Pharmacological Inhibition of mTORC2 Reduces Migration and Metastasis in Melanoma

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Inhibition of mTORC2 Reduces Cell Viability and Proliferation of Melanoma Cells
2.2. Exposure to JR-AB2-011 Leads to Impaired Activation of Akt Signaling
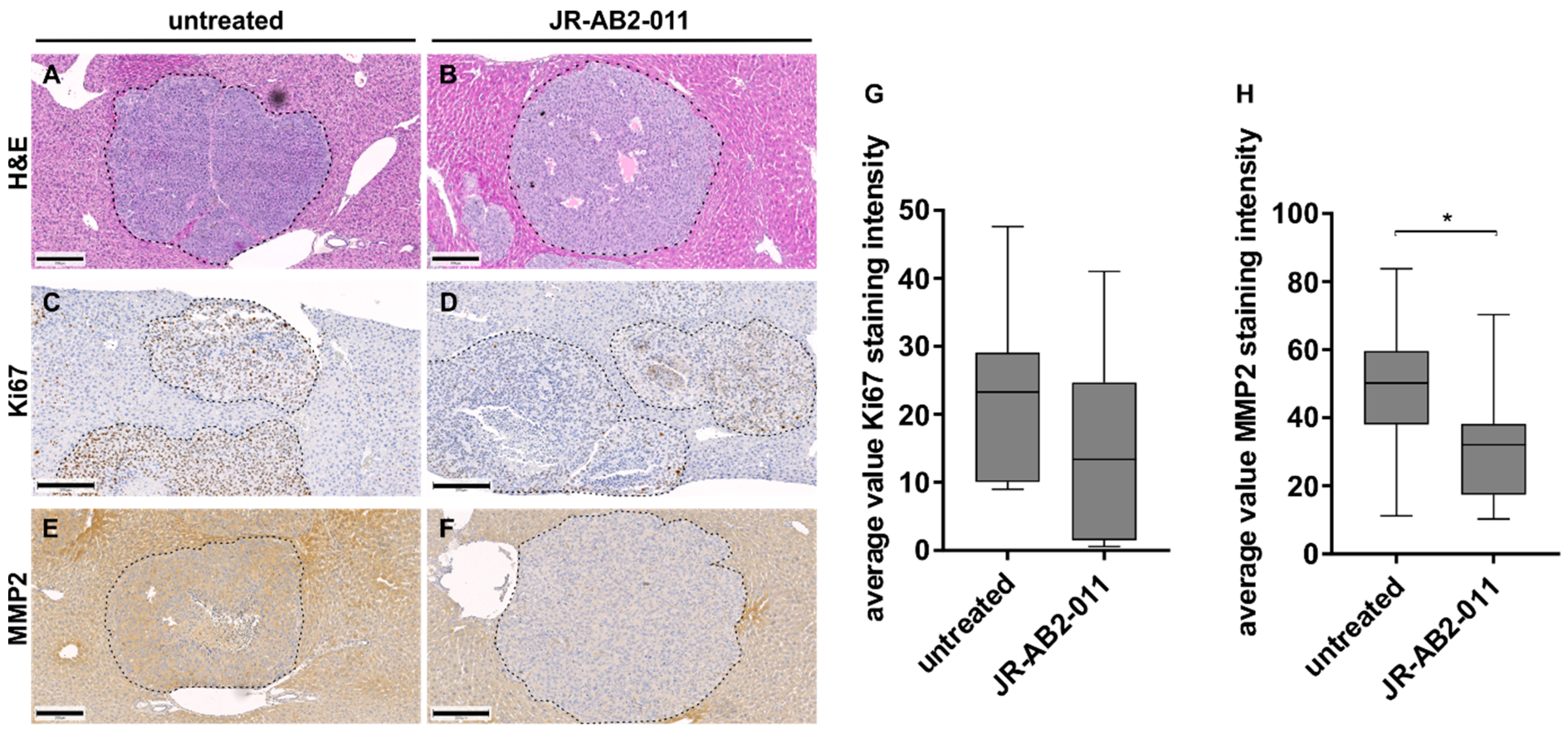
2.3. mTORC2 Blockade Inhibits Migration and Invasion Capacity by Regulation of MMP2
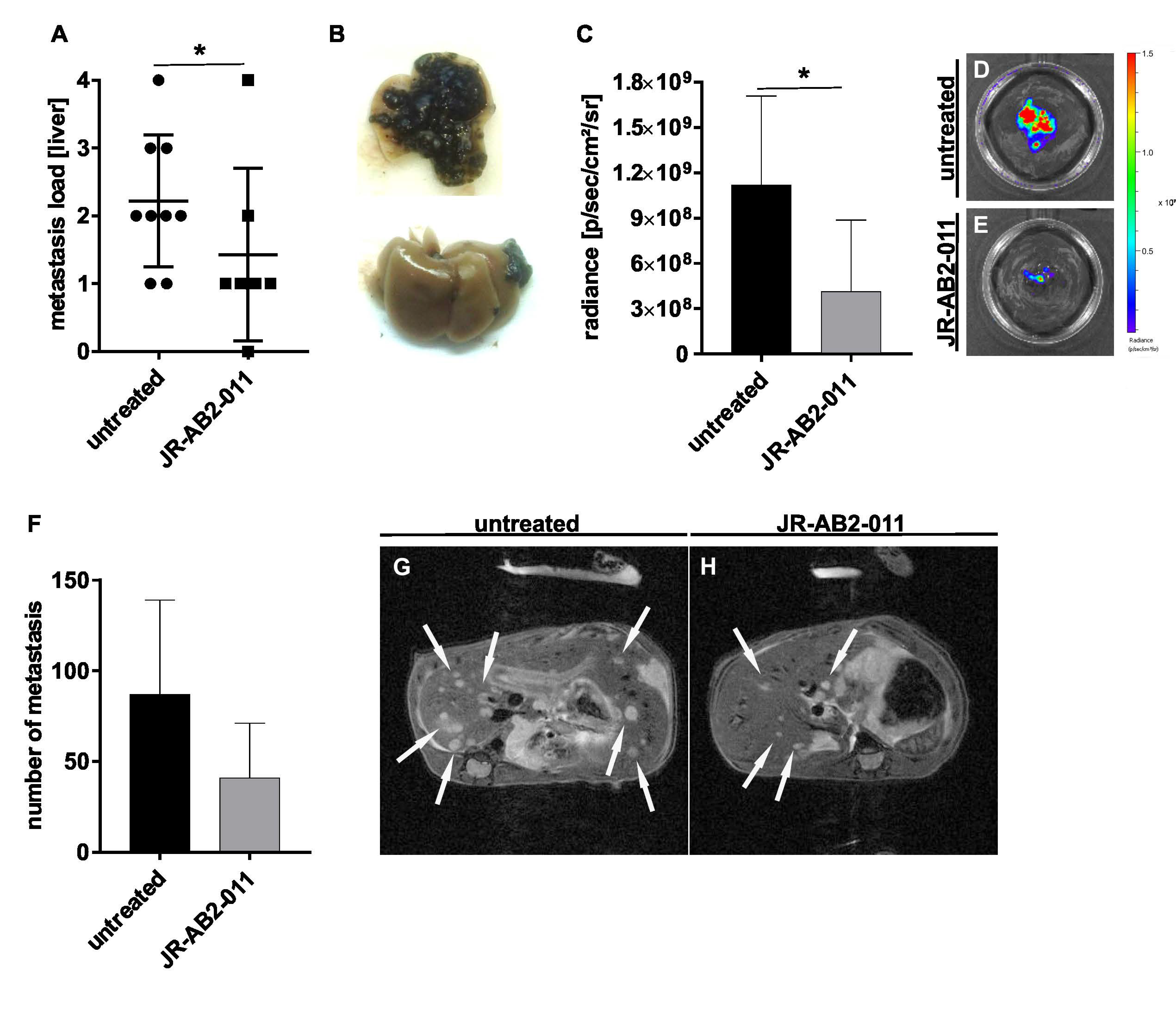
2.4. Pharmacological Inhibition of mTORC2 In Vivo Reduced the Metastasis Load in the Liver
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Viability Assay
4.3. Cell Proliferation Assay
4.4. Migration and Invasion Assay
4.5. Colony Forming Unit Assay
4.6. Western Blotting
4.7. Gelatin Zymography
4.8. In vivo Xenograft Experiments
4.9. Bioluminescence Imaging
4.10. MRI Measurement
4.11. Immunohistochemistry
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BLI | bioluminescence imaging |
| BrdU | Bromdesoxyuridin |
| ECM | extracellular matrix |
| FOXO1/3a | Forkhead box protein O1/3a |
| H and E | hematoxylin and eosin |
| IHC | immunohistochemistry |
| i.p. | intraperitoneal |
| JR-AB2-011 | 3-(3,4-Dichloro-phenyl)-1-(4-fluoro-phenyl)-1-(5-methyl-4,5-dihydro-thiazol-2-yl)-urea |
| MTT | 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromid |
| mTOR | mammalian target of rapamycin |
| mTORC1 | mammalian target of rapamycin complex 1 |
| mTORC2 | mammalian target of rapamycin complex 2 |
| MMP2 | matrix metalloproteinase 2 |
| MRI | magnetic resonance imaging |
| NDGR1 | N-myc downstream regulated 1 |
| PDK1 | 3-phosphoinositide dependent kinase 1 |
| PI3K | phosphoinositide 3-kinase |
| RAPTOR | regulatory associated protein of mTOR |
| RICTOR | rapamycin-insensitive companion of mTOR |
| ROI | region of interest |
| S6K | ribosomal protein S6 kinase |
| Ser | serine |
| SF | surviving fraction |
| SGK1 | serum/glucocorticoid regulated kinase 1 |
| Thr | threonine |
References
- Zhou, H.; Huang, S. Role of mTOR signaling in tumor cell motility, invasion and metastasis. Curr. Protein Pept. Sci. 2011, 12, 30–42. [Google Scholar] [PubMed]
- Zhao, Z.; Wang, S.; Barber, B.L. Treatment Patterns in Patients with Metastatic Melanoma: A Retrospective Analysis. J. Skin Cancer 2014, 2014, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Agarwala, S.S.; Eggermont, A.M.M.; O’Day, S.; Zager, J.S. Metastatic melanoma to the liver: A contemporary and comprehensive review of surgical, systemic, and regional therapeutic options. Cancer 2014, 120, 781–789. [Google Scholar] [CrossRef]
