
Characterization of a Mouse Model of Alzheimer’s Disease Expressing Aβ4-42 and Human Mutant Tau


Abstract
1. Introduction
2. Results
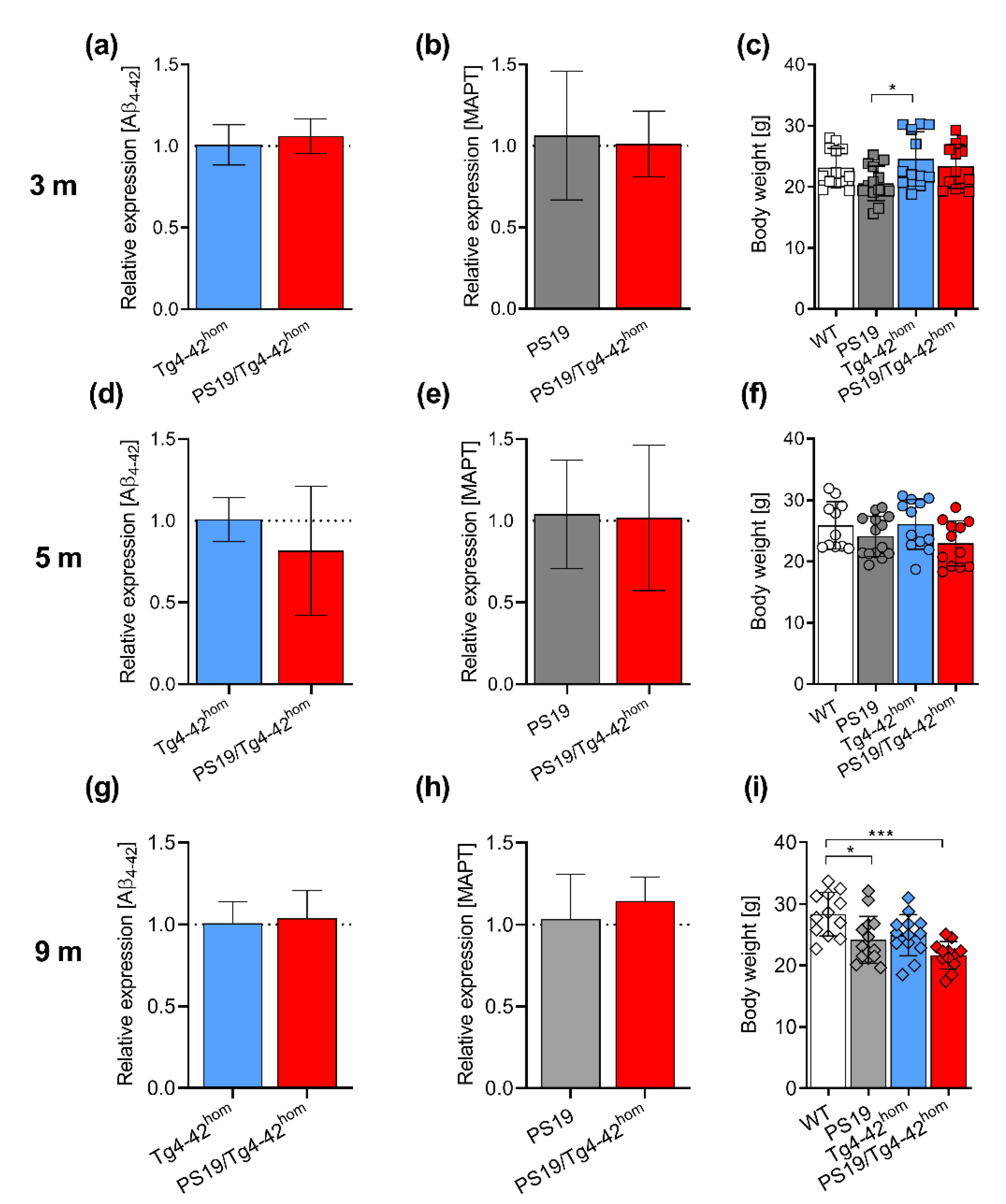
2.1. Transgene Expression and Weight Assessment in PS19/Tg4–42hom
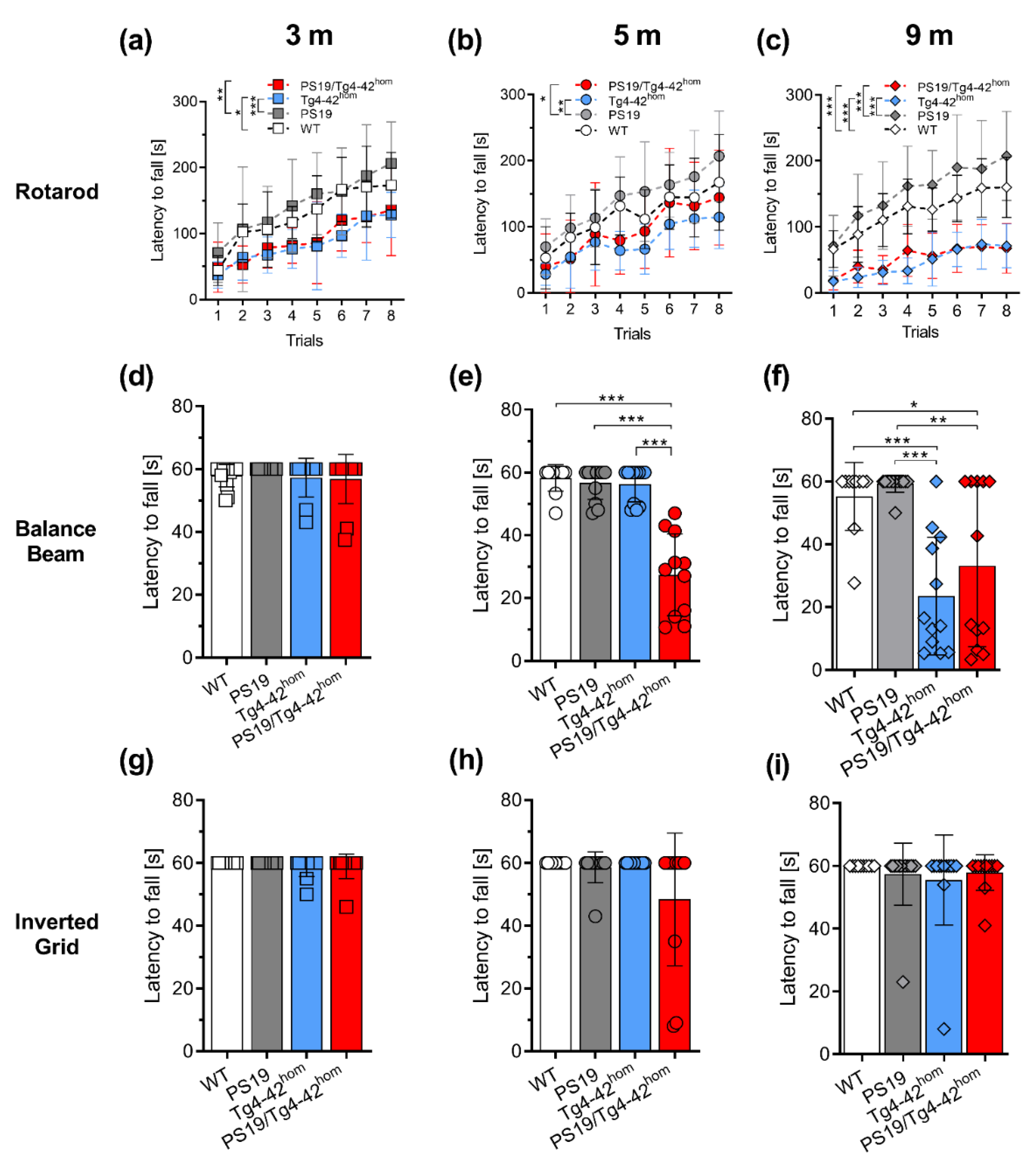
2.2. PS19/Tg4–42hom Mice Display a Partial Worsening of Motor Performance at 5 Months of Age
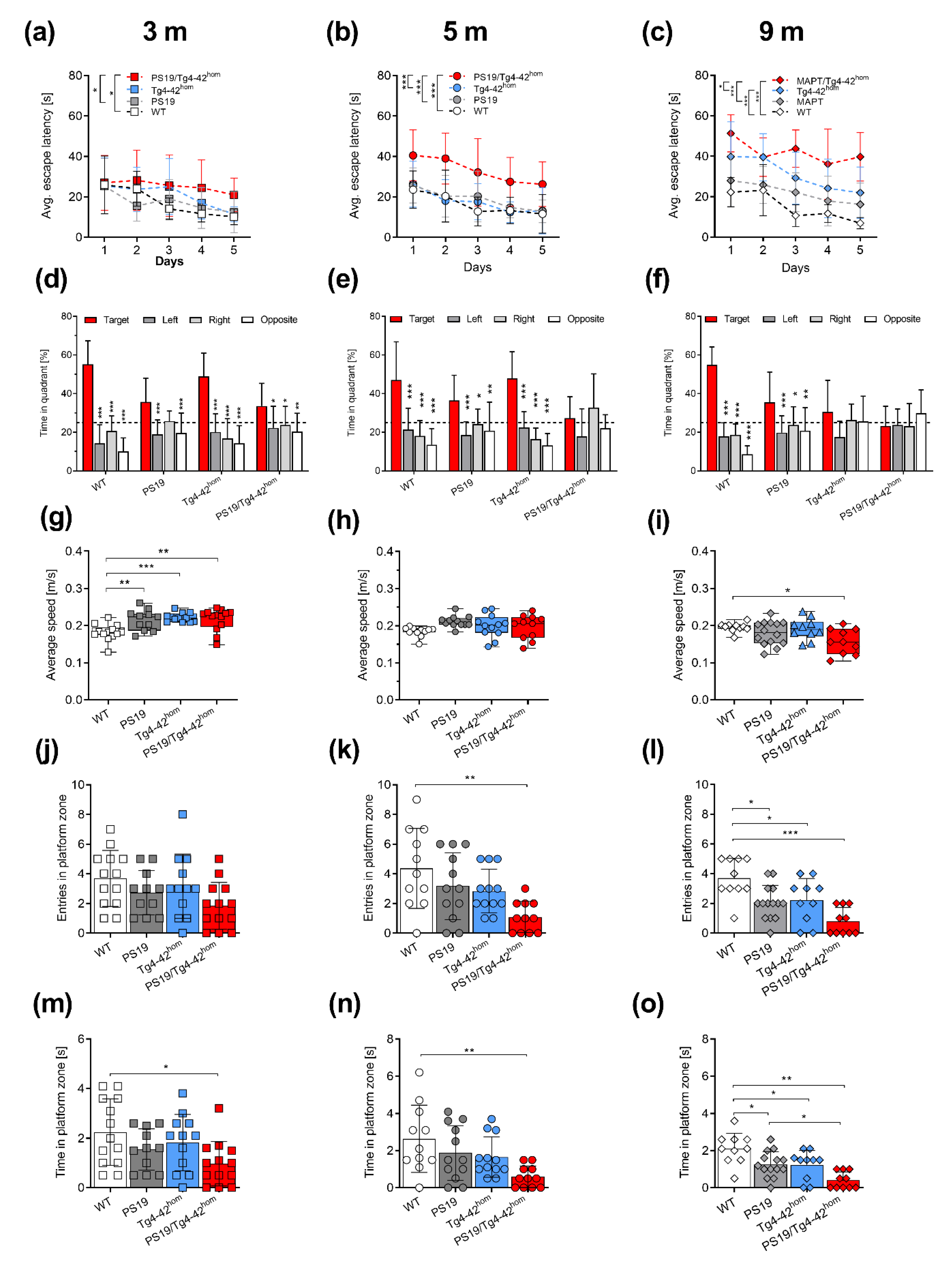
2.3. Co-Expression of Aβ4-42 and Mutant Tau Leads to Spatial Memory Deficits in PS19/Tg4–42hom Mice
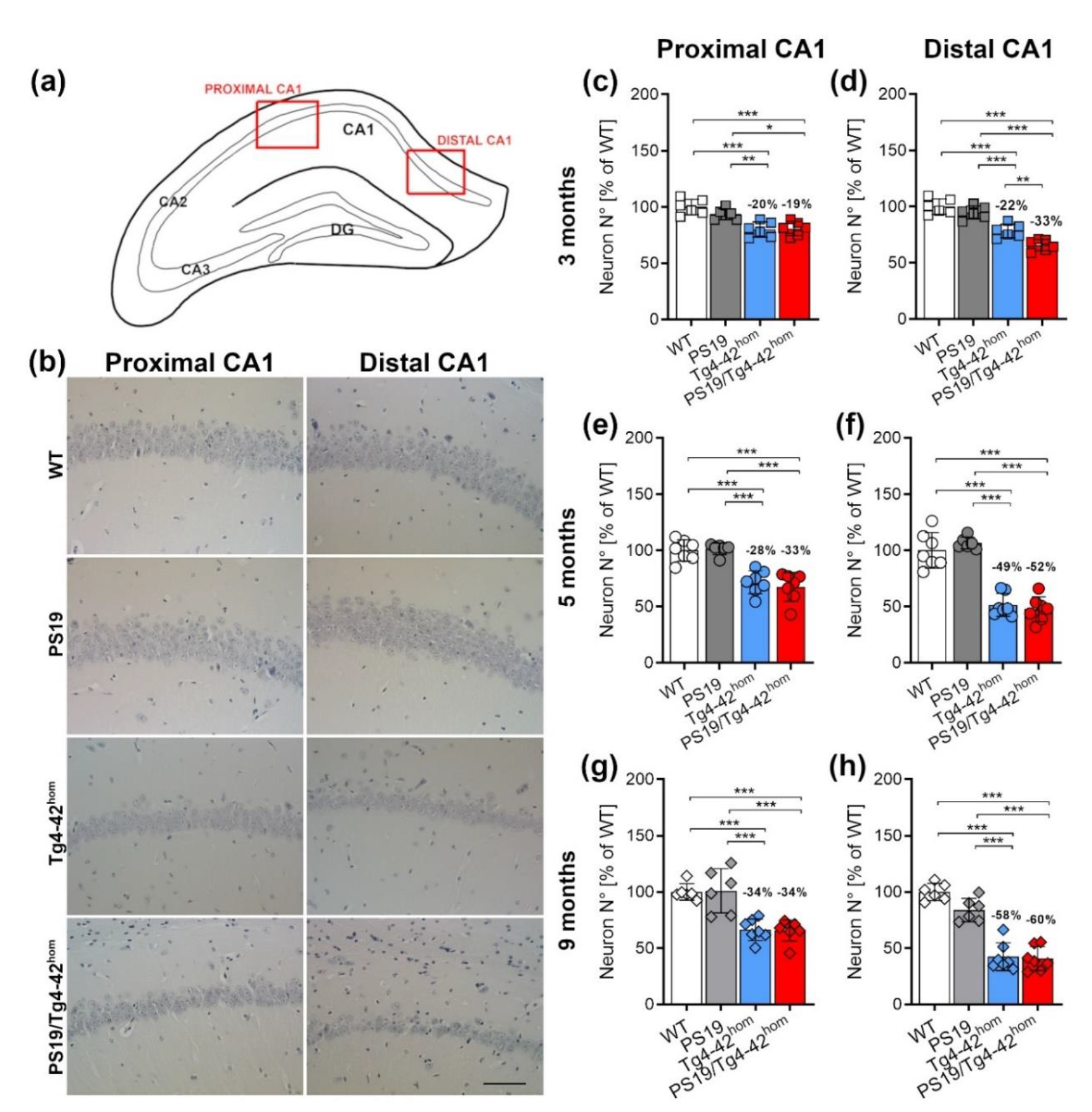
2.4. Increased Distal CA1 Neuron Loss in Young PS19/Tg4–42hom Mice
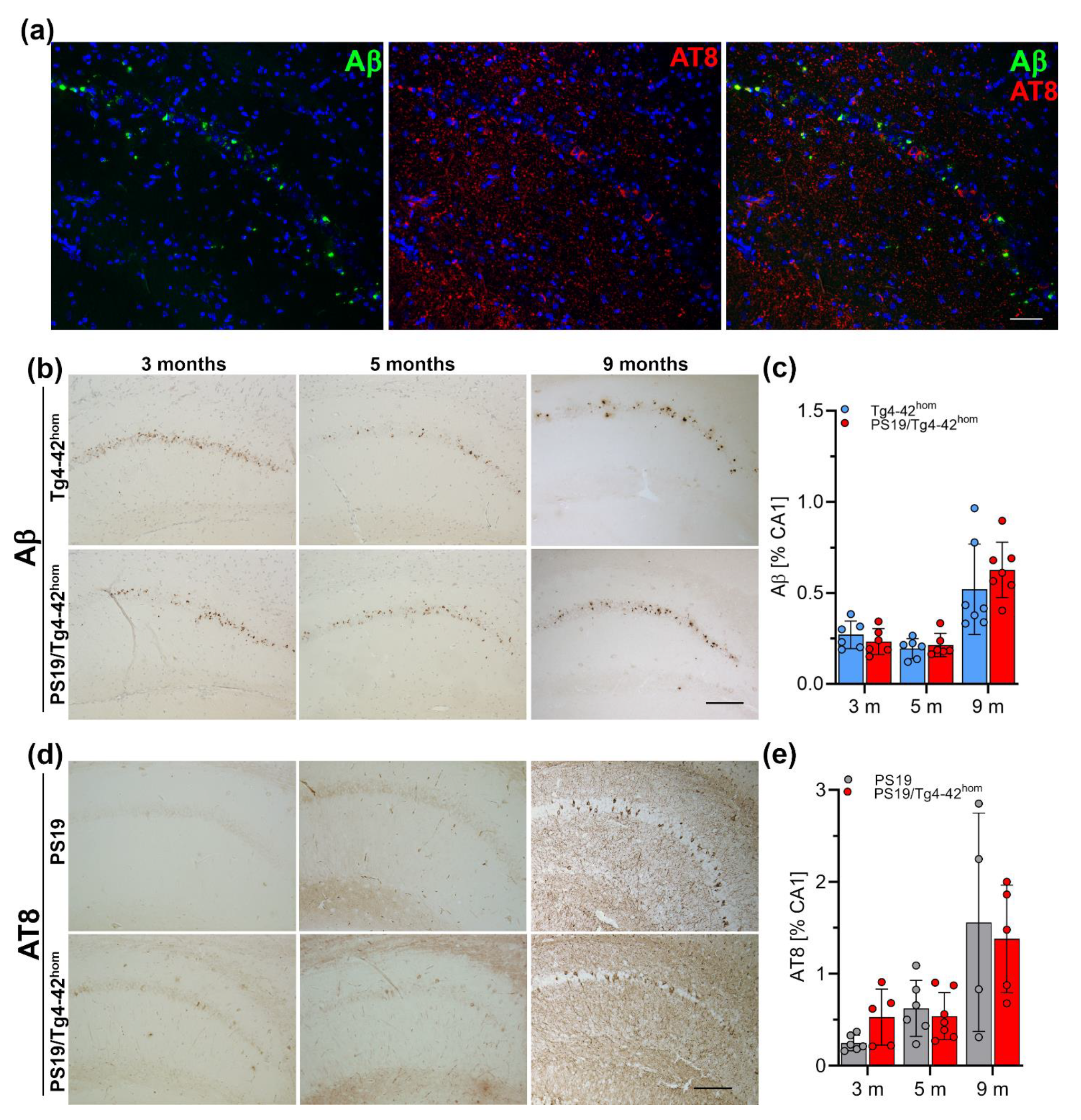
2.5. Hippocampal Aβ Ppathology and Tau Phosphorylation in PS19/Tg4–42hom Mice
3. Discussion
4. Materials and Methods
4.1. Transgenic Mice
4.2. Reverse Transcription and qRT-PCRs
4.3. Behavioral Tasks
4.3.1. Accelerating Rotarod
4.3.2. Balance Beam
4.3.3. Inverted Grid
4.3.4. Morris Water Maze (MWM)
4.3.5. Novel Object Recognition (NOR)
4.4. Tissue Collection and Preservation
4.5. Quantification of CA1 Neuron Number
4.6. Immunohistochemistry
4.7. Quantification of Aβ and Tau Immunoreactivity
4.8. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Masters, C.L.; Simms, G.; Weinman, N.A.; Multhaup, G.; McDonald, B.L.; Beyreuther, K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. USA 1985, 82, 4245–4249. [Google Scholar] [CrossRef] [PubMed]
- Grundke-Iqbal, I.; Iqbal, K.; Quinlan, M.; Tung, Y.C.; Zaidi, M.S.; Wisniewski, H.M. Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J. Biol. Chem. 1986, 261, 6084–6089. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Bloom, G.S. Amyloid-β and tau: The trigger and bullet in alzheimer disease pathogenesis. JAMA Neurol. 2014, 71, 505–508. [Google Scholar] [CrossRef]
