
Ion Channels as New Attractive Targets to Improve Re-Myelination Processes in the Brain




Abstract
1. Introduction
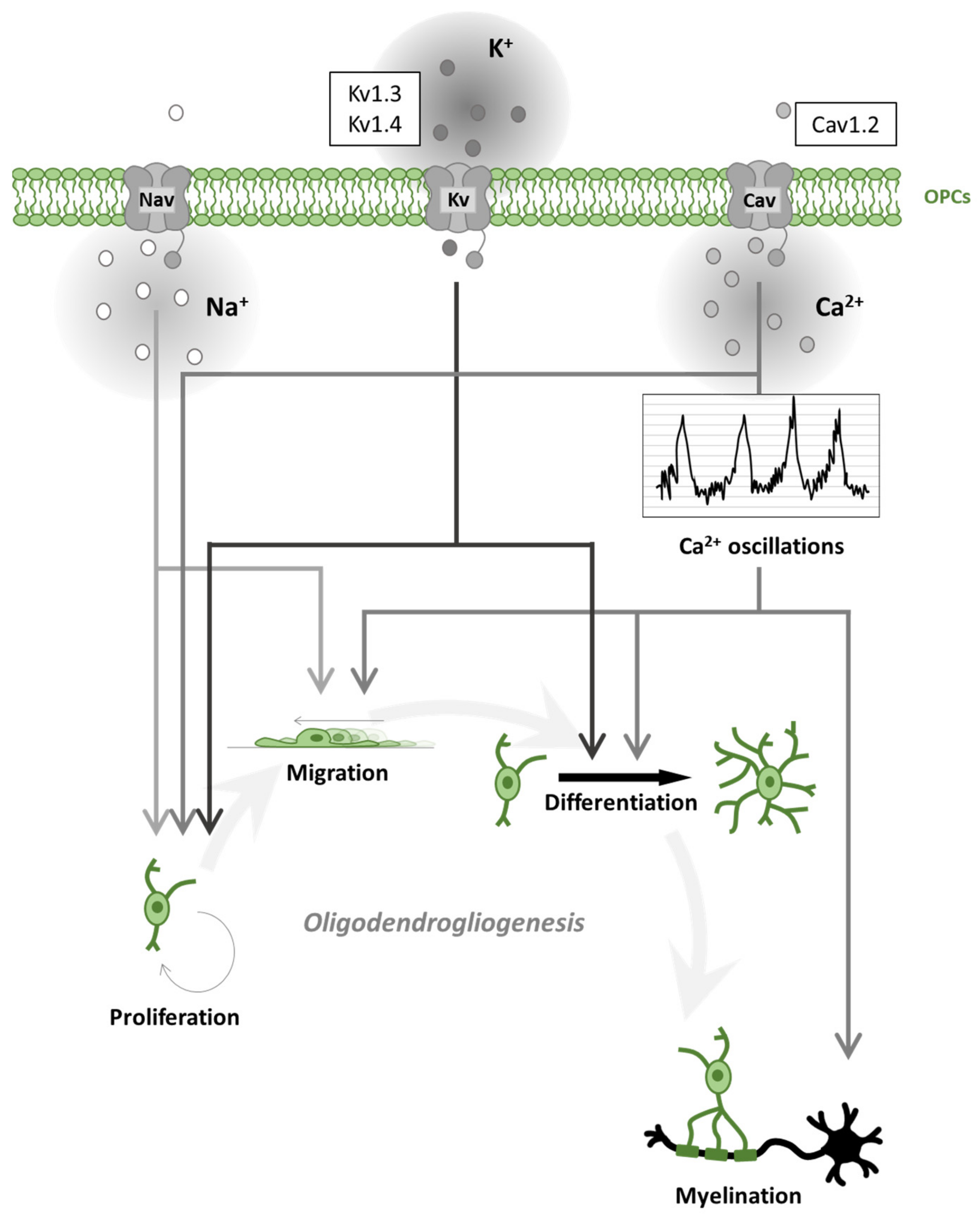
2. Oligodendrogliogenesis
3. Voltage-Gated Channels in Oligodendroglial Cells and Myelination
3.1. Voltage-Gated Na+ Channels in Oligodendroglial Cells
3.2. Voltage-Gated Ca2+ Channels in Oligodendroglial Cells
Cav Channels in Demyelinating Diseases
3.3. Voltage-Gated K+ Channels in Oligodendroglial Cells
Voltage-Gated K+ Channels in Demyelinating Diseases
4. Neurotransmitters in Oligodendroglial Cells and Myelination
4.1. Glutamate
4.2. GABA
4.3. Purines
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cohen, C.C.H.; Popovic, M.A.; Klooster, J.; Weil, M.T.; Möbius, W.; Nave, K.A.; Kole, M.H.P. Saltatory Conduction along Myelinated Axons Involves a Periaxonal Nanocircuit. Cell 2020, 180, 311–322.e15. [Google Scholar] [CrossRef] [PubMed]
- Rosko, L.; Smith, V.N.; Yamazaki, R.; Huang, J.K. Oligodendrocyte Bioenergetics in Health and Disease. Neuroscientist 2019, 25, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Kessaris, N.; Fogarty, M.; Iannarelli, P.; Grist, M.; Wegner, M.; Richardson, W.D. Competing waves of oligodendrocytes in the forebrain and postnatal elimination of an embryonic lineage. Nat. Neurosci. 2006, 9, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Baydyuk, M.; Morrison, V.E.; Gross, P.S.; Huang, J.K. Extrinsic Factors Driving Oligodendrocyte Lineage Cell Progression in CNS Development and Injury. Neurochem. Res. 2020, 45, 630–642. [Google Scholar] [CrossRef] [PubMed]
- Bergles, D.E.; Richardson, W.D. Oligodendrocyte development and plasticity. Cold Spring Harb. Perspect. Biol. 2016, 8, a020453. [Google Scholar] [CrossRef] [PubMed]
- Emery, B. Regulation of oligodendrocyte differentiation and myelination. Science 2010, 330, 779–782. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.R.L.; Polito, A.; Levine, J.M.; Reynolds, R. NG2-expressing glial progenitor cells: An abundant and widespread population of cycling cells in the adult rat CNS. Mol. Cell. Neurosci. 2003, 24, 476–488. [Google Scholar] [CrossRef]
- Kuhn, S.; Gritti, L.; Crooks, D.; Dombrowski, Y. Oligodendrocytes in Development, Myelin Generation and Beyond. Cells 2019, 8, 1424. [Google Scholar] [CrossRef]
- Takase, H.; Regenhardt, R. Motor tract reorganization after acute central nervous system injury: A translational perspective. Neural Regen. Res. 2021, 16, 1144. [Google Scholar] [CrossRef] [PubMed]
- Philips, T.; Rothstein, J.D. Oligodendroglia: Metabolic supporters of neurons. J. Clin. Investig. 2017, 127, 3271–3280. [Google Scholar] [CrossRef] [PubMed]
- Meyer, N.; Richter, N.; Fan, Z.; Siemonsmeier, G.; Pivneva, T.; Jordan, P.; Steinhäuser, C.; Semtner, M.; Nolte, C.; Kettenmann, H. Oligodendrocytes in the Mouse Corpus Callosum Maintain Axonal Function by Delivery of Glucose. Cell Rep. 2018, 22, 2383–2394. [Google Scholar] [CrossRef]
- Takase, H.; Washida, K.; Hayakawa, K.; Arai, K.; Wang, X.; Lo, E.H.; Lok, J. Oligodendrogenesis after traumatic brain injury. Behav. Brain Res. 2018, 340, 205–211. [Google Scholar] [CrossRef]
- Ohtomo, R.; Kinoshita, K.; Ohtomo, G.; Takase, H.; Hamanaka, G.; Washida, K.; Islam, M.R.; Wrann, C.D.; Katsuki, H.; Iwata, A.; et al. Treadmill Exercise Suppresses Cognitive Decline and Increases White Matter Oligodendrocyte Precursor Cells in a Mouse Model of Prolonged Cerebral Hypoperfusion. Transl. Stroke Res. 2020, 11, 496–502. [Google Scholar] [CrossRef]
- Fok-Seang, J.; Miller, R.H. Distribution and differentiation of A2B5+ glial precursors in the developing rat spinal cord. J. Neurosci. Res. 1994, 37, 219–235. [Google Scholar] [CrossRef]
- Klum, S.; Zaouter, C.; Alekseenko, Z.; Björklund, Å.K.; Hagey, D.W.; Ericson, J.; Muhr, J.; Bergsland, M. Sequentially acting SOX proteins orchestrate astrocyte- and oligodendrocyte-specific gene expression. EMBO Rep. 2018, 19, e46635. [Google Scholar] [CrossRef]
- Li, P.; Li, H.X.; Jiang, H.Y.; Zhu, L.; Wu, H.Y.; Li, J.T.; Lai, J.H. Expression of NG2 and platelet-derived growth factor receptor alpha in the developing neonatal rat brain. Neural Regen. Res. 2017, 12, 1843–1852. [Google Scholar] [CrossRef]
- Nishiyama, A.; Boshans, L.; Goncalves, C.M.; Wegrzyn, J.; Patel, K.D. Lineage, fate, and fate potential of NG2-glia. Brain Res. 2016, 1638, 116–128. [Google Scholar] [CrossRef]
- Eugenín-von Bernhardi, J.; Dimou, L. NG2-glia, more than progenitor cells. Adv. Exp. Med. Biol. 2016, 949, 27–45. [Google Scholar] [CrossRef]
- Calver, A.R.; Hall, A.C.; Yu, W.P.; Walsh, F.S.; Heath, J.K.; Betsholtz, C.; Richardson, W.D. Oligodendrocyte population dynamics and the role of PDGF in vivo. Neuron 1998, 20, 869–882. [Google Scholar] [CrossRef]
- Braun, P.E.; Sandillon, F.; Edwards, A.; Matthieu, J.M.; Privat, A. Immunocytochemical localization by electron microscopy of 2’,3’-cyclic nucleotide 3’-phosphodiesterase in developing oligodendrocytes of normal and mutant brain. J. Neurosci. 1988, 8, 3057–3066. [Google Scholar] [CrossRef]
- Jakovcevski, I.; Filipovic, R.; Mo, Z.; Rakic, S.; Zecevic, N. Oligodendrocyte development and the onset of myelination in the human fetal brain. Front. Neuroanat. 2009, 3, 5. [Google Scholar] [CrossRef]
- Brunner, C.; Lassmann, H.; Waehneldt, T.V.; Matthieu, J.-M.; Linington, C. Differential Ultrastructural Localization of Myelin Basic Protein, Myelin/Oligodendroglial Glycoprotein, and 2′,3′-Cyclic Nucleotide 3′-Phosphodiesterase in the CNS of Adult Rats. J. Neurochem. 1989, 52, 296–304. [Google Scholar] [CrossRef]
- Barbarese, E.; Barry, C.; Chou, C.J.; Goldstein, D.J.; Nakos, G.A.; Hyde-DeRuyscher, R.; Scheld, K.; Carson, J.H. Expression and Localization of Myelin Basic Protein in Oligodendrocytes and Transfected Fibroblasts. J. Neurochem. 1988, 51, 1737–1745. [Google Scholar] [CrossRef]
- Michalski, J.-P.; Anderson, C.; Beauvais, A.; De Repentigny, Y.; Kothary, R. The Proteolipid Protein Promoter Drives Expression outside of the Oligodendrocyte Lineage during Embryonic and Early Postnatal Development. PLoS ONE 2011, 6, e19772. [Google Scholar] [CrossRef]
- Trapp, B.D. Myelin-Associated Glycoprotein Location and Potential Functions. Ann. N. Y. Acad. Sci. 1990, 605, 29–43. [Google Scholar] [CrossRef]
- Sun, W.; Matthews, E.A.; Nicolas, V.; Schoch, S.; Dietrich, D. Ng2 glial cells integrate synaptic input in global and dendritic calcium signals. eLife 2016, 5, e16262. [Google Scholar] [CrossRef]
- Krasnow, A.M.; Attwell, D. NMDA Receptors: Power Switches for Oligodendrocytes. Neuron 2016, 91, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Bergles, D.E.; Roberts, J.D.B.; Somogyl, P.; Jahr, C.E. Glutamatergic synapses on oligodendrocyte precursor cells in the hippocampus. Nature 2000, 405, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Gallo, V.; Mangin, J.-M.; Kukley, M.; Dietrich, D. Synapses on NG2-expressing progenitors in the brain: Multiple functions? J. Physiol. 2008, 586, 3767–3781. [Google Scholar] [CrossRef] [PubMed]
- Boulanger, J.J.; Messier, C. Oligodendrocyte progenitor cells are paired with GABA neurons in the mouse dorsal cortex: Unbiased stereological analysis. Neuroscience 2017, 362, 127–140. [Google Scholar] [CrossRef] [PubMed]
