
Prolyl Carboxypeptidase Mediates the C-Terminal Cleavage of (Pyr)-Apelin-13 in Human Umbilical Vein and Aortic Endothelial Cells

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
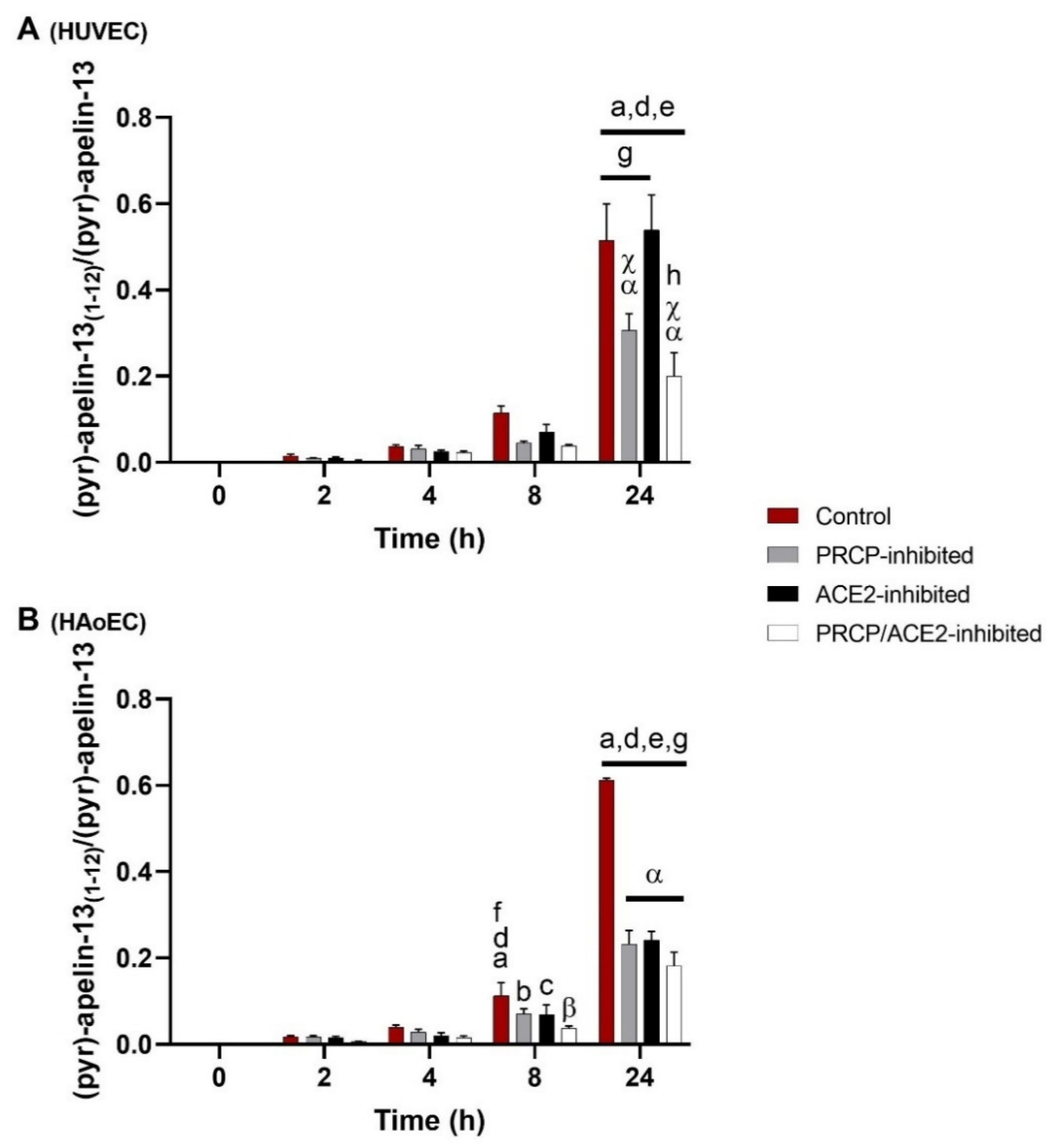
2.1. Cleavage of (Pyr)-Apelin-13 to (Pyr)-Apelin-13(1–12) Is PRCP-Dependent in HUVEC and PRCP- and ACE2-Dependent in HAoEC
2.2. α-MSH 1–13 Is Cleaved at Its C-Terminus in Function of Time in HUVEC and HAoEC
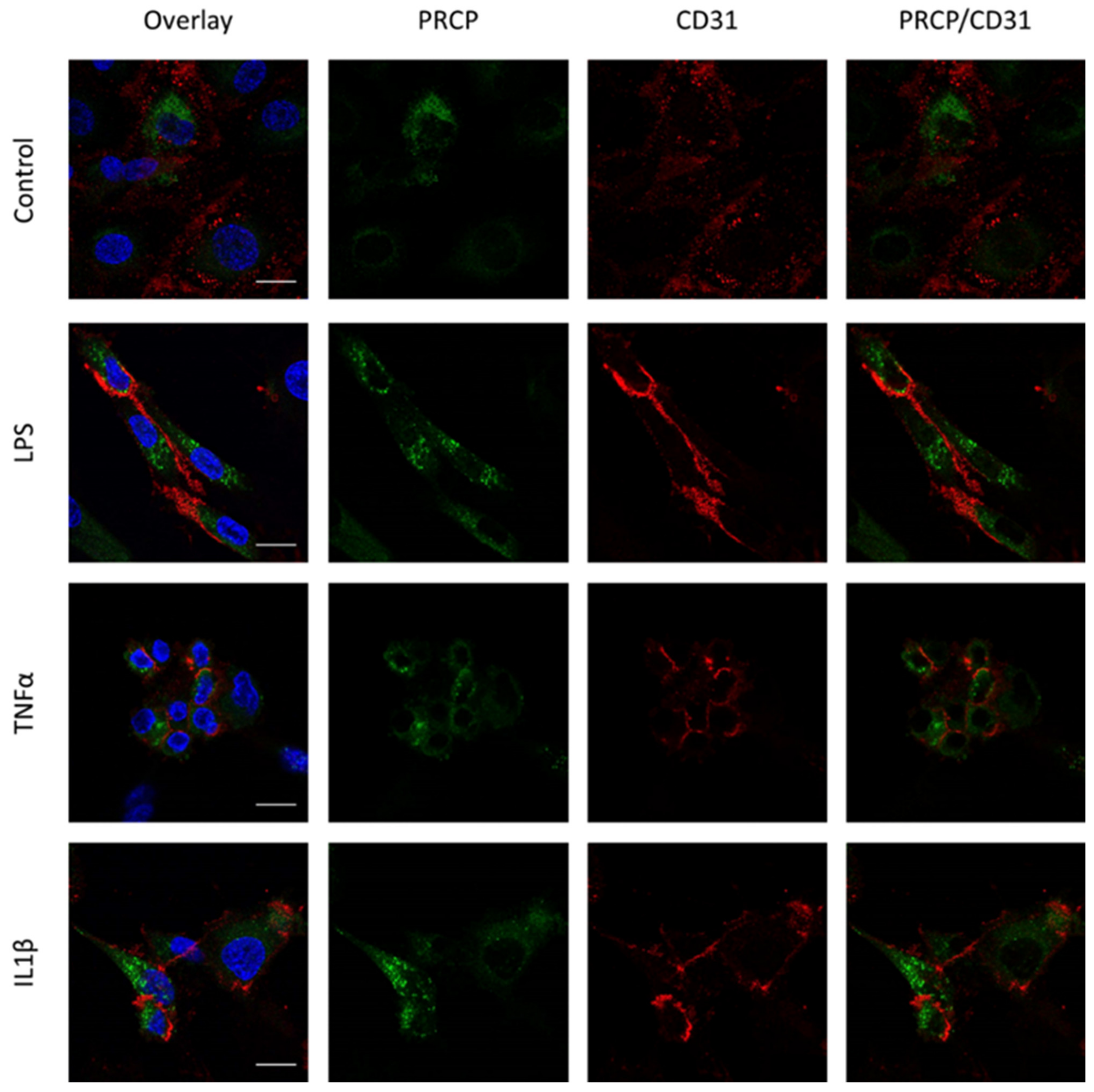
2.3. PRCP Is Observed in Lysosomes of HUVEC and HAoEC
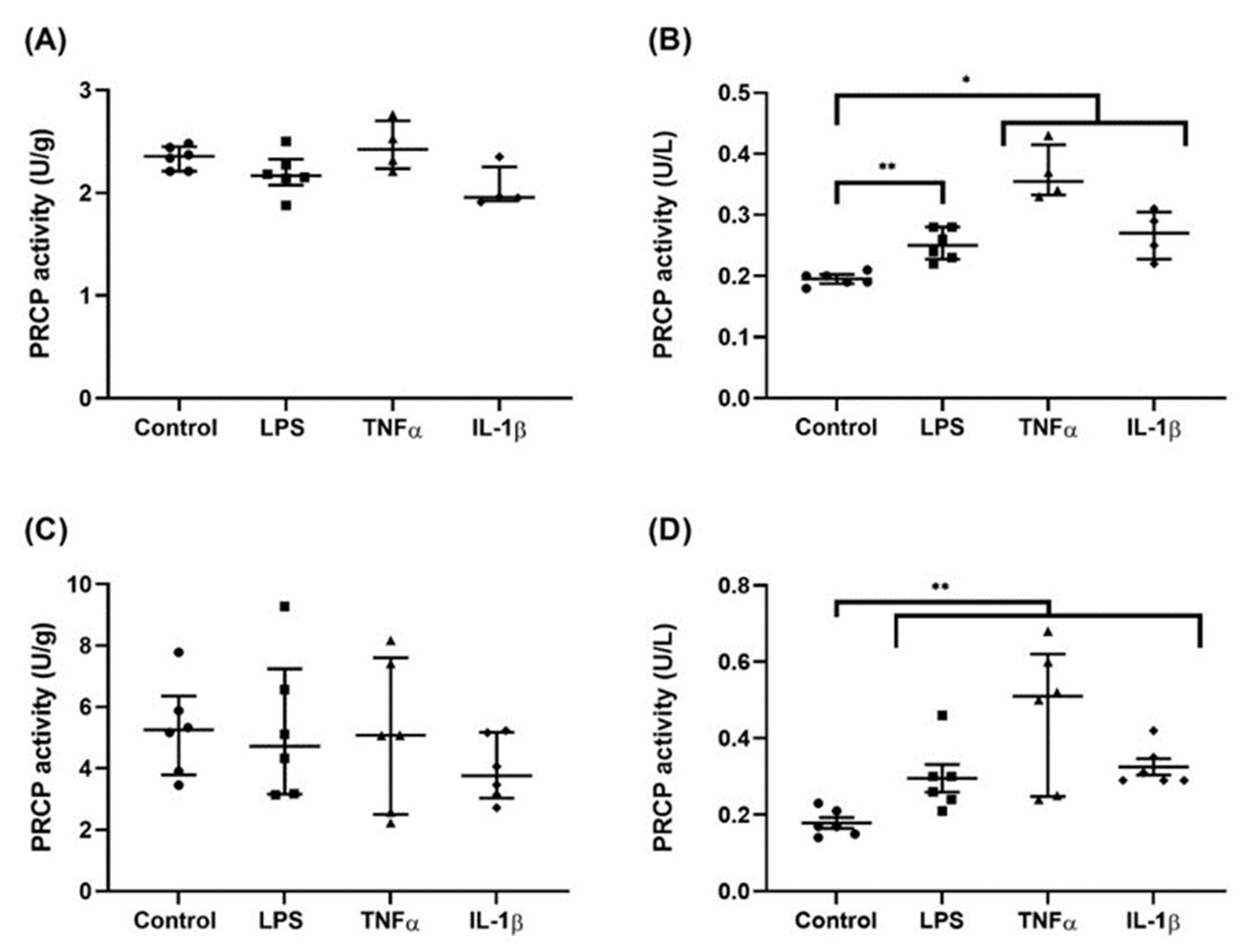
2.4. Stimulation with Pro-Inflammatory Agents Revealed an Increase in PRCP Activity in the Supernatant of HUVEC and HAoEC
3. Discussion
3.1. Cleavage of (Pyr)-Apelin-13 Is Attributed to PRCP and ACE2 and Differs between HUVEC and HAoEC
3.2. Enzyme Activity Levels May Account For Differences in Cleavage Patterns of (Pyr)-Apelin-13 in Both Types of Endothelial Cells
3.3. PRCP’s Location in Endothelial Cells Can Be Related to Its Role in the Cleavage of Circulating Peptides
3.4. The Studied Enzymes Do Not Seem to Be Involved to a Significant Extent in the Cleavage of α-MSH 1–13 in Human Endothelial Cells
3.5. Limitations
4. Materials and Methods
4.1. Inhibitors
4.2. Substrates
4.3. Cell Culture
4.4. Substrate Cleavage in Endothelial Cells
4.5. MALDI-TOF/TOF Analysis
4.6. Immunocytochemistry
4.7. Stimulation of HUVEC and HAoEC with Pro-Inflammatory Stimuli LPS, TNFα or IL-1β
4.8. PRCP Activity Measurement
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yang, H.Y.; Erdös, E.G.; Chiang, T.S. New enzymatic route for the inactivation of angiotensin. Nature 1968, 218, 1224–1226. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.Y.T.; Erdos, E.G.; Chiang, T.S.; Jenssen, T.A.; Rodgers, J.G. Characteristics of an enzyme that inactivates angiotensin II (Angiotensinase C). Biochem. Pharmacol. 1970, 19, 1201–1211. [Google Scholar] [CrossRef]
- Shariat-Madar, Z.; Mahdi, F.; Schmaier, A.H. Identification and characterization of prolylcarboxypeptidase as an endothelial cell prekallikrein activator. J. Biol. Chem. 2002, 277, 17962–17969. [Google Scholar] [CrossRef] [PubMed]
- Odya, C.E.; Marinkovic, D.V.; Hammon, K.J.; Stewart, T.A.; Erdös, E.G. Purification and properties of prolylcarboxypeptidase (angiotensinase C) from human kidney. J. Biol. Chem. 1978, 253, 5927–5931. [Google Scholar] [CrossRef]