- Nisbet, R.M.; Polanco, J.C.; Ittner, L.M.; Gotz, J. Tau aggregation and its interplay with amyloid-beta. Acta Neuropathol. 2015, 129, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Tomiyama, T.; Matsuyama, S.; Iso, H.; Umeda, T.; Takuma, H.; Ohnishi, K.; Ishibashi, K.; Teraoka, R.; Sakama, N.; Yamashita, T.; et al. A mouse model of amyloid-β oligomers: Their contribution to synaptic alteration, abnormal tau phosphorylation, glial activation, and neuronal loss in vivo. J. Neurosci. 2010, 30, 4845–4856. [Google Scholar] [CrossRef]
- Grueninger, F.; Bohrmann, B.; Czech, C.; Ballard, T.M.; Frey, J.R.; Weidensteiner, C.; von Kienlin, M.; Ozmen, L. Phosphorylation of Tau at S422 is enhanced by Abeta in TauPS2APP triple transgenic mice. Neurobiol. Dis. 2010, 37, 294–306. [Google Scholar] [CrossRef]
- Hurtado, D.E.; Molina-Porcel, L.; Iba, M.; Aboagye, A.K.; Paul, S.M.; Trojanowski, J.Q.; Lee, V.M. Aβ accelerates the spatiotemporal progression of tau pathology and augments tau amyloidosis in an Alzheimer mouse model. Am. J. Pathol. 2010, 177, 1977–1988. [Google Scholar] [CrossRef]
- Saul, A.; Sprenger, F.; Bayer, T.A.; Wirths, O. Accelerated tau pathology with synaptic and neuronal loss in a novel triple transgenic mouse model of Alzheimer’s disease. Neurobiol. Aging 2013, 34, 2564–2573. [Google Scholar] [CrossRef]
- Stancu, I.-C.; Ris, L.; Vasconcelos, B.; Marinangeli, C.; Goeminne, L.; Laporte, V.; Haylani, L.E.; Couturier, J.; Schakman, O.; Gailly, P.; et al. Tauopathy contributes to synaptic and cognitive deficits in a murine model for Alzheimer’s disease. FASEB J. 2014, 28, 2620–2631. [Google Scholar] [CrossRef]
- Lewis, J.; Dickson, D.W.; Lin, W.L.; Chisholm, L.; Corral, A.; Jones, G.; Yen, S.H.; Sahara, N.; Skipper, L.; Yager, D.; et al. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science 2001, 293, 1487–1491. [Google Scholar] [CrossRef]
