
Inhibition of the PI3K/mTOR Pathway in Breast Cancer to Enhance Response to Immune Checkpoint Inhibitors in Breast Cancer

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Combination of PI3Kα Isoform-Specific Inhibitor, Alpelisib, and Paclitaxel Induces Granulocyte Responses but Does Not Synergizes with Immunotherapy
2.2. Alpelisib Induces Beneficial, but Not Durable, Granulocyte Responses in Breast Cancer Patients
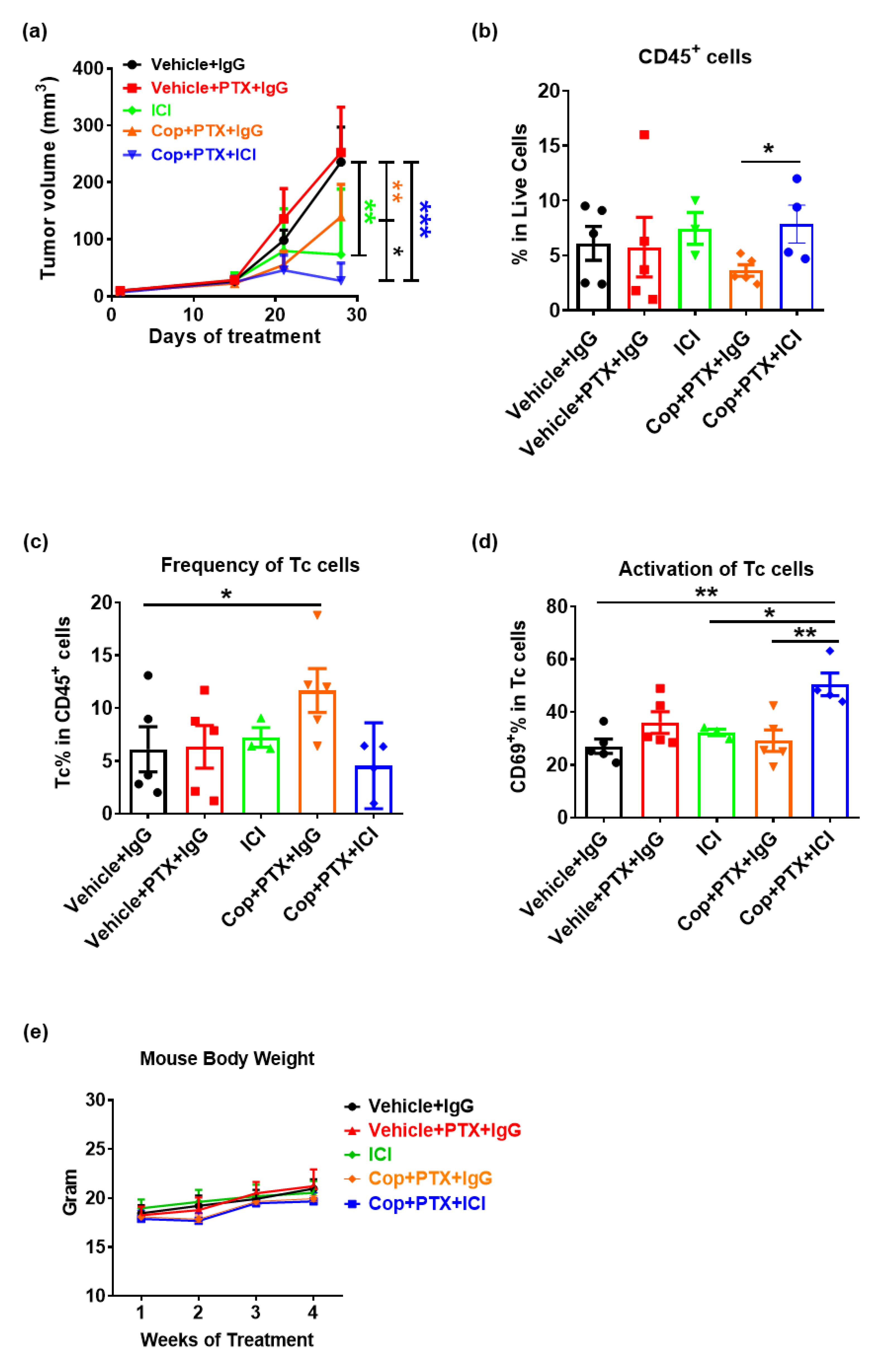
2.3. Pan-PI3K Inhibitor, Copanlisib, Plus Immune Therapy Produced Partial Remission of PyMT Breast Tumor Growth
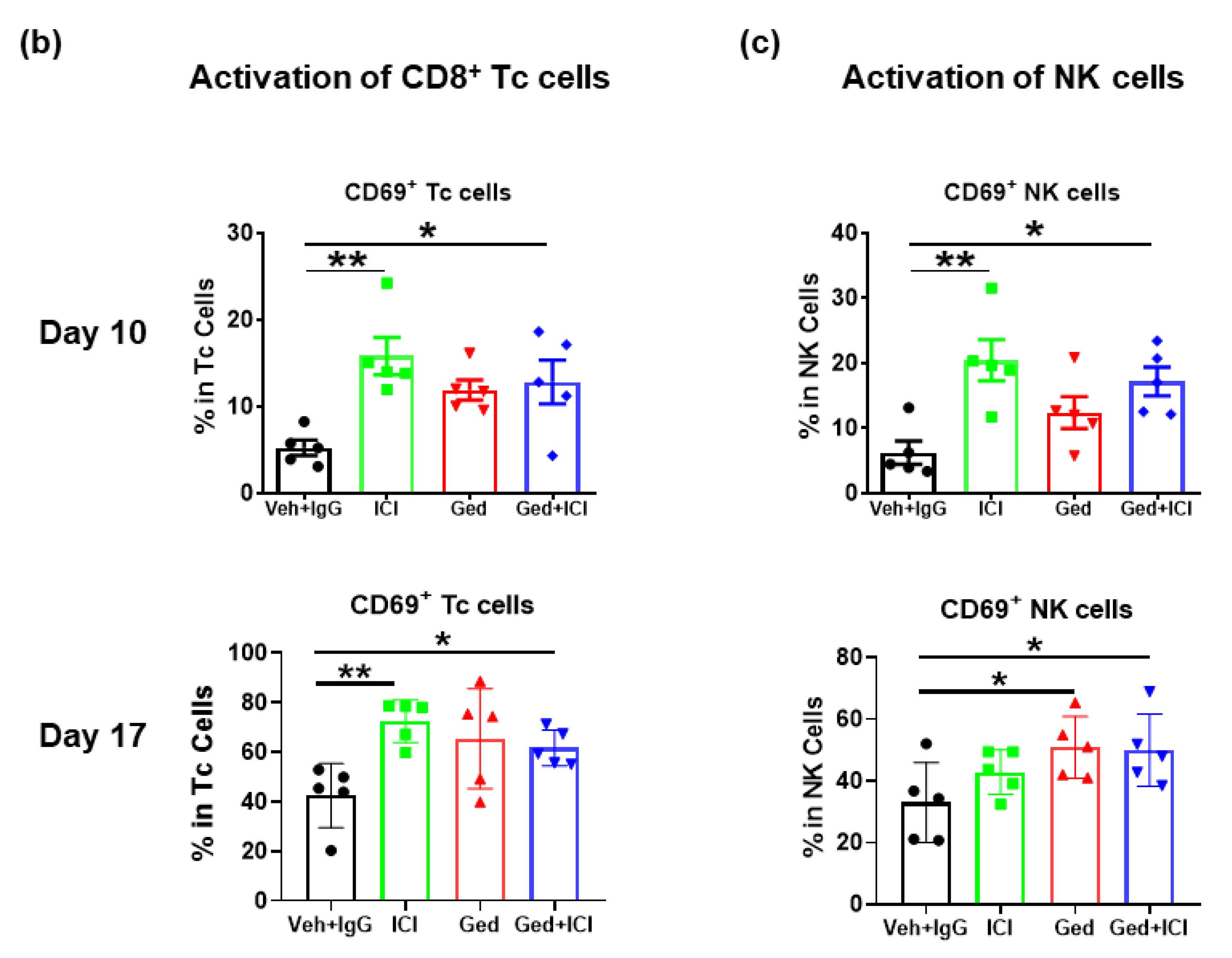
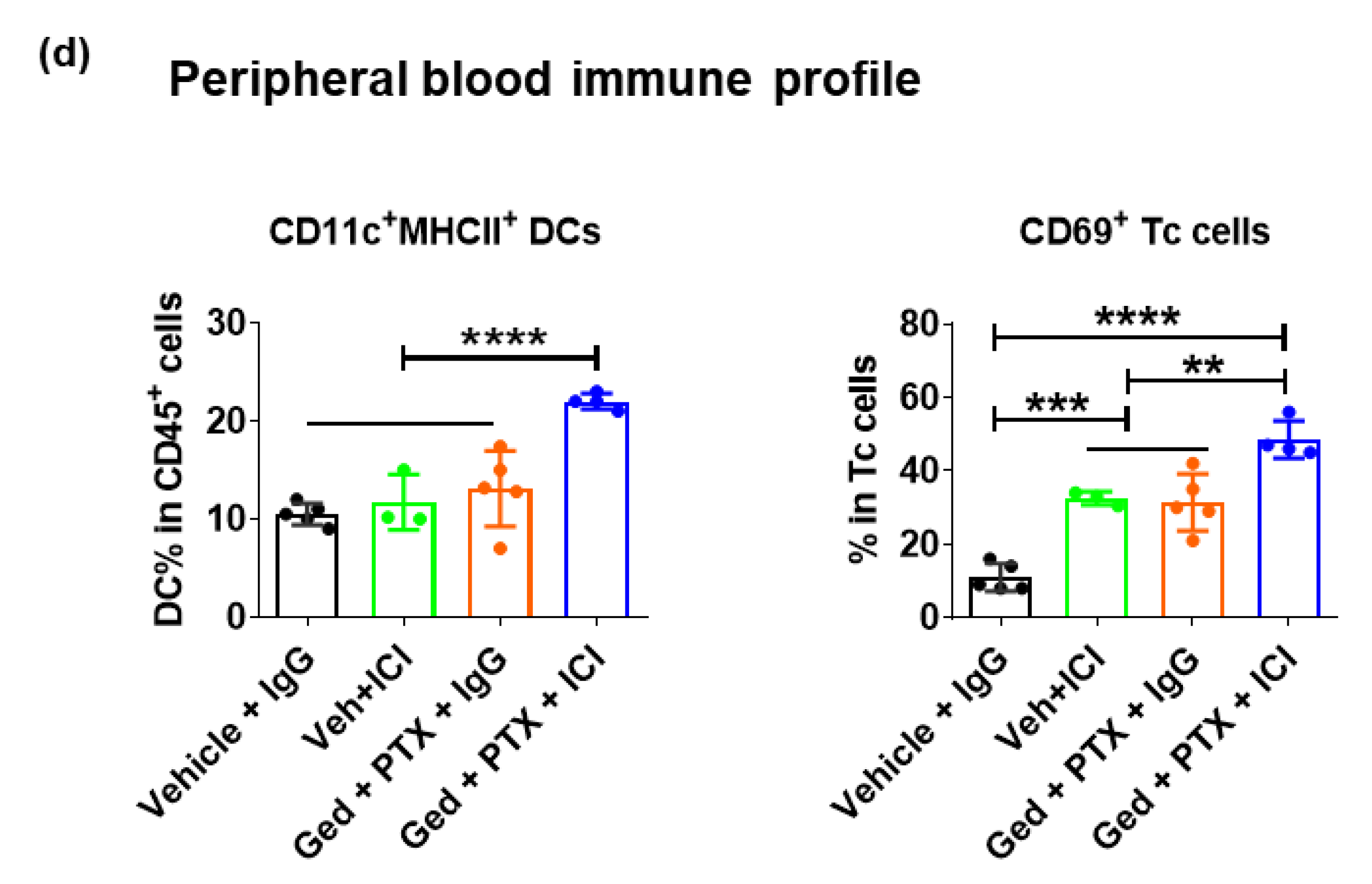
2.4. Dual PI3K/mTOR Inhibitor, Gedatolisib, Plus Immune Therapy Effectively Stopped PyMT Breast Tumor Growth
2.5. Gedatolisib Plus PTX and Immune Therapy Resulted in 60% Complete Regression of PyMT Breast Tumors
2.6. PIK3CD and PIK3CG Are Preferentially Expressed in the Lymphocytes of Triple-Negative BC Patient Tumors
3. Discussion
4. Methods
4.1. Patients and Samples
4.2. Mouse Studies
4.3. Multicolor Flow Cytometry
4.4. Analysis of Publicly Available Dataset
4.5. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ellis, M.J.; Perou, C.M. The genomic landscape of breast cancer as a therapeutic roadmap. Cancer Discov. 2013, 3, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Vanhaesebroeck, B.; Guillermet-Guibert, J.; Graupera, M.; Bilanges, B. The emerging mechanisms of isoform-specific PI3K signalling. Nat. Rev. Mol. Cell Biol. 2010, 11, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Costa, R.L.B.; Han, H.S.; Gradishar, W.J. Targeting the PI3K/AKT/mTOR pathway in triple-negative breast cancer: A review. Breast Cancer Res. Treat 2018, 169, 397–406. [Google Scholar] [CrossRef]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef]