- Adams, G.N.; LaRusch, G.A.; Stavrou, E.; Zhou, Y.; Nieman, M.T.; Jacobs, G.H.; Cui, Y.; Lu, Y.; Jain, M.K.; Mahdi, F.; et al. Murine prolylcarboxypeptidase depletion induces vascular dysfunction with hypertension and faster arterial thrombosis. Blood 2011, 117, 3929–3937. [Google Scholar] [CrossRef]
- Adams, G.N.; Stavrou, E.X.; Fang, C.; Merkulova, A.; Alaiti, M.A.; Nakajima, K.; Morooka, T.; Merkulov, S.; Larusch, G.A.; Simon, D.I.; et al. Prolylcarboxypeptidase promotes angiogenesis and vascular repair. Blood 2013, 122, 1522–1531. [Google Scholar] [CrossRef]
- Wang, L.; Feng, Y.; Zhang, Y.; Zhou, H.; Jiang, S.; Niu, T.; Wei, L.J.; Xu, X.; Xu, X.; Wang, X. Prolylcarboxypeptidase gene, chronic hypertension, and risk of preeclampsia. Am. J. Obstet. Gynecol. 2006, 195, 162–171. [Google Scholar] [CrossRef]
- Zhang, Y.; Hong, X.M.; Xing, H.X.; Li, J.P.; Huo, Y.; Xu, X.P. E112D polymorphism in the prolylcarboxypeptidase gene is associated with blood pressure response to benazepril in Chinese hypertensive patients. Chin. Med. J. 2009, 122, 2461–2465. [Google Scholar] [CrossRef]
- Wu, Y.; Yang, H.; Yang, B.; Yang, K.; Xiao, C. Association of polymorphisms in prolylcarboxypeptidase and chymase genes with essential hypertension in the Chinese Han population. JRAAS J. Renin Angiotensin Aldosterone Syst. 2013, 14, 263–270. [Google Scholar] [CrossRef]
- Wallingford, N.; Perroud, B.; Gao, Q.; Coppola, A.; Gyengesi, E.; Liu, Z.W.; Gao, X.B.; Diament, A.; Haus, K.A.; Shariat-Madar, Z.; et al. Prolylcarboxypeptidase regulates food intake by inactivating alpha-MSH in rodents. J. Clin. Invest. 2009, 119, 2291–2303. [Google Scholar] [CrossRef]
- Bruschetta, G.; Jin, S.; Kim, J.D.; Diano, S. Prolyl carboxypeptidase in Agouti-related Peptide neurons modulates food intake and body weight. Mol. Metab. 2018, 10, 28–38. [Google Scholar] [CrossRef]
- Kehoe, K.; Noels, H.; Theelen, W.; De Hert, E.; Xu, S.; Verrijken, A.; Arnould, T.; Fransen, E.; Hermans, N.; Lambeir, A.; et al. Prolyl carboxypeptidase activity in the circulation and its correlation with body weight and adipose tissue in lean and obese subjects. PLoS ONE 2018, 13, 1–15. [Google Scholar] [CrossRef]