- Kang, S.; Kim, J.; Chang, K.A. Spatial memory deficiency early in 6xTg Alzheimer’s disease mouse model. Sci. Rep. 2021, 11, 1334. [Google Scholar] [CrossRef]
- Héraud, C.; Goufak, D.; Ando, K.; Leroy, K.; Suain, V.; Yilmaz, Z.; De Decker, R.; Authelet, M.; Laporte, V.; Octave, J.-N.; et al. Increased misfolding and truncation of tau in APP/PS1/tau transgenic mice compared to mutant tau mice. Neurobiol. Dis. 2014, 62, 100–112. [Google Scholar] [CrossRef] [PubMed]
- Rapoport, M.; Dawson, H.N.; Binder, L.I.; Vitek, M.P.; Ferreira, A. Tau is essential to beta-amyloid-induced neurotoxicity. Proc. Natl. Acad. Sci. USA 2002, 99, 6364–6369. [Google Scholar] [CrossRef] [PubMed]
- Leroy, K.; Ando, K.; Laporte, V.; Dedecker, R.; Suain, V.; Authelet, M.; Heraud, C.; Pierrot, N.; Yilmaz, Z.; Octave, J.N.; et al. Lack of Tau Proteins Rescues Neuronal Cell Death and Decreases Amyloidogenic Processing of APP in APP/PS1 Mice. Am. J. Pathol. 2012, 181, 1928–1940. [Google Scholar] [CrossRef] [PubMed]
- Shipton, O.A.; Leitz, J.R.; Dworzak, J.; Acton, C.E.; Tunbridge, E.M.; Denk, F.; Dawson, H.N.; Vitek, M.P.; Wade-Martins, R.; Paulsen, O.; et al. Tau protein is required for amyloid {beta}-induced impairment of hippocampal long-term potentiation. J. Neurosci. 2011, 31, 1688–1692. [Google Scholar] [CrossRef]
- Puzzo, D.; Argyrousi, E.K.; Staniszewski, A.; Zhang, H.; Calcagno, E.; Zuccarello, E.; Acquarone, E.; Fa’, M.; Li Puma, D.D.; Grassi, C.; et al. Tau is not necessary for amyloid-β–induced synaptic and memory impairments. J. Clin. Investig. 2020, 130, 4831–4844. [Google Scholar] [CrossRef]
- Jin, M.; Shepardson, N.; Yang, T.; Chen, G.; Walsh, D.; Selkoe, D.J. Soluble amyloid beta-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc. Natl. Acad. Sci. USA 2011, 108, 5819–5824. [Google Scholar] [CrossRef]