- Gordon, S.R.; Maute, R.L.; Dulken, B.W.; Hutter, G.; George, B.M.; McCracken, M.N.; Gupta, R.; Tsai, J.M.; Sinha, R.; Corey, D.; et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 2017, 545, 495–499. [Google Scholar] [CrossRef]
- Bates, J.P.; Derakhshandeh, R.; Jones, L.; Webb, T.J. Mechanisms of immune evasion in breast cancer. BMC Cancer 2018, 18, 556. [Google Scholar] [CrossRef]
- Pfirschke, C.; Engblom, C.; Rickelt, S.; Cortez-Retamozo, V.; Garris, C.; Pucci, F.; Yamazaki, T.; Poirier-Colame, V.; Newton, A.; Redouane, Y.; et al. Immunogenic Chemotherapy Sensitizes Tumors to Checkpoint Blockade Therapy. Immunity 2016, 44, 343–354. [Google Scholar] [CrossRef]
- Schmid, P.; Rugo, H.S.; Adams, S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Dieras, V.; Henschel, V.; Molinero, L.; Chui, S.Y.; et al. Atezolizumab plus nab-paclitaxel as first-line treatment for unresectable, locally advanced or metastatic triple-negative breast cancer (IMpassion130): Updated efficacy results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2020, 21, 44–59. [Google Scholar] [CrossRef]
- Morgillo, F.; Della Corte, C.M.; Diana, A.; Mauro, C.D.; Ciaramella, V.; Barra, G.; Belli, V.; Franzese, E.; Bianco, R.; Maiello, E.; et al. Phosphatidylinositol 3-kinase (PI3Kalpha)/AKT axis blockade with taselisib or ipatasertib enhances the efficacy of anti-microtubule drugs in human breast cancer cells. Oncotarget 2017, 8, 76479–76491. [Google Scholar] [CrossRef]
- Sai, J.; Owens, P.; Novitskiy, S.V.; Hawkins, O.E.; Vilgelm, A.E.; Yang, J.; Sobolik, T.; Lavender, N.; Johnson, A.C.; McClain, C.; et al. PI3K inhibition reduces mammary tumor growth and facilitates antitumor immunity and anti-PD1 responses. Clin. Cancer Res. 2017, 23, 3371–3384. [Google Scholar] [CrossRef]
- Benefit mixed with caution for buparlisib. Cancer Discov. 2017, 7, 121. [CrossRef][Green Version]