- Xu, S.; Lind, L.; Zhao, L.; Lindahl, B.; Venge, P. Plasma prolylcarboxypeptidase (Angiotensinase C) is increased in obesity and diabetes mellitus and related to cardiovascular dysfunction. Clin. Chem. 2012, 58, 1110–1115. [Google Scholar] [CrossRef]
- Bruschetta, G.; Jin, S.; Liu, Z.W.; Kim, J.D.; Diano, S. MC4R Signaling in dorsal raphe nucleus controls feeding, anxiety, and depression. Cell Rep. 2020, 33, 108267. [Google Scholar] [CrossRef] [PubMed]
- Graham, T.H. Prolylcarboxypeptidase (PrCP) inhibitors and the therapeutic uses thereof: A patent review. Expert Opin. Ther. Pat. 2017, 27, 1077–1088. [Google Scholar] [CrossRef]
- Jeong, J.K.; Diano, S. Prolyl carboxypeptidase and its inhibitors in metabolism. Trends Endocrinol. Metab. 2013, 24, 1–14. [Google Scholar] [CrossRef][Green Version]
- Kehoe, K.; Van Elzen, R.; Verkerk, R.; Sim, Y.; Van der Veken, P.; Lambeir, A.-M.; De Meester, I. Prolyl carboxypeptidase purified from human placenta: Its characterization and identification as an apelin-cleaving enzyme. Biochim. Biophys. Acta Proteins Proteomics 2016, 1864, 1481–1488. [Google Scholar] [CrossRef]
- Tatemoto, K.; Hosoya, M.; Habata, Y.; Fujii, R.; Kakegawa, T.; Zou, M.X.; Kawamata, Y.; Fukusumi, S.; Hinuma, S.; Kitada, C.; et al. Isolation and characterization of a novel endogenous peptide ligand for the human APJ receptor. Biochem. Biophys. Res. Commun. 1998, 251, 471–476. [Google Scholar] [CrossRef]
- Maguire, J.J.; Kleinz, M.J.; Pitkin, S.L.; Davenport, A.P. [Pyr1]apelin-13 identified as the predominant apelin isoform in the human heart: Vasoactive mechanisms and inotropic action in disease. Hypertension 2009, 54, 598–604. [Google Scholar] [CrossRef]
- Zhen, E.Y.; Higgs, R.E.; Gutierrez, J.A. Pyroglutamyl apelin-13 identified as the major apelin isoform in human plasma. Anal. Biochem. 2013, 442, 1–9. [Google Scholar] [CrossRef]
- Cheng, J.; Luo, X.; Huang, Z.; Chen, L. Apelin/APJ system: A potential therapeutic target for endothelial dysfunction-related diseases. J. Cell. Physiol. 2019, 234, 12149–12160. [Google Scholar] [CrossRef] [PubMed]
- Murza, A.; Trân, K.; Bruneau-Cossette, L.; Lesur, O.; Auger-Messier, M.; Lavigne, P.; Sarret, P.; Marsault, É. Apelins, ELABELA, and their derivatives: Peptidic regulators of the cardiovascular system and beyond. Pept. Sci. 2018, 111, e24064. [Google Scholar] [CrossRef]