- Hu, X.; Li, X.; Zhao, M.; Gottesdiener, A.; Luo, W.; Paul, S. Tau pathogenesis is promoted by Abeta1-42 but not Abeta1-40. Mol. Neurodegen. 2014, 9, 52. [Google Scholar] [CrossRef]
- Gomes, L.A.; Hipp, S.A.; Upadhaya, A.R.; Balakrishnan, K.; Ospitalieri, S.; Koper, M.J.; Largo-Barrientos, P.; Uytterhoeven, V.; Reichwald, J.; Rabe, S.; et al. Aβ-induced acceleration of Alzheimer-related τ-pathology spreading and its association with prion protein. Acta Neuropathol. 2019, 138, 913–941. [Google Scholar] [CrossRef]
- Bouter, Y.; Dietrich, K.; Wittnam, J.L.; Rezaei-Ghaleh, N.; Pillot, T.; Papot-Couturier, S.; Lefebvre, T.; Sprenger, F.; Wirths, O.; Zweckstetter, M.; et al. N-truncated amyloid β (Aβ) 4–42 forms stable aggregates and induces acute and long-lasting behavioral deficits. Acta Neuropathol. 2013, 126, 189–205. [Google Scholar] [CrossRef]
- Portelius, E.; Bogdanovic, N.; Gustavsson, M.K.; Volkmann, I.; Brinkmalm, G.; Zetterberg, H.; Winblad, B.; Blennow, K. Mass spectrometric characterization of brain amyloid beta isoform signatures in familial and sporadic Alzheimer’s disease. Acta Neuropathol. 2010, 120, 185–193. [Google Scholar] [CrossRef]
- Wirths, O.; Zampar, S. Emerging roles of N- and C-terminally truncated Aβ species in Alzheimer’s disease. Expert Opin. Ther. Targets 2019, 23, 991–1004. [Google Scholar] [CrossRef] [PubMed]
- Hüttenrauch, M.; Brauss, A.; Kurdakova, A.; Borgers, H.; Klinker, F.; Liebetanz, D.; Salinas-Riester, G.; Wiltfang, J.; Klafki, H.W.; Wirths, O. Physical activity delays hippocampal neurodegeneration and rescues memory deficits in an Alzheimer disease mouse model. Transl. Psych. 2016, 6, e800. [Google Scholar] [CrossRef] [PubMed]
- Stazi, M.; Wirths, O. Chronic Memantine Treatment Ameliorates Behavioral Deficits, Neuron Loss, and Impaired Neurogenesis in a Model of Alzheimer’s Disease. Mol. Neurobiol. 2021, 58, 204–216. [Google Scholar] [CrossRef] [PubMed]