- Jovanovic, B.; Mayer, I.A.; Mayer, E.L.; Abramson, V.G.; Bardia, A.; Sanders, M.E.; Kuba, M.G.; Estrada, M.V.; Beeler, J.S.; Shaver, T.M.; et al. A randomized phase II neoadjuvant study of cisplatin, paclitaxel with or without everolimus in patients with stage II/III triple-negative breast cancer (TNBC): Responses and long-term outcome correlated with increased frequency of DNA damage response gene mutations, TNBC subtype, AR status, and Ki67. Clin. Cancer Res. 2017, 23, 4035–4045. [Google Scholar] [CrossRef] [PubMed]
- Juric, D.; Janku, F.; Rodon, J.; Burris, H.A.; Mayer, I.A.; Schuler, M.; Seggewiss-Bernhardt, R.; Gil-Martin, M.; Middleton, M.R.; Baselga, J.; et al. Alpelisib plus fulvestrant in PIK3CA-altered and PIK3CA-wild-type estrogen receptor-positive advanced breast cancer: A phase 1b clinical trial. JAMA Oncol. 2019, 5, e184475. [Google Scholar] [CrossRef] [PubMed]
- Teo, Z.L.; Versaci, S.; Dushyanthen, S.; Caramia, F.; Savas, P.; Mintoff, C.P.; Zethoven, M.; Virassamy, B.; Luen, S.J.; McArthur, G.A.; et al. Combined CDK4/6 and PI3Kalpha inhibition is synergistic and immunogenic in triple-negative breast cancer. Cancer Res. 2017, 77, 6340–6352. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.B.; Nebhan, C.A.; Yang, J.; Starnes, L.S.; Yan, C.; Vilgelm, A.E.; Chen, S.C.; Dan Ayers, G.; Abramson, V.; Mayer, I.A.; et al. Correlative studies investigating effects of PI3K inhibition on peripheral leukocytes in metastatic breast cancer: Potential implications for immunotherapy. Breast Cancer Res. Treat 2020, 184, 357–364. [Google Scholar] [CrossRef] [PubMed]
- De Henau, O.; Rausch, M.; Winkler, D.; Campesato, L.F.; Liu, C.; Cymerman, D.H.; Budhu, S.; Ghosh, A.; Pink, M.; Tchaicha, J.; et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kgamma in myeloid cells. Nature 2016, 539, 443–447. [Google Scholar] [CrossRef]