- Shin, K.; Kenward, C.; Rainey, J.K. Apelinergic system structure and function. Compr. Physiol. 2018, 8, 407–450. [Google Scholar] [CrossRef]
- Wysocka, M.B.; Pietraszek-Gremplewicz, K.; Nowak, D. The role of apelin in cardiovascular diseases, obesity and cancer. Front. Physiol. 2018, 9, 1–15. [Google Scholar] [CrossRef]
- Kidoya, H.; Takakura, N. Biology of the apelin-APJ axis in vascular formation. J. Biochem. 2012, 152, 125–131. [Google Scholar] [CrossRef]
- Chaves-Almagro, C.; Castan-Laurell, I.; Dray, C.; Knauf, C.; Valet, P.; Masri, B. Apelin receptors: From signaling to antidiabetic strategy. Eur. J. Pharmacol. 2015, 763, 149–159. [Google Scholar] [CrossRef]
- Murza, A.; Belleville, K.; Longpré, J.M.; Sarret, P.; Marsault, É. Stability and degradation patterns of chemically modified analogs of apelin-13 in plasma and cerebrospinal fluid. Biopolym. Pept. Sci. Sect. 2014, 102, 297–303. [Google Scholar] [CrossRef]
- Vickers, C.; Hales, P.; Kaushik, V.; Dick, L.; Gavin, J.; Tang, J.; Godbout, K.; Parsons, T.; Baronas, E.; Hsieh, F.; et al. Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J. Biol. Chem. 2002, 277, 14838–14843. [Google Scholar] [CrossRef]
- Yang, P.; Kuc, R.E.; Brame, A.L.; Dyson, A.; Singer, M.; Maguire, J.J. [Pyr1]Apelin-13(1–12) Is a biologically active ACE2 metabolite of the endogenous cardiovascular peptide [Pyr1]Apelin-13. Front. Neurosci. 2017, 11, 1–14. [Google Scholar] [CrossRef]
- Wang, W.; McKinnie, S.M.K.; Farhan, M.; Paul, M.; McDonald, T.; McLean, B.; Llorens-Cortes, C.; Hazra, S.; Murray, A.G.; Vederas, J.C.; et al. Angiotensin-converting enzyme 2 metabolizes and partially inactivates Pyr-Apelin-13 and Apelin-17: Physiological effects in the cardiovascular system. Hypertension 2016, 68, 365–377. [Google Scholar] [CrossRef]
- Medhurst, A.D.; Jennings, C.A.; Robbins, M.J.; Davis, R.P.; Ellis, C.; Winborn, K.Y.; Lawrie, K.W.M.; Hervieu, G.; Riley, G.; Bolaky, J.E.; et al. Pharmacological and immunohistochemical characterization of the APJ receptor and its endogenous ligand apelin. J. Neurochem. 2003, 84, 1162–1172. [Google Scholar] [CrossRef]