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Huang, S.M.; Iwata, N.; Saido, T.C.; Maeda, J.; Suhara, T.; Trojanowski, J.Q.; Lee, V.M. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 2007, 53, 337–351. [Google Scholar] [CrossRef]
- Antonios, G.; Borgers, H.; Richard, B.C.; Brauß, A.; Meißner, J.; Weggen, S.; Pena, V.; Pillot, T.; Davies, S.L.; Bakrania, P.; et al. Alzheimer therapy with an antibody against N-terminal Abeta 4-X and pyroglutamate Abeta 3-X. Sci. Rep. 2015, 5, 17338. [Google Scholar] [CrossRef]
- Takeuchi, H.; Iba, M.; Inoue, H.; Higuchi, M.; Takao, K.; Tsukita, K.; Karatsu, Y.; Iwamoto, Y.; Miyakawa, T.; Suhara, T.; et al. P301S Mutant Human Tau Transgenic Mice Manifest Early Symptoms of Human Tauopathies with Dementia and Altered Sensorimotor Gating. PLoS ONE 2011, 6, e21050. [Google Scholar] [CrossRef]
- Sun, Y.; Guo, Y.; Feng, X.; Jia, M.; Ai, N.; Dong, Y.; Zheng, Y.; Fu, L.; Yu, B.; Zhang, H.; et al. The behavioural and neuropathologic sexual dimorphism and absence of MIP-3α in tau P301S mouse model of Alzheimer’s disease. J. Neuroinflamm. 2020, 17, 72. [Google Scholar] [CrossRef]
- Götz, J.; Chen, F.; van Dorpe, J.; Nitsch, R.M. Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science 2001, 293, 1491–1495. [Google Scholar] [CrossRef]
- Bolmont, T.; Clavaguera, F.; Meyer-Luehmann, M.; Herzig, M.C.; Radde, R.; Staufenbiel, M.; Lewis, J.; Hutton, M.; Tolnay, M.; Jucker, M. Induction of tau pathology by intracerebral infusion of amyloid-beta -containing brain extract and by amyloid-beta deposition in APP x Tau transgenic mice. Am. J. Pathol. 2007, 171, 2012–2020. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos, B.; Stancu, I.-C.; Buist, A.; Bird, M.; Wang, P.; Vanoosthuyse, A.; Van Kolen, K.; Verheyen, A.; Kienlen-Campard, P.; Octave, J.-N.; et al. Heterotypic seeding of Tau fibrillization by pre-aggregated Abeta provides potent seeds for prion-like seeding and propagation of Tau-pathology in vivo. Acta Neuropathol. 2016, 131, 549–569. [Google Scholar] [CrossRef]