- Magagnoli, M.; Carlo-Stella, C.; Santoro, A. Copanlisib for the treatment of adults with relapsed follicular lymphoma. Expert Rev. Clin. Pharmacol. 2020, 13, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Mallon, R.; Feldberg, L.R.; Lucas, J.; Chaudhary, I.; Dehnhardt, C.; Santos, E.D.; Chen, Z.; dos Santos, O.; Ayral-Kaloustian, S.; Venkatesan, A.; et al. Antitumor efficacy of PKI-587, a highly potent dual PI3K/mTOR kinase inhibitor. Clin. Cancer Res. 2011, 17, 3193–3203. [Google Scholar] [CrossRef]
- Colombo, I.; Genta, S.; Martorana, F.; Guidi, M.; Samartzis, E.S.P.; Brandt, S.; Gaggetta, S.; Moser, L.; Pascale, M.R.; Terrot, T.; et al. Phase I dose-escalation study of the dual PI3K/mTORC1/2 inhibitor Gedatolisib (PF-05212384) in combination with paclitaxel (P) and carboplatin (C) in patients (pts) with advanced solid tumours. Ann. Oncol. 2020, 31, S487. [Google Scholar] [CrossRef]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Sorlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef]
- Duraiswamy, J.; Ibegbu, C.C.; Masopust, D.; Miller, J.D.; Araki, K.; Doho, G.H.; Tata, P.; Gupta, S.; Zilliox, M.J.; Nakaya, H.I.; et al. Phenotype, function, and gene expression profiles of programmed death-1(hi) CD8 T cells in healthy human adults. J. Immunol. 2011, 186, 4200–4212. [Google Scholar] [CrossRef]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef]
- Nicolas-Avila, J.A.; Adrover, J.M.; Hidalgo, A. Neutrophils in homeostasis, immunity, and cancer. Immunity 2017, 46, 15–28. [Google Scholar] [CrossRef]
- Mantovani, A.; Cassatella, M.A.; Costantini, C.; Jaillon, S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat. Rev. Immunol. 2011, 11, 519–531. [Google Scholar] [CrossRef] [PubMed]
- Ali, K.; Soond, D.R.; Pineiro, R.; Hagemann, T.; Pearce, W.; Lim, E.L.; Bouabe, H.; Scudamore, C.L.; Hancox, T.; Maecker, H.; et al. Inactivation of PI(3)K p110delta breaks regulatory T-cell-mediated immune tolerance to cancer. Nature 2014, 510, 407–411. [Google Scholar] [CrossRef]