- El Messari, S.; Iturrioz, X.; Fassot, C.; De Mota, N.; Roesch, D.; Llorens-Cortes, C. Functional dissociation of apelin receptor signaling and endocytosis: Implications for the effects of apelin on arterial blood pressure. J. Neurochem. 2004, 90, 1290–1301. [Google Scholar] [CrossRef]
- Lee, D.K.; Saldivia, V.R.; Nguyen, T.; Cheng, R.; George, S.R.; O’Dowd, B.F. Modification of the terminal residue of apelin-13 antagonizes its hypotensive action. Endocrinology 2005, 146, 231–236. [Google Scholar] [CrossRef]
- Iturrioz, X.; Gerbier, R.; Leroux, V.; Alvear-perez, R.; Maigret, B.; Llorens-Cortes, C. By Interacting with the C-terminal Phe of Apelin, Phe255 and Trp259 in Helix VI of the Apelin receptor are critical for internalization. J. Biol. Chem. 2010, 285, 32627–32637. [Google Scholar] [CrossRef]
- Ceraudo, E.; Galanth, C.; Carpentier, E.; Banegas-Font, I.; Schonegge, A.M.; Alvear-Perez, R.; Iturrioz, X.; Bouvier, M.; Llorens-Cortes, C. Biased signaling favoring G i over β-arrestin promoted by an apelin fragment lacking the C-terminal phenylalanine. J. Biol. Chem. 2014, 289, 24599–24610. [Google Scholar] [CrossRef]
- Gerbier, R.; Leroux, V.; Couvineau, P.; Alvear-perez, R. New structural insights into the apelin receptor: Identification of key residues for apelin binding. FASEB J. 2015, 29, 314–322. [Google Scholar] [CrossRef]
- Murza, A.; Besserer-Offroy, E.; Côté, J.; Bérubé, P.; Longpré, J.M.; Dumaine, R.; Lesur, O.; Auger-Messier, M.; Leduc, R.; Sarret, P.; et al. C-terminal modifications of apelin-13 significantly change ligand binding, receptor signaling, and hypotensive action. J. Med. Chem. 2015, 58, 2431–2440. [Google Scholar] [CrossRef]
- Besserer-Offroy, É.; Bérubé, P.; Côté, J.; Murza, A.; Longpré, J.M.; Dumaine, R.; Lesur, O.; Auger-Messier, M.; Leduc, R.; Marsault, É.; et al. The hypotensive effect of activated apelin receptor is correlated with β-arrestin recruitment. Pharmacol. Res. 2018, 131, 7–16. [Google Scholar] [CrossRef]
- Murza, A.; Parent, A.; Besserer-Offroy, E.; Tremblay, H.; Karadereye, F.; Beaudet, N.; Leduc, R.; Sarret, P.; Marsault, É. Elucidation of the structure-activity relationships of apelin: Influence of unnatural amino acids on binding, signaling, and plasma stability. ChemMedChem 2012, 7, 318–325. [Google Scholar] [CrossRef]
- Shariat-Madar, Z.; Mahdi, F.; Schmaier, A.H. Recombinant prolylcarboxypeptidase activates plasma prekallikrein. Blood 2004, 103, 4554–4561. [Google Scholar] [CrossRef]