- Benamer, N.; Vidal, M.; Angulo, M.C. The cerebral cortex is a substrate of multiple interactions between GABAergic interneurons and oligodendrocyte lineage cells. Neurosci. Lett. 2020, 715, 134615. [Google Scholar] [CrossRef]
- Xiao, L.; Ohayon, D.; Mckenzie, I.A.; Sinclair-Wilson, A.; Wright, J.L.; Fudge, A.D.; Emery, B.; Li, H.; Richardson, W.D. Rapid production of new oligodendrocytes is required in the earliest stages of motor-skill learning. Nat. Neurosci. 2016, 19, 1210–1217. [Google Scholar] [CrossRef]
- Bacmeister, C.M.; Barr, H.J.; McClain, C.R.; Thornton, M.A.; Nettles, D.; Welle, C.G.; Hughes, E.G. Motor learning promotes remyelination via new and surviving oligodendrocytes. bioRxiv 2020. [Google Scholar] [CrossRef]
- Steadman, P.E.; Xia, F.; Ahmed, M.; Mocle, A.J.; Penning, A.R.; Geraghty, A.C.; Steenland, H.W.; Monje, M.; Josselyn, S.A.; Frankland, P.W. Disruption of Oligodendrogenesis Impairs Memory Consolidation in Adult Mice. Neuron 2020, 105, 150–164. [Google Scholar] [CrossRef]
- Makinodan, M.; Ikawa, D.; Miyamoto, Y.; Yamauchi, J.; Yamamuro, K.; Yamashita, Y.; Toritsuka, M.; Kimoto, S.; Okumura, K.; Yamauchi, T.; et al. Social isolation impairs remyelination in mice through modulation of IL-6. FASEB J. 2016, 30, 4267–4274. [Google Scholar] [CrossRef]
- Shimizu, T.; Ishida, A.; Hagiwara, M.; Ueda, Y.; Hattori, A.; Tajiri, N.; Hida, H. Social Defeat Stress in Adolescent Mice Induces Depressive-like Behaviors with Reduced Oligodendrogenesis. Neuroscience 2020, 443, 218–232. [Google Scholar] [CrossRef]
- Forbes, T.A.; Gallo, V. All Wrapped Up: Environmental Effects on Myelination. Trends Neurosci. 2017, 40, 572–587. [Google Scholar] [CrossRef]
- Suminaite, D.; Lyons, D.A.; Livesey, M.R. Myelinated axon physiology and regulation of neural circuit function. Glia 2019, 67, 2050–2062. [Google Scholar] [CrossRef]
- Schmidt, H.; Knösche, T.R. Action potential propagation and synchronisation in myelinated axons. PLoS Comput. Biol. 2019, 15, e1007004. [Google Scholar] [CrossRef]
- Salami, M.; Itami, C.; Tsumoto, T.; Kimura, F. Change of conduction velocity by regional myelination yields constant latency irrespective of distance between thalamus and cortex. Proc. Natl. Acad. Sci. USA 2003, 100, 6174–6179. [Google Scholar] [CrossRef]
- Long, P.; Wan, G.; Roberts, M.T.; Corfas, G. Myelin development, plasticity, and pathology in the auditory system. Dev. Neurobiol. 2018, 78, 80–92. [Google Scholar] [CrossRef]
- Bengtsson, S.L.; Nagy, Z.; Skare, S.; Forsman, L.; Forssberg, H.; Ullén, F. Extensive piano practicing has regionally specific effects on white matter development. Nat. Neurosci. 2005, 8, 1148–1150. [Google Scholar] [CrossRef]
- Scholz, J.; Klein, M.C.; Behrens, T.E.J.; Johansen-Berg, H. Training induces changes in white-matter architecture. Nat. Neurosci. 2009, 12, 1370–1371. [Google Scholar] [CrossRef]
- Ontaneda, D.; Thompson, A.J.; Fox, R.J.; Cohen, J.A. Progressive multiple sclerosis: Prospects for disease therapy, repair, and restoration of function. Lancet 2017, 389, 1357–1366. [Google Scholar] [CrossRef]
- Di Filippo, M.; Portaccio, E.; Mancini, A.; Calabresi, P. Multiple sclerosis and cognition: Synaptic failure and network dysfunction. Nat. Rev. Neurosci. 2018, 19, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Correale, J.; Marrodan, M.; Benarroch, E.E. What is the role of axonal ion channels in multiple sclerosis? Neurology 2020, 95, 120–123. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.; Ursu, O.; Gaulton, A.; Patrícia Bento, A.; Donadi, R.S.; Bologa, C.G.; Karlsson, A.; Al-Lazikani, B.; Hersey, A.; Oprea, T.I.; et al. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 2017, 16, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Duncan, G.J.; Simkins, T.J.; Emery, B. Neuron-Oligodendrocyte Interactions in the Structure and Integrity of Axons. Front. Cell Dev. Biol. 2021, 9, 460. [Google Scholar] [CrossRef]
- Imming, P.; Sinning, C.; Meyer, A. Drugs, their targets and the nature and number of drug targets. Nat. Rev. Drug Discov. 2006, 5, 821–834. [Google Scholar] [CrossRef]
- Schattling, B.; Eggert, B.; Friese, M.A. Acquired channelopathies as contributors to development and progression of multiple sclerosis. Exp. Neurol. 2014, 262, 28–36. [Google Scholar] [CrossRef]
- Bittner, S.; Meuth, S.G.; Göbel, K.; Melzer, N.; Herrmann, A.M.; Simon, O.J.; Weishaupt, A.; Budde, T.; Bayliss, D.A.; Bendszus, M.; et al. TASK1 modulates inflammation and neurodegeneration in autoimmune inflammation of the central nervous system. Brain 2009, 132, 2501–2516. [Google Scholar] [CrossRef]
- Criscuolo, C.; Cianflone, A.; Lanzillo, R.; Carrella, D.; Carissimo, A.; Napolitano, F.; de Cegli, R.; de Candia, P.; La Rocca, C.; Petrozziello, T.; et al. Glatiramer Acetate modulates ion channels expression and calcium homeostasis in B cell of patients with relapsing-remitting multiple sclerosis. Sci. Rep. 2019, 9, 1–8. [Google Scholar] [CrossRef]
- Shields, S.D.; Butt, R.P.; Dib-Hajj, S.D.; Waxman, S.G. Oral administration of PF-01247324, a subtype-selective Nav1.8 blocker, reverses cerebellar deficits in a mouse model of multiple sclerosis. PLoS ONE 2015, 10, e0119067. [Google Scholar] [CrossRef]
- Morsali, D.; Bechtold, D.; Lee, W.; Chauhdry, S.; Palchaudhuri, U.; Hassoon, P.; Snell, D.M.; Malpass, K.; Piers, T.; Pocock, J.; et al. Safinamide and flecainide protect axons and reduce microglial activation in models of multiple sclerosis. Brain 2013, 136, 1067–1082. [Google Scholar] [CrossRef] [PubMed]
- Wasan, H.; Singh, D.; KH, R. Safinamide in neurological disorders and beyond: Evidence from preclinical and clinical studies. Brain Res. Bull. 2021, 168, 165–177. [Google Scholar] [CrossRef]
- Bechtold, D.A.; Kapoor, R.; Smith, K.J. Axonal Protection Using Flecainide in Experimental Autoimmune Encephalomyelitis. Ann. Neurol. 2004, 55, 607–616. [Google Scholar] [CrossRef]
- Kapoor, R. Sodium channel blockers and neuroprotection in multiple sclerosis using lamotrigine. J. Neurol. Sci. 2008, 274, 54–56. [Google Scholar] [CrossRef]
- Faissner, S.; Plemel, J.R.; Gold, R.; Yong, V.W. Progressive multiple sclerosis: From pathophysiology to therapeutic strategies. Nat. Rev. Drug Discov. 2019, 18, 905–922. [Google Scholar] [CrossRef]
- Raftopoulos, R.; Hickman, S.J.; Toosy, A.; Sharrack, B.; Mallik, S.; Paling, D.; Altmann, D.R.; Yiannakas, M.C.; Malladi, P.; Sheridan, R.; et al. Phenytoin for neuroprotection in patients with acute optic neuritis: A randomised, placebo-controlled, phase 2 trial. Lancet Neurol. 2016, 15, 259–269. [Google Scholar] [CrossRef]
- Kapoor, R.; Furby, J.; Hayton, T.; Smith, K.J.; Altmann, D.R.; Brenner, R.; Chataway, J.; Hughes, R.A.; Miller, D.H. Lamotrigine for neuroprotection in secondary progressive multiple sclerosis: A randomised, double-blind, placebo-controlled, parallel-group trial. Lancet Neurol. 2010, 9, 681–688. [Google Scholar] [CrossRef]
- Freiha, J.; Riachi, N.; Chalah, M.A.; Zoghaib, R.; Ayache, S.S.; Ahdab, R. Paroxysmal Symptoms in Multiple Sclerosis—A Review of the Literature. J. Clin. Med. 2020, 9, 3100. [Google Scholar] [CrossRef]