- Oddo, S.; Billings, L.; Kesslak, J.P.; Cribbs, D.H.; LaFerla, F.M. Abeta Immunotherapy Leads to Clearance of Early, but Not Late, Hyperphosphorylated Tau Aggregates via the Proteasome. Neuron 2004, 43, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Ribe, E.M.; Perez, M.; Puig, B.; Gich, I.; Lim, F.; Cuadrado, M.; Sesma, T.; Catena, S.; Sanchez, B.; Nieto, M.; et al. Accelerated amyloid deposition, neurofibrillary degeneration and neuronal loss in double mutant APP/tau transgenic mice. Neurobiol. Dis. 2005, 20, 814–822. [Google Scholar] [CrossRef] [PubMed]
- Abramowski, D.; Rabe, S.; Upadhaya, A.R.; Reichwald, J.; Danner, S.; Staab, D.; Capetillo-Zarate, E.; Yamaguchi, H.; Saido, T.C.; Wiederhold, K.-H.; et al. Transgenic Expression of Intraneuronal Aβ42 But Not Aβ40 Leads to Cellular Aβ Lesions, Degeneration, and Functional Impairment without Typical Alzheimer’s Disease Pathology. J. Neurosci. 2012, 32, 1273–1283. [Google Scholar] [CrossRef] [PubMed]
- Chabrier, M.A.; Blurton-Jones, M.; Agazaryan, A.A.; Nerhus, J.L.; Martinez-Coria, H.; Laferla, F.M. Soluble abeta promotes wild-type tau pathology in vivo. J. Neurosci. 2012, 32, 17345–17350. [Google Scholar] [CrossRef]
- Takahashi, M.; Miyata, H.; Kametani, F.; Nonaka, T.; Akiyama, H.; Hisanaga, S.-i.; Hasegawa, M. Extracellular association of APP and tau fibrils induces intracellular aggregate formation of tau. Acta Neuropathol. 2015, 129, 895–907. [Google Scholar] [CrossRef] [PubMed]
- Puzzo, D.; Piacentini, R.; Fá, M.; Gulisano, W.; Li Puma, D.D.; Staniszewski, A.; Zhang, H.; Tropea, M.R.; Cocco, S.; Palmeri, A.; et al. LTP and memory impairment caused by extracellular Aβ and Tau oligomers is APP-dependent. eLife 2017, 6, e26991. [Google Scholar] [CrossRef]