- Kleinz, M.J.; Skepper, J.N.; Davenport, A.P. Immunocytochemical localisation of the apelin receptor, APJ, to human cardiomyocytes, vascular smooth muscle and endothelial cells. Regul. Pept. 2005, 126, 233–240. [Google Scholar] [CrossRef]
- Strohbach, A.; Pennewitz, M.; Glaubitz, M.; Palankar, R.; Groß, S.; Lorenz, F.; Materzok, I.; Rong, A.; Busch, M.C.; Felix, S.B.; et al. The apelin receptor influences biomechanical and morphological properties of endothelial cells. J. Cell. Physiol. 2018, 233, 6250–6261. [Google Scholar] [CrossRef] [PubMed]
- Odya, C.E.; Erdos, E.G. Human prolylcarboxypeptidase. Methods Enzymol. 1981, 80, 460–466. [Google Scholar]
- Yin, X.; Bern, M.; Xing, Q.; Ho, J.; Viner, R.; Mayr, M. Glycoproteomic analysis of the secretome of human endothelial cells. Mol. Cell. Proteomics 2013, 12, 956–978. [Google Scholar] [CrossRef] [PubMed]
- Ngo, M.-L.; Mahdi, F.; Kolte, D.; Shariat-Madar, Z. Upregulation of prolylcarboxypeptidase (PRCP) in lipopolysaccharide (LPS) treated endothelium promotes inflammation. J. Inflamm. 2009, 6, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Garcia-Calvo, M.; Pinto, S.; Lombardo, M.; Feng, Z.; Bender, K.; Pryor, K.D.; Bhatt, U.R.; Chabin, R.M.; Geissler, W.M.; et al. Design and synthesis of prolylcarboxypeptidase (PrCP) inhibitors to validate PrCP as a potential target for obesity. J. Med. Chem. 2010, 53, 7251–7263. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Sexton, D.J.; Skogerson, K.; Devlin, M.; Smith, R.; Sanyal, I.; Parry, T.; Kent, R.; Enright, J.; Wu, Q.; et al. Novel Peptide Inhibitors of Angiotensin-converting Enzyme 2. J. Biol. Chem. 2003, 278, 15532–15540. [Google Scholar] [CrossRef] [PubMed]
- Van Elzen, R.; Schoenmakers, E.; Brandt, I.; Van Der Veken, P.; Lambeir, A.M. Ligand-induced conformational changes in prolyl oligopeptidase: A kinetic approach. Protein Eng. Des. Sel. 2017, 30, 219–226. [Google Scholar] [CrossRef]
- Nyimanu, D.; Kay, R.G.; Sulentic, P.; Kuc, R.E.; Ambery, P.; Jermutus, L.; Reimann, F.; Gribble, F.M.; Cheriyan, J.; Maguire, J.J.; et al. Development and validation of an LC-MS/MS method for detection and quantification of in vivo derived metabolites of [Pyr1]apelin-13 in humans. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef]