- Schmid, M.C.; Avraamides, C.J.; Dippold, H.C.; Franco, I.; Foubert, P.; Ellies, L.G.; Acevedo, L.M.; Manglicmot, J.R.; Song, X.; Wrasidlo, W.; et al. Receptor tyrosine kinases and TLR/IL1Rs unexpectedly activate myeloid cell PI3kgamma, a single convergent point promoting tumor inflammation and progression. Cancer Cell 2011, 19, 715–727. [Google Scholar] [CrossRef]
- Carnevalli, L.S.; Sinclair, C.; Taylor, M.A.; Gutierrez, P.M.; Langdon, S.; Coenen-Stass, A.M.L.; Mooney, L.; Hughes, A.; Jarvis, L.; Staniszewska, A.; et al. PI3Kalpha/delta inhibition promotes anti-tumor immunity through direct enhancement of effector CD8(+) T-cell activity. J. Immunother. Cancer 2018, 6, 158. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef]
- Ooms, L.M.; Binge, L.C.; Davies, E.M.; Rahman, P.; Conway, J.R.; Gurung, R.; Ferguson, D.T.; Papa, A.; Fedele, C.G.; Vieusseux, J.L.; et al. The inositol polyphosphate 5-phosphatase PIPP regulates AKT1-dependent breast cancer growth and metastasis. Cancer Cell 2015, 28, 155–169. [Google Scholar] [CrossRef] [PubMed]
- Miles, D.W.; Gligorov, J.; André, F.; Cameron, D.; Schneeweiss, A.; Barrios, C.H.; Xu, B.; Wardley, A.M.; Kaen, D.; Andrade, L.; et al. LBA15 Primary results from IMpassion131, a double-blind placebo-controlled randomised phase III trial of first-line paclitaxel (PAC) ± atezolizumab (atezo) for unresectable locally advanced/metastatic triple-negative breast cancer (mTNBC). Ann. Oncol. 2020, 31, S1147–S1148. [Google Scholar] [CrossRef]
- Swamydas, M.; Lionakis, M.S. Isolation, purification and labeling of mouse bone marrow neutrophils for functional studies and adoptive transfer experiments. J. Vis. Exp. 2013, 10, e50586. [Google Scholar] [CrossRef]