- Joshi, I.; Taylor, C.P. Pregabalin action at a model synapse: Binding to presynaptic calcium channel α2-δ subunit reduces neurotransmission in mice. Eur. J. Pharmacol. 2006, 553, 82–88. [Google Scholar] [CrossRef]
- Hundehege, P.; Fernandez-Orth, J.; Römer, P.; Ruck, T.; Müntefering, T.; Eichler, S.; Cerina, M.; Epping, L.; Albrecht, S.; Menke, A.F.; et al. Targeting Voltage-Dependent Calcium Channels with Pregabalin Exerts a Direct Neuroprotective Effect in an Animal Model of Multiple Sclerosis. NeuroSignals 2019, 26, 77–93. [Google Scholar] [CrossRef] [PubMed]
- Myhre, M.; Jacobsen, H.B.; Andersson, S.; Stubhaug, A. Cognitive effects of perioperative pregabalin: Secondary exploratory analysis of a randomized placebo-controlled study. Anesthesiology 2019, 130, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, A.; Emam, M. Cognitive impairment and pregabalin dependence. Egypt. J. Psychiatry 2020, 41, 14. [Google Scholar] [CrossRef]
- Brand-Schieber, E.; Werner, P. Calcium channel blockers ameliorate disease in a mouse model of multiple sclerosis. Exp. Neurol. 2004, 189, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Ingwersen, J.; De Santi, L.; Wingerath, B.; Graf, J.; Koop, B.; Schneider, R.; Hecker, C.; Schröter, F.; Bayer, M.; Engelke, A.D.; et al. Nimodipine confers clinical improvement in two models of experimental autoimmune encephalomyelitis. J. Neurochem. 2018, 146, 86–98. [Google Scholar] [CrossRef]
- Leisz, S.; Simmermacher, S.; Prell, J.; Strauss, C.; Scheller, C. Nimodipine-dependent protection of schwann cells, astrocytes and neuronal cells from osmotic, oxidative and heat stress is associated with the activation of AKT and CREB. Int. J. Mol. Sci. 2019, 20, 4578. [Google Scholar] [CrossRef]
- Singh, A.; Verma, P.; Raju, A.; Mohanakumar, K.P. Nimodipine attenuates the parkinsonian neurotoxin, MPTP-induced changes in the calcium binding proteins, calpain and calbindin. J. Chem. Neuroanat. 2019, 95, 89–94. [Google Scholar] [CrossRef]
- Liu, Y.; Lo, Y.C.; Qian, L.; Crews, F.T.; Wilson, B.; Chen, H.L.; Wu, H.M.; Chen, S.H.; Wei, K.; Lu, R.B.; et al. Verapamil protects dopaminergic neuron damage through a novel anti-inflammatory mechanism by inhibition of microglial activation. Neuropharmacology 2011, 60, 373–380. [Google Scholar] [CrossRef]
- Hashioka, S.; Klegeris, A.; McGeer, P.L. Inhibition of human astrocyte and microglia neurotoxicity by calcium channel blockers. Neuropharmacology 2012, 63, 685–691. [Google Scholar] [CrossRef]
- Schampel, A.; Volovitch, O.; Koeniger, T.; Scholz, C.J.; Jörg, S.; Linker, R.A.; Wischmeyer, E.; Wunsch, M.; Hell, J.W.; Ergün, S.; et al. Nimodipine fosters remyelination in a mouse model of multiple sclerosis and induces microglia-specific apoptosis. Proc. Natl. Acad. Sci. USA 2017, 114, E3295–E3304. [Google Scholar] [CrossRef]
- Li, Y.; Hu, X.; Liu, Y.; Bao, Y.; An, L. Nimodipine protects dopaminergic neurons against inflammation-mediated degeneration through inhibition of microglial activation. Neuropharmacology 2009, 56, 580–589. [Google Scholar] [CrossRef]
- Zamora, N.N.; Cheli, V.T.; Santiago González, D.A.; Wan, R.; Paez, P.M. Deletion of voltage-gated calcium channels in astrocytes during demyelination reduces brain inflammation and promotes myelin regeneration in mice. J. Neurosci. 2020, 40, 3332–3347. [Google Scholar] [CrossRef]
- Beraud, E.; Viola, A.; Regaya, I.; Confort-Gouny, S.; Siaud, P.; Ibarrola, D.; Le Fur, Y.; Barbaria, J.; Pellissier, J.F.; Sabatier, J.M.; et al. Block of neural Kv1.1 potassium channels for neuroinflammatory disease therapy. Ann. Neurol. 2006, 60, 586–596. [Google Scholar] [CrossRef]
- Göbel, K.; Wedell, J.H.; Herrmann, A.M.; Wachsmuth, L.; Pankratz, S.; Bittner, S.; Budde, T.; Kleinschnitz, C.; Faber, C.; Wiendl, H.; et al. 4-Aminopyridine ameliorates mobility but not disease course in an animal model of multiple sclerosis. Exp. Neurol. 2013, 248, 62–71. [Google Scholar] [CrossRef]
- Zhang, E.; Tian, X.; Li, R.; Chen, C.; Li, M.; Ma, L.; Wei, R.; Zhou, Y.; Cui, Y. Dalfampridine in the treatment of multiple sclerosis: A meta-analysis of randomised controlled trials. Orphanet J. Rare Dis. 2021, 16, 87. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, K. Exploiting the Diversity of Ion Channels: Modulation of Ion Channels for Therapeutic Indications. In Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2019; Volume 260, pp. 187–205. [Google Scholar]
- Sontheimer, H.; Trotter, J.; Schachner, M.; Kettenmann, H. Channel expression correlates with differentiation stage during the development of Oligodendrocytes from their precursor cells in culture. Neuron 1989, 2, 1135–1145. [Google Scholar] [CrossRef]
- Barres, B.A.; Koroshetz, W.J.; Swartz, K.J.; Chun, L.L.Y.; Corey, D.P. Ion channel expression by white matter glia: The O-2A glial progenitor cell. Neuron 1990, 4, 507–524. [Google Scholar] [CrossRef]
- Williamson, A.V.; Compston, D.A.S.; Randall, A.D. Analysis of the Ion Channel Complement of the Rat Oligodendrocyte Progenitor in a Commonly Studied In vitro Preparation. Eur. J. Neurosci. 1997, 9, 706–720. [Google Scholar] [CrossRef]
- Borges, K.; Kettenmann, H. Blockade of K+ channels induced by AMPA/kainate receptor activation in mouse oligodendrocyte precursor cells is mediated by NA+ entry. J. Neurosci. Res. 1995, 42, 579–593. [Google Scholar] [CrossRef]
- Berger, T.; Schnitzer, J.; Orkand, P.M.; Kettenmann, H. Sodium and Calcium Currents in Glial Cells of the Mouse Corpus Callosum Slice. Eur. J. Neurosci. 1992, 4, 1271–1284. [Google Scholar] [CrossRef]
- Spitzer, S.O.; Sitnikov, S.; Kamen, Y.; De Faria, O. Oligodendrocyte Progenitor Cells Become Regionally Diverse and Heterogeneous with Age. Neuron 2019, 101, 459–471.e5. [Google Scholar] [CrossRef]
- De Biase, L.M.; Nishiyama, A.; Bergles, D.E. Excitability and synaptic communication within the oligodendrocyte lineage. J. Neurosci. 2010, 30, 3600–3611. [Google Scholar] [CrossRef]
- Paez, P.M.; Fulton, D.; Colwell, C.S.; Campagnoni, A.T. Voltage-operated Ca 2+ and Na + channels in the oligodendrocyte lineage. J. Neurosci. Res. 2009, 87, 3259–3266. [Google Scholar] [CrossRef]
- Coppi, E.; Maraula, G.; Fumagalli, M.; Failli, P.; Cellai, L.; Bonfanti, E.; Mazzoni, L.; Coppini, R.; Abbracchio, M.P.; Pedata, F.; et al. UDP-glucose enhances outward K + currents necessary for cell differentiation and stimulates cell migration by activating the GPR17 receptor in oligodendrocyte precursors. Glia 2013, 61, 1155–1171. [Google Scholar] [CrossRef]
- Mallon, B.S.; Elizabeth Shick, H.; Kidd, G.J.; Macklin, W.B. Proteolipid promoter activity distinguishes two populations of NG2-positive cells throughout neonatal cortical development. J. Neurosci. 2002, 22, 876–885. [Google Scholar] [CrossRef]
