covid-19) (accessed on 13 May 2020).












{kind=link}
{kind=link}
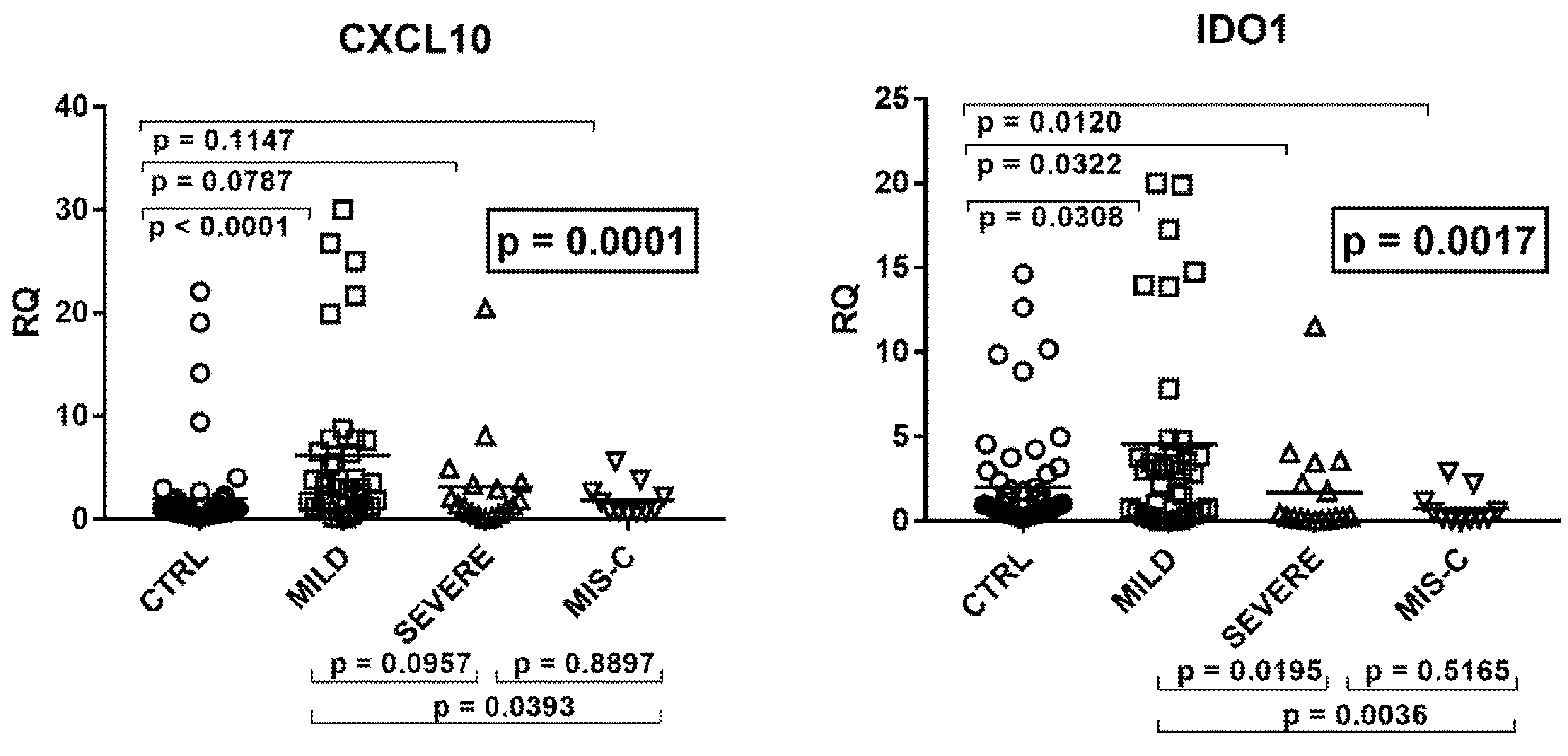{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total (n = 64) | Group A (n = 36) | Group B (n = 17) | Group C (n = 11) | |
|---|---|---|---|---|
| Median age (IQR) | 7.8 yrs (2.3; 12.5) | 8.0 yrs (1.3; 11.7) | 10.1 yrs (3.8; 13.8) | 6.8 yrs (4.7; 8.7) |
| Males (%) | 37 (58.7) | 25 (69.4) | 7 (41.2) | 5 (45.5) |
| Comorbidities, n (%) | 27 (42.9) | 16 (44.4) | 10 (58.8) | 1 (9.9) |
| Mean interval (+SD) from symptom onset and sampling | 6.2 days (5.5) | 5.8 days (5.6) | 5.9 days (2.9) | 7.6 days (3.3) |
| CRP > 10 mg/L, n (%) | 18 (28.6) | 3 (8.3) | 7 (41.2) | 8 (72.7) |
| Leucocytosis, n (%) * | 6 (9.5) | 3 (8.3) | 1 (5.9) | 2 (18.2) |
| Lymphopenia, n (%) ** | 16 (25.4) | 4 (11.1) | 7 (41.2) | 5 (45.5) |
| Steroid treatment, n (%) | 23 (35.9) | 1 (2.8) | 11 (64.7) | 11 (100) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tovo, P.-A.; Garazzino, S.; Daprà, V.; Pruccoli, G.; Calvi, C.; Mignone, F.; Alliaudi, C.; Denina, M.; Scolfaro, C.; Zoppo, M.; et al. COVID-19 in Children: Expressions of Type I/II/III Interferons, TRIM28, SETDB1, and Endogenous Retroviruses in Mild and Severe Cases. Int. J. Mol. Sci. 2021, 22, 7481. https://doi.org/10.3390/ijms22147481
Tovo P-A, Garazzino S, Daprà V, Pruccoli G, Calvi C, Mignone F, Alliaudi C, Denina M, Scolfaro C, Zoppo M, et al. COVID-19 in Children: Expressions of Type I/II/III Interferons, TRIM28, SETDB1, and Endogenous Retroviruses in Mild and Severe Cases. International Journal of Molecular Sciences. 2021; 22(14):7481. https://doi.org/10.3390/ijms22147481
Chicago/Turabian StyleTovo, Pier-Angelo, Silvia Garazzino, Valentina Daprà, Giulia Pruccoli, Cristina Calvi, Federica Mignone, Carla Alliaudi, Marco Denina, Carlo Scolfaro, Marisa Zoppo, and et al. 2021. "COVID-19 in Children: Expressions of Type I/II/III Interferons, TRIM28, SETDB1, and Endogenous Retroviruses in Mild and Severe Cases" International Journal of Molecular Sciences 22, no. 14: 7481. https://doi.org/10.3390/ijms22147481
APA StyleTovo, P.-A., Garazzino, S., Daprà, V., Pruccoli, G., Calvi, C., Mignone, F., Alliaudi, C., Denina, M., Scolfaro, C., Zoppo, M., Licciardi, F., Ramenghi, U., Galliano, I., & Bergallo, M. (2021). COVID-19 in Children: Expressions of Type I/II/III Interferons, TRIM28, SETDB1, and Endogenous Retroviruses in Mild and Severe Cases. International Journal of Molecular Sciences, 22(14), 7481. https://doi.org/10.3390/ijms22147481