- Tipnis, S.R.; Hooper, N.M.; Hyde, R.; Karran, E.; Christie, G.; Turner, A.J. A human homolog of angiotensin-converting enzyme: Cloning and functional expression as a captopril-insensitive carboxypeptidase. J. Biol. Chem. 2000, 275, 33238–33243. [Google Scholar] [CrossRef]
- Lee, D.K.; Ferguson, S.S.G.; George, S.R.; O’Dowd, B.F. The fate of the internalized apelin receptor is determined by different isoforms of apelin mediating differential interaction with β-arrestin. Biochem. Biophys. Res. Commun. 2010, 395, 185–189. [Google Scholar] [CrossRef]
- Pope, G.R.; Tilve, S.; McArdle, C.A.; Lolait, S.J.; O’Carroll, A.M. Agonist-induced internalization and desensitization of the apelin receptor. Mol. Cell. Endocrinol. 2016, 437, 108–119. [Google Scholar] [CrossRef]
- Inpanathan, S.; Botelho, R.J. The lysosome signaling platform: Adapting with the times. Front. Cell Dev. Biol. 2019, 7, 1–22. [Google Scholar] [CrossRef]
- Perroud, B.; Alvarado, R.J.; Espinal, G.M.; Morado, A.R.; Phinney, B.S.; Warden, C.H. In vivo multiplex quantitative analysis of 3 forms of alpha melanocyte stimulating hormone in pituitary of prolyl endopeptidase deficient mice. Mol. Brain 2009, 2. [Google Scholar] [CrossRef] [PubMed]
- Venäläinen, J.I.; Garcia-Horsman, J.A.; Forsberg, M.M.; Jalkanen, A.; Wallén, E.A.A.; Jarho, E.M.; Christiaans, J.A.M.; Gynther, J.; Männistö, P.T. Binding kinetics and duration of in vivo action of novel prolyl oligopeptidase inhibitors. Biochem. Pharmacol. 2006, 71, 683–692. [Google Scholar] [CrossRef]
- Svarcbahs, R.; Jäntti, M.; Kilpeläinen, T.; Julku, U.H.; Urvas, L.; Kivioja, S.; Norrbacka, S.; Myöhänen, T.T. Prolyl oligopeptidase inhibition activates autophagy via protein phosphatase 2A. Pharmacol. Res. 2020, 151, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Matheeussen, V.; Lambeir, A.M.; Jungraithmayr, W.; Gomez, N.; Mc Entee, K.; Van der Veken, P.; Scharpé, S.; De Meester, I. Method comparison of dipeptidyl peptidase IV activity assays and their application in biological samples containing reversible inhibitors. Clin. Chim. Acta 2012, 413, 456–462. [Google Scholar] [CrossRef]
- Strohalm, M.; Hassman, M.; Kosata, B.; Kodicek, M. mMass data miner: An open source alternative for mass spectrometric data analysis. Rapid Commun. Mass Spectrom. 2008, 22, 905–908. [Google Scholar] [CrossRef]