- Masurkar, A.V. Towards a circuit-level understanding of hippocampal CA1 dysfunction in Alzheimer’s disease across anatomical axes. J. Alzheimer’s Dis. Park. 2018, 8, 412. [Google Scholar] [CrossRef]
- Beer, Z.; Vavra, P.; Atucha, E.; Rentzing, K.; Heinze, H.-J.; Sauvage, M.M. The memory for time and space differentially engages the proximal and distal parts of the hippocampal subfields CA1 and CA3. PLoS Biol. 2018, 16, e2006100. [Google Scholar] [CrossRef]
- Eckenweber, F.; Medina-Luque, J.; Blume, T.; Sacher, C.; Biechele, G.; Wind, K.; Deussing, M.; Briel, N.; Lindner, S.; Boening, G.; et al. Longitudinal TSPO expression in tau transgenic P301S mice predicts increased tau accumulation and deteriorated spatial learning. J. Neuroinflamm. 2020, 17, 208. [Google Scholar] [CrossRef] [PubMed]
- Chalermpalanupap, T.; Schroeder, J.P.; Rorabaugh, J.M.; Liles, L.C.; Lah, J.J.; Levey, A.I.; Weinshenker, D. Locus Coeruleus Ablation Exacerbates Cognitive Deficits, Neuropathology, and Lethality in P301S Tau Transgenic Mice. J. Neurosci. 2018, 38, 74–92. [Google Scholar] [CrossRef]
- Liu, M.; Wang, L.; Gao, J.; Dong, Q.; Perry, G.; Ma, X.; Wang, X. Inhibition of Calpain Protects Against Tauopathy in Transgenic P301S Tau Mice. J. Alzheimer’s Dis. 2019, 69, 1077–1087. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Shiotsuki, H.; Yoshimi, K.; Shimo, Y.; Funayama, M.; Takamatsu, Y.; Ikeda, K.; Takahashi, R.; Kitazawa, S.; Hattori, N. A rotarod test for evaluation of motor skill learning. J. Neurosci. Methods 2010, 189, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Luong, T.N.; Carlisle, H.J.; Southwell, A.; Patterson, P.H. Assessment of motor balance and coordination in mice using the balance beam. J. Vis. Exp. 2011, 49, 2376. [Google Scholar] [CrossRef]