- Trapp, B.D.; Nishiyama, A.; Cheng, D.; Macklin, W. Differentiation and death of premyelinating oligodendrocytes in developing rodent brain. J. Cell Biol. 1997, 137, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Black, J.A.; Waxman, S.G.; Smith, K.J. Remyelination of dorsal column axons by endogenous Schwann cells restores the normal pattern of Na v1.6 and K v1.2 at nodes of Ranvier. Brain 2006, 129, 1319–1329. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Huang, J.; Waxman, S.G. Sodium channel Nav1.8: Emerging links to human disease. Neurology 2016, 86, 473–783. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Ou, S.W.; Wang, Y.J. Distribution and function of voltage-gated sodium channels in the nervous system. Channels 2017, 11, 534–554. [Google Scholar] [CrossRef]
- Craner, M.J.; Hains, B.C.; Lo, A.C.; Black, J.A.; Waxman, S.G. Co-localization of sodium channel Nav1.6 and the sodium-calcium exchanger at sites of axonal injury in the spinal cord in EAE. Brain 2004, 127, 294–303. [Google Scholar] [CrossRef]
- Káradóttir, R.; Hamilton, N.B.; Bakiri, Y.; Attwell, D. Spiking and nonspiking classes of oligodendrocyte precursor glia in CNS white matter. Nat. Neurosci. 2008, 11, 450–456. [Google Scholar] [CrossRef]
- Xie, M.; Lynch, D.T.; Schools, G.P.; Feustel, P.J.; Kimelberg, H.K.; Zhou, M. Sodium channel currents in rat hippocampal NG2 glia: Characterization and contribution to resting membrane potential. Neuroscience 2007, 150, 853–862. [Google Scholar] [CrossRef]
- Cheli, V.T.; Santiago González, D.A.; Spreuer, V.; Paez, P.M. Voltage-gated Ca++ entry promotes oligodendrocyte progenitor cell maturation and myelination in vitro. Exp. Neurol. 2015, 265, 69–83. [Google Scholar] [CrossRef]
- Fulton, D.; Paez, P.M.; Fisher, R.; Handley, V.; Colwell, C.S.; Campagnoni, A.T. Regulation of L-type Ca++ currents and process morphology in white matter oligodendrocyte precursor cells by golli-myelin proteins. Glia 2010, 58, 1292–1303. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef]
- Agrawal, S.K.; Nashmi, R.; Fehlings, M.G. Role of L- and N-type calcium channels in the pathophysiology of traumatic spinal cord white matter injury. Neuroscience 2000, 99, 179–188. [Google Scholar] [CrossRef]
- Cheli, V.T.; González, D.A.S.; Lama, T.N.; Spreuer, V.; Handley, V.; Murphy, G.G.; Paez, P.M. Conditional deletion of the L-type calcium channel cav1.2 in oligodendrocyte progenitor cells affects postnatal myelination in mice. J. Neurosci. 2016, 36, 10853–10869. [Google Scholar] [CrossRef]
- González, D.A.S.; Cheli, V.T.; Zamora, N.N.; Lama, T.N.; Spreuer, V.; Murphy, G.G.; Paez, P.M. Conditional deletion of the L-type calcium channel Cav1.2 in NG2-positive cells impairs remyelination in mice. J. Neurosci. 2017, 37, 10038–10051. [Google Scholar] [CrossRef]
- Paez, P.M.; Cheli, V.T.; Ghiani, C.A.; Spreuer, V.; Handley, V.W.; Campagnoni, A.T. Golli myelin basic proteins stimulate oligodendrocyte progenitor cell proliferation and differentiation in remyelinating adult mouse brain. Glia 2012, 60, 1078–1093. [Google Scholar] [CrossRef]
- Kornek, B.; Storch, M.K.; Weissert, R.; Wallstroem, E.; Stefferl, A.; Olsson, T.; Linington, C.; Schmidbauer, M.; Lassmann, H. Multiple sclerosis and chronic autoimmune encephalomyelitis: A comparative quantitative study of axonal injury in active, inactive, and remyelinated lesions. Am. J. Pathol. 2000, 157, 267–276. [Google Scholar] [CrossRef]
- Paez, P.M.; Fulton, D.J.; Spreur, V.; Handley, V.; Campagnoni, A.T. Multiple kinase pathways regulate voltage-dependent Ca2+ influx and migration in oligodendrocyte precursor cells. J. Neurosci. 2010, 30, 6422–6433. [Google Scholar] [CrossRef]
- Lohr, C.; Heil, J.E.; Deitmer, J.W. Blockage of voltage-gated calcium signaling impairs migration of glial cells in vivo. Glia 2005, 50, 198–211. [Google Scholar] [CrossRef] [PubMed]
- Oland, L.A.; Tolbert, L.P. Glial patterns during early development of antennal lobes ofManduca sexta: A comparison between normal lobes and lobes deprived of antennal axons. J. Comp. Neurol. 1987, 255, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Gudz, T.I.; Komuro, H.; Macklin, W.B. Glutamate stimulates oligodendrocyte progenitor migration mediated via an αv integrin/myelin proteolipid protein complex. J. Neurosci. 2006, 26, 2458–2466. [Google Scholar] [CrossRef] [PubMed]
- Tsai, F.C.; Kuo, G.H.; Chang, S.W.; Tsai, P.J. Ca2+ signaling in cytoskeletal reorganization, cell migration, and cancer metastasis. Biomed Res. Int. 2015, 2015, 409245. [Google Scholar] [CrossRef]
- Gallo, V.; Zhou, J.M.; McBain, C.J.; Wright, P.; Knutson, P.L.; Armstrong, R.C. Oligodendrocyte progenitor cell proliferation and lineage progression are regulated by glutamate receptor-mediated K+ channel block. J. Neurosci. 1996, 16, 2659–2670. [Google Scholar] [CrossRef] [PubMed]
- Larson, V.A.; Zhang, Y.; Bergles, D.E. Electrophysiological properties of NG2+ cells: Matching physiological studies with gene expression profiles. Brain Res. 2016, 1638, 138–160. [Google Scholar] [CrossRef] [PubMed]
- Papanikolaou, M.; Butt, A.M.; Lewis, A. A critical role for the inward rectifying potassium channel Kir7.1 in oligodendrocytes of the mouse optic nerve. Brain Struct. Funct. 2020, 225, 925–934. [Google Scholar] [CrossRef] [PubMed]
- Brasko, C.; Butt, A. Expression of Kir2.1 Inward Rectifying Potassium Channels in Optic Nerve Glia: Evidence for Heteromeric Association with Kir4.1 and Kir5.1. Neuroglia 2018, 1, 176–1873. [Google Scholar] [CrossRef]
- Butt, A.M.; Papanikolaou, M.; Rivera, A. Physiology of oligodendroglia. In Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2019; Volume 1175, pp. 117–128. [Google Scholar]
- Larson, V.A.; Mironova, Y.; Vanderpool, K.G.; Waisman, A.; Rash, J.E.; Agarwal, A.; Bergles, D.E. Oligodendrocytes control potassium accumulation in white matter and seizure susceptibility. eLife 2018, 7, e34829. [Google Scholar] [CrossRef]
- Schirmer, L.; Möbius, W.; Zhao, C.; Cruz-Herranz, A.; Ben Haim, L.; Cordano, C.; Shiow, L.R.; Kelley, K.W.; Sadowski, B.; Timmons, G.; et al. Oligodendrocyte-encoded kir4.1 function is required for axonal integrity. eLife 2018, 7, e36428. [Google Scholar] [CrossRef]
- Srivastava, R.; Aslam, M.; Kalluri, S.R.; Schirmer, L.; Buck, D.; Tackenberg, B.; Rothhammer, V.; Chan, A.; Gold, R.; Berthele, A.; et al. Potassium Channel KIR4.1 as an Immune Target in Multiple Sclerosis. N. Engl. J. Med. 2012, 367, 115–123. [Google Scholar] [CrossRef]
- Coppi, E.; Cellai, L.; Maraula, G.; Pugliese, A.M.; Pedata, F. Adenosine A2A receptors inhibit delayed rectifier potassium currents and cell differentiation in primary purified oligodendrocyte cultures. Neuropharmacology 2013, 73, 301–310. [Google Scholar] [CrossRef]
- Coppi, E.; Cherchi, F.; Fusco, I.; Dettori, I.; Gaviano, L.; Magni, G.; Catarzi, D.; Colotta, V.; Varano, F.; Rossi, F.; et al. Adenosine A2B receptors inhibit K+ currents and cell differentiation in cultured oligodendrocyte precursor cells and modulate sphingosine-1-phosphate signaling pathway. Biochem. Pharmacol. 2020, 177, 113956. [Google Scholar] [CrossRef]
- Attali, B.; Wang, N.; Kolot, A.; Sobko, A.; Cherepanov, V.; Soliven, B. Characterization of delayed rectifier Kv channels in oligodendrocytes and progenitor cells. J. Neurosci. 1997, 17, 8234–8245. [Google Scholar] [CrossRef]
- Chittajallu, R.; Chen, Y.; Wang, H.; Yuan, X.; Ghiani, C.A.; Heckman, T.; McBain, C.J.; Gallo, V. Regulation of Kv1 subunit expression in oligodendrocyte progenitor cells and their role in G 1/S phase progression of the cell cycle. Proc. Natl. Acad. Sci. USA 2002, 99, 2350–2355. [Google Scholar] [CrossRef]