- Makó, V.; Czúcz, J.; Weiszhár, Z.; Herczenik, E.; Matkó, J.; Prohászka, Z.; Cervenak, L. Proinflammatory activation pattern of human umbilical vein endothelial cells induced by IL-1β, TNF-α, and LPS. Cytom. Part A 2010, 77, 962–970. [Google Scholar] [CrossRef] [PubMed]
- Kulhankova, K.; Kinney, K.J.; Stach, J.M.; Gourronc, F.A.; Grumbach, I.M.; Klingelhutz, A.J.; Salgado-Pabon, W. The Superantigen Toxic Shock Syndrome Toxin 1 Alters Human Aortic Endothelial Cell Function. Infect. Immun. 2018, 86, 1–16. [Google Scholar] [CrossRef]
- Kehoe, K.; Verkerk, R.; Sim, Y.; Waumans, Y.; Van Der Veken, P.; Lambeir, A.M.; De Meester, I. Validation of a specific prolylcarboxypeptidase activity assay and its suitability for plasma and serum measurements. Anal. Biochem. 2013, 443, 232–239. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Hert, E.; Bracke, A.; Pintelon, I.; Janssens, E.; Lambeir, A.-M.; Van Der Veken, P.; De Meester, I. Prolyl Carboxypeptidase Mediates the C-Terminal Cleavage of (Pyr)-Apelin-13 in Human Umbilical Vein and Aortic Endothelial Cells. Int. J. Mol. Sci. 2021, 22, 6698. https://doi.org/10.3390/ijms22136698
De Hert E, Bracke A, Pintelon I, Janssens E, Lambeir A-M, Van Der Veken P, De Meester I. Prolyl Carboxypeptidase Mediates the C-Terminal Cleavage of (Pyr)-Apelin-13 in Human Umbilical Vein and Aortic Endothelial Cells. International Journal of Molecular Sciences. 2021; 22(13):6698. https://doi.org/10.3390/ijms22136698
Chicago/Turabian StyleDe Hert, Emilie, An Bracke, Isabel Pintelon, Eline Janssens, Anne-Marie Lambeir, Pieter Van Der Veken, and Ingrid De Meester. 2021. "Prolyl Carboxypeptidase Mediates the C-Terminal Cleavage of (Pyr)-Apelin-13 in Human Umbilical Vein and Aortic Endothelial Cells" International Journal of Molecular Sciences 22, no. 13: 6698. https://doi.org/10.3390/ijms22136698
APA StyleDe Hert, E., Bracke, A., Pintelon, I., Janssens, E., Lambeir, A.-M., Van Der Veken, P., & De Meester, I. (2021). Prolyl Carboxypeptidase Mediates the C-Terminal Cleavage of (Pyr)-Apelin-13 in Human Umbilical Vein and Aortic Endothelial Cells. International Journal of Molecular Sciences, 22(13), 6698. https://doi.org/10.3390/ijms22136698