- Yang, J.; Yan, C.; Vilgelm, A.E.; Chen, S.C.; Ayers, G.D.; Johnson, C.A.; Richmond, A. Targeted deletion of CXCR2 in myeloid cells alters the tumor immune environment to improve antitumor immunity. Cancer Immunol. Res. 2021, 9, 200–213. [Google Scholar] [CrossRef]
- Chung, W.; Eum, H.H.; Lee, H.O.; Lee, K.M.; Lee, H.B.; Kim, K.T.; Ryu, H.S.; Kim, S.; Lee, J.E.; Park, Y.H.; et al. Single-cell RNA-seq enables comprehensive tumour and immune cell profiling in primary breast cancer. Nat. Commun. 2017, 8, 15081. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, C.; Yang, J.; Saleh, N.; Chen, S.-C.; Ayers, G.D.; Abramson, V.G.; Mayer, I.A.; Richmond, A. Inhibition of the PI3K/mTOR Pathway in Breast Cancer to Enhance Response to Immune Checkpoint Inhibitors in Breast Cancer. Int. J. Mol. Sci. 2021, 22, 5207. https://doi.org/10.3390/ijms22105207
Yan C, Yang J, Saleh N, Chen S-C, Ayers GD, Abramson VG, Mayer IA, Richmond A. Inhibition of the PI3K/mTOR Pathway in Breast Cancer to Enhance Response to Immune Checkpoint Inhibitors in Breast Cancer. International Journal of Molecular Sciences. 2021; 22(10):5207. https://doi.org/10.3390/ijms22105207
Chicago/Turabian StyleYan, Chi, Jinming Yang, Nabil Saleh, Sheau-Chiann Chen, Gregory D. Ayers, Vandana G. Abramson, Ingrid A. Mayer, and Ann Richmond. 2021. "Inhibition of the PI3K/mTOR Pathway in Breast Cancer to Enhance Response to Immune Checkpoint Inhibitors in Breast Cancer" International Journal of Molecular Sciences 22, no. 10: 5207. https://doi.org/10.3390/ijms22105207
APA StyleYan, C., Yang, J., Saleh, N., Chen, S.-C., Ayers, G. D., Abramson, V. G., Mayer, I. A., & Richmond, A. (2021). Inhibition of the PI3K/mTOR Pathway in Breast Cancer to Enhance Response to Immune Checkpoint Inhibitors in Breast Cancer. International Journal of Molecular Sciences, 22(10), 5207. https://doi.org/10.3390/ijms22105207