- Coman, I.; Aigrot, M.S.; Seilhean, D.; Reynolds, R.; Girault, J.A.; Zalc, B.; Lubetzki, C. Nodal, paranodal and juxtaparanodal axonal proteins during demyelination and remyelination in multiple sclerosis. Brain 2006, 129, 3186–3195. [Google Scholar] [CrossRef]
- Jukkola, P.I.; Lovett-Racke, A.E.; Zamvil, S.S.; Gu, C. K+ channel alterations in the progression of experimental autoimmune encephalomyelitis. Neurobiol. Dis. 2012, 47, 280–293. [Google Scholar] [CrossRef][Green Version]
- Vautier, F.; Belachew, S.; Chittajallu, R.; Gallo, V. Shaker-type potassium channel subunits differentially control oligodendrocyte progenitor proliferation. Glia 2004, 48, 337–345. [Google Scholar] [CrossRef]
- Chittajallu, R.; Aguirre, A.; Gallo, V. NG2-positive cells in the mouse white and grey matter display distinct physiological properties. J. Physiol. 2004, 561, 109–122. [Google Scholar] [CrossRef]
- Tong, X.P.; Li, X.Y.; Zhou, B.; Shen, W.; Zhang, Z.J.; Xu, T.L.; Duan, S. Ca2+ signaling evoked by activation of Na+ channels and Na+/Ca2+ exchangers is required for GABA-induced NG2 cell migration. J. Cell Biol. 2009, 186, 113–128. [Google Scholar] [CrossRef]
- Kukley, M.; Kiladze, M.; Tognatta, R.; Hans, M.; Swandulla, D.; Schramm, J.; Dietrich, D. Glial cells are born with synapses. FASEB J. 2008, 22, 2957–2969. [Google Scholar] [CrossRef]
- Ziskin, J.L.; Nishiyama, A.; Rubio, M.; Fukaya, M.; Bergles, D.E. Vesicular release of glutamate from unmyelinated axons in white matter. Nat. Neurosci. 2007, 10, 321–330. [Google Scholar] [CrossRef]
- Schattling, B.; Fazeli, W.; Engeland, B.; Liu, Y.; Lerche, H.; Isbrandt, D.; Friese, M.A. Activity of NaV1.2 promotes neurodegeneration in an animal model of multiple sclerosis. JCI Insight 2016, 1, e89810. [Google Scholar] [CrossRef]
- Alrashdi, B.; Dawod, B.; Schampel, A.; Tacke, S.; Kuerten, S.; Marshall, J.S.; Côté, P.D. Nav1.6 promotes inflammation and neuronal degeneration in a mouse model of multiple sclerosis. J. Neuroinflammation 2019, 16, 215. [Google Scholar] [CrossRef]
- Boscia, F.; D’Avanzo, C.; Pannaccione, A.; Secondo, A.; Casamassa, A.; Formisano, L.; Guida, N.; Annunziato, L. Silencing or knocking out the Na +/Ca2+ exchanger-3 (NCX3) impairs oligodendrocyte differentiation. Cell Death Differ. 2012, 19, 562–572. [Google Scholar] [CrossRef] [PubMed]
- Boscia, F.; de Rosa, V.; Cammarota, M.; Secondo, A.; Pannaccione, A.; Annunziato, L. The Na+/Ca2+ exchangers in demyelinating diseases. Cell Calcium 2020, 85, 102130. [Google Scholar] [CrossRef] [PubMed]
- Hammann, J.; Bassetti, D.; White, R.; Luhmann, H.J.; Kirischuk, S. α2 isoform of Na +,K + -ATPase via Na +,Ca2+ exchanger modulates myelin basic protein synthesis in oligodendrocyte lineage cells in vitro. Cell Calcium 2018, 73, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Black, J.A.; Waxman, S.G. Phenytoin protects central axons in experimental autoimmune encephalomyelitis. J. Neurol. Sci. 2008, 274, 57–63. [Google Scholar] [CrossRef]
- Black, J.A.; Liu, S.; Carrithers, M.; Carrithers, L.M.; Waxman, S.G. Exacerbation of experimental autoimmune encephalomyelitis after withdrawal of phenytoin and carbamazepine. Ann. Neurol. 2007, 62, 21–33. [Google Scholar] [CrossRef]
- Keppel Hesselink, J.M.; Kopsky, D.J. Phenytoin: Neuroprotection or neurotoxicity? Neurol. Sci. 2017, 38, 1137–1141. [Google Scholar] [CrossRef]
- Paez, P.M.; Lyons, D.A. Calcium Signaling in the Oligodendrocyte Lineage: Regulators and Consequences. Annu. Rev. Neurosci. 2020, 43, 163–186. [Google Scholar] [CrossRef]
- Baraban, M.; Koudelka, S.; Lyons, D.A. Ca2+ activity signatures of myelin sheath formation and growth in vivo. Nat. Neurosci. 2018, 21, 19–25. [Google Scholar] [CrossRef]
- Krasnow, A.M.; Ford, M.C.; Valdivia, L.E.; Wilson, S.W.; Attwell, D. Regulation of developing myelin sheath elongation by oligodendrocyte calcium transients in vivo. Nat. Neurosci. 2018, 21, 24–30. [Google Scholar] [CrossRef]
- Battefeld, A.; Popovic, M.A.; de Vries, S.I.; Kole, M.H.P. High-Frequency Microdomain Ca2+ Transients and Waves during Early Myelin Internode Remodeling. Cell Rep. 2019, 26, 182–191. [Google Scholar] [CrossRef]
- Kirchhoff, F.; Kettenmann, H. GABA Triggers a [Ca2+]i Increase in Murine Precursor Cells of the Oligodendrocyte Lineage. Eur. J. Neurosci. 1992, 4, 1049–1058. [Google Scholar] [CrossRef]
- Barron, T.; Kim, J.H. Neuronal input triggers Ca2+ influx through AMPA receptors and voltage-gated Ca2+ channels in oligodendrocytes. Glia 2019, 67, 1922–1932. [Google Scholar] [CrossRef]
- Li, T.; Wang, L.; Ma, T.; Wang, S.; Niu, J.; Li, H.; Xiao, L. Dynamic Calcium Release From Endoplasmic Reticulum Mediated by Ryanodine Receptor 3 Is Crucial for Oligodendroglial Differentiation. Front. Mol. Neurosci. 2018, 11, 162. [Google Scholar] [CrossRef]
- Rui, Y.; Pollitt, S.L.; Myers, K.R.; Feng, Y.; Zheng, J.Q. Spontaneous local calcium transients regulate oligodendrocyte development in culture through store-operated ca2+ entry and release. eNeuro 2020, 7, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Feldman, D.H.; Horiuchi, M.; Keachie, K.; Mccauley, E.; Bannerman, P.; Itoh, A.; Itoh, T.; Pleasure, D. Characterization of acid-sensing ion channel expression in oligodendrocyte-lineage cells. Glia 2008, 56, 1238–1249. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Ren, Y.Q.; Bing, R.; Hillman, D.E. Alpha 1E subunit of the R-type calcium channel is associated with myelinogenesis. J. Neurocytol. 2000, 29, 719–728. [Google Scholar] [CrossRef] [PubMed]
- Gopalasingam, G.; Bartlett, C.A.; McGonigle, T.; Majimbi, M.; Warnock, A.; Ford, A.; Gough, A.; Toomey, L.M.; Fitzgerald, M. The effects of a combination of ion channel inhibitors on pathology in a model of demyelinating disease. Mult. Scler. Relat. Disord. 2019, 34, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Kornek, B.; Storch, M.K.; Bauer, J.; Djamshidian, A.; Weissert, R.; Wallstroem, E.; Stefferl, A.; Zimprich, F.; Olsson, T.; Linington, C.; et al. Distribution of a calcium channel subunit in dystrophic axons in multiple sclerosis and experimental autoimmune encephalomyelitis. Brain 2001, 124, 1114–1124. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Liu, Z.; Lv, P.; Wang, H.; Zhu, Y.; Qi, Q.; Xu, J.; Gao, L. Nimodipine activates neuroprotective signaling events and inactivates autophages in the VCID rat hippocampus. Neurol. Res. 2017, 39, 904–909. [Google Scholar] [CrossRef]
- Ma, M.; Ferguson, T.A.; Schoch, K.M.; Li, J.; Qian, Y.; Shofer, F.S.; Saatman, K.E.; Neumar, R.W. Calpains mediate axonal cytoskeleton disintegration during Wallerian degeneration. Neurobiol. Dis. 2013, 56, 34–46. [Google Scholar] [CrossRef]
- Stebbing, M.J.; Cottee, J.M.; Rana, I. The role of ion channels in microglial activation and proliferation—A complex interplay between ligand-gated ion channels, K+ channels, and intracellular Ca2+. Front. Immunol. 2015, 6, 497. [Google Scholar] [CrossRef]
- Enders, M.; Heider, T.; Ludwig, A.; Kuerten, S. Strategies for neuroprotection in multiple sclerosis and the role of calcium. Int. J. Mol. Sci. 2020, 21, 1663. [Google Scholar] [CrossRef]