- Wirths, O.; Breyhan, H.; Schafer, S.; Roth, C.; Bayer, T.A. Deficits in working memory and motor performance in the APP/PS1ki mouse model for Alzheimer’s disease. Neurobiol. Aging 2008, 29, 891–901. [Google Scholar] [CrossRef]
- Morris, R. Developments of a water-maze procedure for studying spatial learning in the rat. J. Neurosci. Methods 1984, 11, 47–60. [Google Scholar] [CrossRef]
- Wirths, O.; Multhaup, G.; Czech, C.; Feldmann, N.; Blanchard, V.; Tremp, G.; Beyreuther, K.; Pradier, L.; Bayer, T.A. Intraneuronal APP/A beta trafficking and plaque formation in beta-amyloid precursor protein and presenilin-1 transgenic mice. Brain Pathol. 2002, 12, 275–286. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Transgene/Gene | Forward Primer | Reverse Primer |
|---|---|---|
| Human Aβ4–42 | TCCGGCCAGAACGTCGATTC | GGAGAAGCAAGACCTCTG |
| Human MAPT | CCAAGTGTGGCTCATTAGGCA | CCAATCTTCGACTGGACTCTGT |
| Murine β-Actin | ATGGAGGGGAATACAGCCC | ATGGAGGGGAATACAGCCC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zampar, S.; Wirths, O. Characterization of a Mouse Model of Alzheimer’s Disease Expressing Aβ4-42 and Human Mutant Tau. Int. J. Mol. Sci. 2021, 22, 5191. https://doi.org/10.3390/ijms22105191
Zampar S, Wirths O. Characterization of a Mouse Model of Alzheimer’s Disease Expressing Aβ4-42 and Human Mutant Tau. International Journal of Molecular Sciences. 2021; 22(10):5191. https://doi.org/10.3390/ijms22105191
Chicago/Turabian StyleZampar, Silvia, and Oliver Wirths. 2021. "Characterization of a Mouse Model of Alzheimer’s Disease Expressing Aβ4-42 and Human Mutant Tau" International Journal of Molecular Sciences 22, no. 10: 5191. https://doi.org/10.3390/ijms22105191
APA StyleZampar, S., & Wirths, O. (2021). Characterization of a Mouse Model of Alzheimer’s Disease Expressing Aβ4-42 and Human Mutant Tau. International Journal of Molecular Sciences, 22(10), 5191. https://doi.org/10.3390/ijms22105191