- Maldonado, P.P.; Vélez-Fort, M.; Levavasseur, F.; Angulo, M.C. Oligodendrocyte precursor cells are accurate sensors of local K+ in mature gray matter. J. Neurosci. 2013, 33, 2432–2442. [Google Scholar] [CrossRef]
- Buttigieg, J.; Karimi-Abdolrezaee, S.; Fehlings, M.G. Molecular and electrophysiological evidence for the expression of BK channels in oligodendroglial precursor cells. Eur. J. Neurosci. 2011, 34, 538–547. [Google Scholar] [CrossRef]
- Calvo, M.; Richards, N.; Schmid, A.B.; Barroso, A.; Zhu, L.; Ivulic, D.; Zhu, N.; Anwandter, P.; Bhat, M.A.; Court, F.A.; et al. Altered potassium channel distribution and composition in myelinated axons suppresses hyperexcitability following injury. eLife 2016, 5, 12661. [Google Scholar] [CrossRef]
- Waxman, S.G.; Utzschneider, D.A.; Kocsis, J.D. Enhancement of action potential conduction following demyelination: Experimental approaches to restoration of function in multiple sclerosis and spinal cord injury. Prog. Brain Res. 1994, 100, 233–243. [Google Scholar] [CrossRef]
- Yu, S.P. Regulation and critical role of potassium homeostasis in apoptosis. Prog. Neurobiol. 2003, 70, 363–386. [Google Scholar] [CrossRef]
- González-Alvarado, M.N.; Rötger, C.; Berger, L.; London, B.; Haase, S.; Kuhbandner, K.; Lee, D.H.; Linker, R.A. Functional role of endogenous Kv1.4 in experimental demyelination. J. Neuroimmunol. 2020, 343, 577227. [Google Scholar] [CrossRef]
- Rothhammer, V.; Mascanfroni, I.D.; Bunse, L.; Takenaka, M.C.; Kenison, J.E.; Mayo, L.; Chao, C.C.; Patel, B.; Yan, R.; Blain, M.; et al. Type i interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat. Med. 2016, 22, 586–597. [Google Scholar] [CrossRef]
- Rothhammer, V.; Borucki, D.M.; Tjon, E.C.; Takenaka, M.C.; Chao, C.C.; Ardura-Fabregat, A.; De Lima, K.A.; Gutiérrez-Vázquez, C.; Hewson, P.; Staszewski, O.; et al. Microglial control of astrocytes in response to microbial metabolites. Nature 2018, 557, 724–728. [Google Scholar] [CrossRef]
- Mitew, S.; Gobius, I.; Fenlon, L.R.; McDougall, S.J.; Hawkes, D.; Xing, Y.L.; Bujalka, H.; Gundlach, A.L.; Richards, L.J.; Kilpatrick, T.J.; et al. Pharmacogenetic stimulation of neuronal activity increases myelination in an axon-specific manner. Nat. Commun. 2018, 9, 1–16. [Google Scholar] [CrossRef]
- Nagy, B.; Hovhannisyan, A.; Barzan, R.; Chen, T.J.; Kukley, M. Different patterns of neuronal activity trigger distinct responses of oligodendrocyte precursor cells in the corpus callosum. PLoS Biol. 2017, 15, e2001993. [Google Scholar] [CrossRef]
- Thornton, M.A.; Hughes, E.G. Neuron-oligodendroglia interactions: Activity-dependent regulation of cellular signaling. Neurosci. Lett. 2020, 727, 134916. [Google Scholar] [CrossRef]
- Ortiz, F.C.; Habermacher, C.; Graciarena, M.; Houry, P.Y.; Nishiyama, A.; Oumesmar, B.N.; Angulo, M.C. Neuronal activity in vivo enhances functional myelin repair. JCI Insight 2019, 4, 123434. [Google Scholar] [CrossRef] [PubMed]
- Ronzano, R.; Thetiot, M.; Lubetzki, C.; Desmazieres, A. Myelin Plasticity and Repair: Neuro-Glial Choir Sets the Tuning. Front. Cell. Neurosci. 2020, 14, 42. [Google Scholar] [CrossRef]
- Zonouzi, M.; Scafidi, J.; Li, P.; McEllin, B.; Edwards, J.; Dupree, J.L.; Harvey, L.; Sun, D.; Hübner, C.A.; Cull-Candy, S.G.; et al. GABAergic regulation of cerebellar NG2 cell development is altered in perinatal white matter injury. Nat. Neurosci. 2015, 18, 674–682. [Google Scholar] [CrossRef]
- Friess, M.; Hammann, J.; Unichenko, P.; Luhmann, H.J.; White, R.; Kirischuk, S. Intracellular ion signaling influences myelin basic protein synthesis in oligodendrocyte precursor cells. Cell Calcium 2016, 60, 322–330. [Google Scholar] [CrossRef]
- Maas, D.A.; Angulo, M.C. Can Enhancing Neuronal Activity Improve Myelin Repair in Multiple Sclerosis? Front. Cell. Neurosci. 2021, 15, 38. [Google Scholar] [CrossRef]
- Micu, I.; Plemel, J.R.; Lachance, C.; Proft, J.; Jansen, A.J.; Cummins, K.; van Minnen, J.; Stys, P.K. The molecular physiology of the axo-myelinic synapse. Exp. Neurol. 2016, 276, 41–50. [Google Scholar] [CrossRef]
- Marinelli, C.; Bertalot, T.; Zusso, M.; Skaper, S.D.; Giusti, P. Systematic review of pharmacological properties of the oligodendrocyte lineage. Front. Cell. Neurosci. 2016, 10, 27. [Google Scholar] [CrossRef]
- Cherchi, F.; Pugliese, A.A.M.; Coppi, E. Oligodendrocyte precursor cell maturation: Role of adenosine receptors. Neural Regen. Res. 2021, 16, 1686. [Google Scholar] [CrossRef]
- Bechler, M.E.; Swire, M.; ffrench-Constant, C. Intrinsic and adaptive myelination—A sequential mechanism for smart wiring in the brain. Dev. Neurobiol. 2018, 78, 68–79. [Google Scholar] [CrossRef]
- Wake, H.; Ortiz, F.C.; Woo, D.H.; Lee, P.R.; Angulo, M.C.; Fields, R.D. Nonsynaptic junctions on myelinating glia promote preferential myelination of electrically active axons. Nat. Commun. 2015, 6, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Eisen, A.M.; McBain, C.J.; Gallo, V. A role for glutamate and its receptors in the regulation of oligodendrocyte development in cerebellar tissue slices. Development 1998, 125, 2901–2914. [Google Scholar] [CrossRef] [PubMed]
- De Faria, O.; Gonsalvez, D.G.; Nicholson, M.; Xiao, J. Activity-dependent central nervous system myelination throughout life. J. Neurochem. 2019, 148, 447–461. [Google Scholar] [CrossRef] [PubMed]
- Fannon, J.; Tarmier, W.; Fulton, D. Neuronal activity and AMPA-type glutamate receptor activation regulates the morphological development of oligodendrocyte precursor cells. Glia 2015, 63, 1021–1035. [Google Scholar] [CrossRef] [PubMed]
- Gautier, H.O.B.; Evans, K.A.; Volbracht, K.; James, R.; Sitnikov, S.; Lundgaard, I.; James, F.; Lao-Peregrin, C.; Reynolds, R.; Franklin, R.J.M.; et al. Neuronal activity regulates remyelination via glutamate signalling to oligodendrocyte progenitors. Nat. Commun. 2015, 6, 1–15. [Google Scholar] [CrossRef]
- Kougioumtzidou, E.; Shimizu, T.; Hamilton, N.B.; Tohyama, K.; Sprengel, R.; Monyer, H.; Attwell, D.; Richardson, W.D. Signalling through AMPA receptors on oligodendrocyte precursors promotes myelination by enhancing oligodendrocyte survival. eLife 2017, 6, e28080. [Google Scholar] [CrossRef]
- Li, C.; Xiao, L.; Liu, X.; Yang, W.; Shen, W.; Hu, C.; Yang, G.; He, C. A functional role of nmda receptor in regulating the differentiation of oligodendrocyte precursor cells and remyelination. Glia 2013, 61, 732–749. [Google Scholar] [CrossRef]
- Hamilton, N.B.; Clarke, L.E.; Arancibia-Carcamo, I.L.; Kougioumtzidou, E.; Matthey, M.; Káradóttir, R.; Whiteley, L.; Bergersen, L.H.; Richardson, W.D.; Attwell, D. Endogenous GABA controls oligodendrocyte lineage cell number, myelination, and CNS internode length. Glia 2017, 65, 309–321. [Google Scholar] [CrossRef]
- Cisneros-Mejorado, A.J.; Garay, E.; Ortiz-Retana, J.; Concha, L.; Moctezuma, J.P.; Romero, S.; Arellano, R.O. Demyelination–Remyelination of the Rat Caudal Cerebellar Peduncle Evaluated with Magnetic Resonance Imaging. Neuroscience 2020, 439, 255–267. [Google Scholar] [CrossRef]
- Luyt, K.; Slade, T.P.; Dorward, J.J.; Durant, C.F.; Wu, Y.; Shigemoto, R.; Mundell, S.J.; Váradi, A.; Molnár, E. Developing oligodendrocytes express functional GABAB receptors that stimulate cell proliferation and migration. J. Neurochem. 2007, 100, 822–840. [Google Scholar] [CrossRef]
- Serrano-Regal, M.P.; Luengas-Escuza, I.; Bayón-Cordero, L.; Ibarra-Aizpurua, N.; Alberdi, E.; Pérez-Samartín, A.; Matute, C.; Sánchez-Gómez, M.V. Oligodendrocyte Differentiation and Myelination Is Potentiated via GABAB Receptor Activation. Neuroscience 2020, 439, 163–180. [Google Scholar] [CrossRef]
- Feng, J.F.; Gao, X.F.; Pu, Y.Y.; Burnstock, G.; Xiang, Z.; He, C. P2X7 receptors and Fyn kinase mediate ATP-induced oligodendrocyte progenitor cell migration. Purinergic Signal. 2015, 11, 361–369. [Google Scholar] [CrossRef]
- Domercq, M.; Perez-Samartin, A.; Aparicio, D.; Alberdi, E.; Pampliega, O.; Matute, C. P2X7 receptors mediate ischemic damage to oligodendrocytes. Glia 2010, 58, 730–740. [Google Scholar] [CrossRef]
- Illes, P. P2X7 Receptors Amplify CNS Damage in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 5996. [Google Scholar] [CrossRef]
- Matute, C.; Torre, I.; Pérez-Cerdá, F.; Pérez-Samartín, A.; Alberdi, E.; Etxebarria, E.; Arranz, A.M.; Ravid, R.; Rodríguez-Antigüedad, A.; Sánchez-Gómez, M.V.; et al. P2X7 receptor blockade prevents ATP excitotoxicity in oligodendrocytes and ameliorates experimental autoimmune encephalomyelitis. J. Neurosci. 2007, 27, 9525–9533. [Google Scholar] [CrossRef]
- Coppi, E.; Pedata, F.; Gibb, A.J. P2Y1 receptor modulation of Ca2+-activated K+ currents in medium-sized neurons from neonatal rat striatal slices. J. Neurophysiol. 2012, 107, 1009–1021. [Google Scholar] [CrossRef]
- Agresti, C.; Meomartini, M.E.; Amadio, S.; Ambrosini, E.; Serafini, B.; Franchini, L.; Volonté, C.; Aloisi, F.; Visentin, S. Metabotropic P2 receptor activation regulates oligodendrocyte progenitor migration and development. Glia 2005, 50, 132–144. [Google Scholar] [CrossRef]
- Amadio, S.; Montilli, C.; Magliozzi, R.; Bernardi, G.; Reynolds, R.; Volonté, C. P2Y12 receptor protein in cortical gray matter lesions in multiple sclerosis. Cereb. Cortex 2010, 20, 1263–1273. [Google Scholar] [CrossRef]
- Angelini, J.; Marangon, D.; Raffaele, S.; Lecca, D.; Abbracchio, M. The Distribution of GPR17-Expressing Cells Correlates with White Matter Inflammation Status in Brain Tissues of Multiple Sclerosis Patients. Int. J. Mol. Sci. 2021, 22, 4574. [Google Scholar] [CrossRef]
- Fumagalli, M.; Daniele, S.; Lecca, D.; Lee, P.R.; Parravicini, C.; Douglas Fields, R.; Rosa, P.; Antonucci, F.; Verderio, C.; Letizia Trincavelli, M.; et al. Phenotypic changes, signaling pathway, and functional correlates of GPR17-expressing neural precursor cells during oligodendrocyte differentiation. J. Biol. Chem. 2011, 286, 10593–10604. [Google Scholar] [CrossRef]
- Stevens, B.; Porta, S.; Haak, L.L.; Gallo, V.; Fields, R.D. Adenosine: A neuron-glial transmitter promoting myelination in the CNS in response to action potentials. Neuron 2002, 36, 855–868. [Google Scholar] [CrossRef]
- Spitzer, S.; Volbracht, K.; Lundgaard, I.; Káradóttir, R.T. Glutamate signalling: A multifaceted modulator of oligodendrocyte lineage cells in health and disease. Neuropharmacology 2016, 110, 574–585. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, P.P.; Angulo, M.C. Multiple modes of communication between neurons and oligodendrocyte precursor cells. Neuroscientist 2015, 21, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Pitman, K.A.; Young, K.M. Activity-dependent calcium signalling in oligodendrocyte generation. Int. J. Biochem. Cell Biol. 2016, 77, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Cheli, V.T.; Santiago González, D.A.; Zamora, N.N.; Lama, T.N.; Spreuer, V.; Rasmusson, R.L.; Bett, G.C.; Panagiotakos, G.; Paez, P.M. Enhanced oligodendrocyte maturation and myelination in a mouse model of Timothy syndrome. Glia 2018, 66, 2324–2339. [Google Scholar] [CrossRef] [PubMed]
- Mangin, J.M.; Li, P.; Scafidi, J.; Gallo, V. Experience-dependent regulation of NG2 progenitors in the developing barrel cortex. Nat. Neurosci. 2012, 15, 1192–1194. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.J.; Kula, B.; Nagy, B.; Barzan, R.; Gall, A.; Ehrlich, I.; Kukley, M. In Vivo Regulation of Oligodendrocyte Precursor Cell Proliferation and Differentiation by the AMPA-Receptor Subunit GluA2. Cell Rep. 2018, 25, 852–861.e7. [Google Scholar] [CrossRef]
- Casaccia-Bonnefil, P.; Liu, A. Relationship between cell cycle molecules and onset of oligodendrocyte differentiation. J. Neurosci. Res. 2003, 72, 1–11. [Google Scholar] [CrossRef]
- Saab, A.S.; Tzvetavona, I.D.; Trevisiol, A.; Baltan, S.; Dibaj, P.; Kusch, K.; Möbius, W.; Goetze, B.; Jahn, H.M.; Huang, W.; et al. Oligodendroglial NMDA Receptors Regulate Glucose Import and Axonal Energy Metabolism. Neuron 2016, 91, 119–132. [Google Scholar] [CrossRef]
- Micu, I.; Plemel, J.R.; Caprariello, A.V.; Nave, K.A.; Stys, P.K. Axo-myelinic neurotransmission: A novel mode of cell signalling in the central nervous system. Nat. Rev. Neurosci. 2018, 19, 49–57. [Google Scholar] [CrossRef]
- Habermacher, C.; Angulo, M.C.; Benamer, N. Glutamate versus GABA in neuron–oligodendroglia communication. Glia 2019, 67, 2092–2106. [Google Scholar] [CrossRef]
- De Biase, L.M.; Kang, S.H.; Baxi, E.G.; Fukaya, M.; Pucak, M.L.; Mishina, M.; Calabresi, P.A.; Bergles, D.E. NMDA receptor signaling in oligodendrocyte progenitors is not required for oligodendrogenesis and myelination. J. Neurosci. 2011, 31, 12650–12662. [Google Scholar] [CrossRef]
- López-Chávez, A.; Miledi, R.; Martínez-Torres, A. Cloning and functional expression of the bovine GABAC ρ2 subunit: Molecular evidence of a widespread distribution in the CNS. Neurosci. Res. 2005, 53, 421–427. [Google Scholar] [CrossRef]
- Arellano, R.O.; Sánchez-Gómez, M.V.; Alberdi, E.; Canedo-Antelo, M.; Chara, J.C.; Palomino, A.; Pérez-Samartín, A.; Matute, C. Axon-to-glia interaction regulates gabaa receptor expression in oligodendrocytes. Mol. Pharmacol. 2016, 89, 63–74. [Google Scholar] [CrossRef]
- Serrano-Regal, M.P.; Bayón-Cordero, L.; Ordaz, R.P.; Garay, E.; Limon, A.; Arellano, R.O.; Matute, C.; Sánchez-Gómez, M.V. Expression and Function of GABA Receptors in Myelinating Cells. Front. Cell. Neurosci. 2020, 14, 256. [Google Scholar] [CrossRef]
- Rivera, A.; Vanzulli, I.; Butt, A. A Central Role for ATP Signalling in Glial Interactions in the CNS. Curr. Drug Targets 2016, 17, 1829–1833. [Google Scholar] [CrossRef]
- Abbracchio, M.P.; Burnstock, G. Purinoceptors: Are there families of P2X and P2Y purinoceptors? Pharmacol. Ther. 1994, 64, 445–475. [Google Scholar] [CrossRef]
- Fredholm, B.B.; IJzerman, A.P.; Jacobson, K.A.; Linden, J.; Muller, C.E. International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and Classification of Adenosine Receptors--An Update. Pharmacol. Rev. 2011, 63, 1–34. [Google Scholar] [CrossRef]
- Welsh, T.G.; Kucenas, S. Purinergic signaling in oligodendrocyte development and function. J. Neurochem. 2018, 145, 6–18. [Google Scholar] [CrossRef]
- Di Virgilio, F.; Schmalzing, G.; Markwardt, F. The Elusive P2X7 Macropore. Trends Cell Biol. 2018, 28, 392–404. [Google Scholar] [CrossRef]
- Parravicini, C.; Lecca, D.; Marangon, D.; Coppolino, G.T.; Daniele, S.; Bonfanti, E.; Fumagalli, M.; Raveglia, L.; Martini, C.; Gianazza, E.; et al. Development of the first in vivo GPR17 ligand through an iterative drug discovery pipeline: A novel disease-modifying strategy for multiple sclerosis. PLoS ONE 2020, 15, e0231483. [Google Scholar] [CrossRef]
- Pérez-García, M.T.; Cidad, P.; López-López, J.R. The secret life of ion channels: Kv1.3 potassium channels and proliferation. Am. J. Physiol. Cell Physiol. 2018, 314, C27–C40. [Google Scholar] [CrossRef]
- Coppi, E.; Dettori, I.; Cherchi, F.; Bulli, I.; Venturini, M.; Lana, D.; Giovannini, M.G.; Pedata, F.; Pugliese, A.M. A2B Adenosine Receptors: When Outsiders May Become an Attractive Target to Treat Brain Ischemia or Demyelination. Int. J. Mol. Sci. 2020, 21, 9697. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Drug/s | Ion Channel/s | Preclinical/Clinical Trials | Effects |
|---|---|---|---|
| PF-01247324 | Nav1.8-selective blocker | EAE | Improves motor coordination and cerebellar-like symptoms [53] |
| Safinamide | Unselective Nav blocker | EAE | Protects from neurological deficit and prevents microglial activation [54,55] |
| Flecainide | Nav blocker | EAE | Preserves axonal integrity and electrical conduction, reduces disability scores [56,57] |
| Phenytoin | Nav blocker | EAE Phase II | Preserves axonal integrity and electrical conduction, reduces disability scores [56,57] |
| Neuroprotective in MS and related optic neuritis demyelination [58,59] | |||
| Lamotrigine | Nav blocker | EAE Phase II | Preserves axonal integrity and electrical conduction, reduces disability scores [56,57] |
| Protective effects in preclinical models but no effect on cerebral volume changes [58,60] | |||
| Side effects: reduction in white matter volume in secondary progressive MS patients [58,60] | |||
| Carbamazepine | Nav blocker | EAE Phase II | Improves paroximal dysarthria and ataxia in MS patients [61] |
| Pregabalin | Cav blocker | EAE Phase II | Reduces neuropathic pain in MS patients or EAE model. |
| Neuroprotective during excitotoxicity or neuroinflammation in EAE. | |||
| Side effects: reduces long-term potentiation in EAE mice and impairs memory function in MS patients [58,62,63,64,65] | |||
| Bepridil, Nitrendipine | L-type Cav1.x blocker | EAE | Reduces neuroinflammation and axonal pathology in EAE [66] |
| Nimodipine | L-type Cav1.2 blocker | EAE | Reduces EAE severity and demyelination [67] |
| Antiapoptotic effect by preventing intracellular Ca2+ overload [68,69] Anti-inflammatory effect by preventing microglial activation [70,71,72,73] | |||
| BgK-F6A | Kv1.1 selective blocker | CPZ model EAE | Enhances remyelination in CPZ model [74] |
| Reduces EAE severity [75] | |||
| Dalfampridine | IA blocker | EAE FDA approved in 2010 | Enhances axonal conduction in EAE [76] |
| Improves motor activity (walking) in MS patients [77] | |||
| Glatiramer Acetate | Modulate K+, Cl−, Ca2+ and TRP channels [52] | FDA approved in 1996 | Inhibits B lymphocytes maturation |
| Ion Channel | OPC/OL Culture In Vitro | In Vivo MS Animal Models | MS Patients | |
|---|---|---|---|---|
| Nav | Expression | TTX-sensitive Nav expressed in OPC, downregulated in OL [28,79,80,81,82,83,84,85,86,87,88] | Nav1.2 and Nav1.6 overexpressed in demyelinated sites (EAE) [53,90] Nav1.8 upregulated in cerebellar Purkinje cells (EAE) [91] | Nav1.8 upregulated in cerebellar Purkinje cells [91] Nav1.5 expressed in reactive astrocytes [92] Nav1.6 colocalizes with amyloid precursor protein in post-mortem tissues from secondary progressive MS brain [93] |
| Function | ↑ Migration and proliferation [94,95] | |||
| Cav | Expression | L-type (Cav1.2, Cav1.3) in OPC (downregulated in OL) [81,83,96,97,98] N-type (Cav2.2) in OL [99] | Cav1.2 involved in remyelination (CPZ) [100,101] Increased activity of L-type in demyelinated corpus callosum (CPZ) [102] Overexpression of N-type (Cav2.2) in active lesions (EAE) [103] | Overexpression of N-type (Cav2.2) in active lesion [103] |
| Function | L-type: ↑ Migration [104,105,106,107,108] ↑ Maturation [96,109]↑ Proliferation [86,96] | |||
| Kir | Expression | Kir4.1 OPC > OL [98,106,110] Kir2.1, Kir7.1 and TASK1 in OL [111,112,113] | Kir4.1: ↑ Myelination [114,115] | Kir4.1: High level in serum from MS patients [116] |
| Function | ↑ Maturation [115] | |||
| Kv | Expression | IA and IK OPC > OL [79,80,81,82,117,118] Kv1.2-6, Kv2.1, Kv7.2 in OPC [110,119,120] Kv4.2, Kv4.3, and Kv3.3. in OPC KCa1.1 in OPC [110] | Kv mislocalization in EAE animals [121] Kv1.2, Kv1.4, and Kv2.1 downregulated in EAE [122] | Kv mislocalization in post-mortem human MS lesions [121] |
| Function | ↑ Maturation [87,123] Kv1.3, Kv1.4: ↑ proliferation Kv1.6: ↓ proliferation | |||
| Ligand | Receptor | In Vitro OPC Cultures | In Vivo MS Animal Models/MS Paitens |
|---|---|---|---|
| Glutamate | AMPAR | ↓ proliferation and ↑ maturation [109,173,174,175] | ↑ myelination [176,177] |
| ↑ migration [107] | |||
| NMDAR | ↑ migration and ↑ differentiation [178] | ? | |
| GABA | GABAAR | ↓ myelination [179] | ↑ OPC proliferation |
| ↓ OPC differentiation [165] | |||
| ↑ myelination [165,180] | |||
| GABABR | ↑ proliferation and ↑ migration [181] | ? | |
| ↑ differentiation [182] | |||
| ATP/ADP | P2X7R | ↓ proliferation and ↑ migration [183] | OL damage and myelin loss during ischemia or neuroinflammation [184,185] |
| EAE-induced OL death by Ca2+ overloading [186] | |||
| P2Y1R | ↑ migration [187,188] | ? | |
| ↓ proliferation [188] | |||
| P2Y12R | ? | Downregulated in the cerebral cortex of post-mortem MS brains [113,189] | |
| Uracil-nucleotides | GPR17R | ↑ migration [87] | Overexpressed in active lesion of post-mortem MS brains [190] |
| ↑ differentiation [87,191] | |||
| Adenosine | A2AR/A2BR | ↓ differentiation [117,118] | ? |
| A1R | ↓ proliferation [192] | ↑ myelination [170] | |
| ↑ differentiation [192] | |||
| A3R | Induces OL apoptosis [170] | ? |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cherchi, F.; Bulli, I.; Venturini, M.; Pugliese, A.M.; Coppi, E. Ion Channels as New Attractive Targets to Improve Re-Myelination Processes in the Brain. Int. J. Mol. Sci. 2021, 22, 7277. https://doi.org/10.3390/ijms22147277
Cherchi F, Bulli I, Venturini M, Pugliese AM, Coppi E. Ion Channels as New Attractive Targets to Improve Re-Myelination Processes in the Brain. International Journal of Molecular Sciences. 2021; 22(14):7277. https://doi.org/10.3390/ijms22147277
Chicago/Turabian StyleCherchi, Federica, Irene Bulli, Martina Venturini, Anna Maria Pugliese, and Elisabetta Coppi. 2021. "Ion Channels as New Attractive Targets to Improve Re-Myelination Processes in the Brain" International Journal of Molecular Sciences 22, no. 14: 7277. https://doi.org/10.3390/ijms22147277
APA StyleCherchi, F., Bulli, I., Venturini, M., Pugliese, A. M., & Coppi, E. (2021). Ion Channels as New Attractive Targets to Improve Re-Myelination Processes in the Brain. International Journal of Molecular Sciences, 22(14), 7277. https://doi.org/10.3390/ijms22147277