- Larkin, J.; Ascierto, P.A.; Dréno, B.; Atkinson, V.; Liszkay, G.; Maio, M.; Mandalà, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N. Engl. J. Med. 2014, 371, 1867–1876. [Google Scholar] [CrossRef]
- Kulkarni, A.; Al-Hraishawi, H.; Simhadri, S.; Hirshfield, K.M.; Chen, S.; Pine, S.; Jeyamohan, C.; Sokol, L.; Ali, S.; Teo, M.L.; et al. BRAF fusion as a novel mechanism of acquired resistance to vemurafenib in BRAFV600E mutant melanoma. Clin. Cancer Res. 2017, 23, 5631–5638. [Google Scholar] [CrossRef]
- Kaushik, S.; Pickup, M.W.; Weaver, V.M. From transformation to metastasis: Deconstructing the extracellular matrix in breast cancer. Cancer Metastasis Rev. 2016, 35, 655–667. [Google Scholar] [CrossRef]
- Leber, M.F.; Efferth, T. Molecular principles of cancer invasion and metastasis. Int. J. Hyperth. 2009, 34, 881–895. [Google Scholar]
- Sarna, M.; Zadlo, A.; Hermanowicz, P.; Madeja, Z.; Burda, K.; Sarna, T. Cell elasticity is an important indicator of the metastatic phenotype of melanoma cells. Exp. Dermatol. 2014, 23, 813–818. [Google Scholar] [CrossRef]
- Sarna, M.; Krzykawska-Serda, M.; Jakubowska, M.; Zadlo, A.; Urbanska, K. Melanin presence inhibits melanoma cell spread in mice in a unique mechanical fashion. Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef]
- Guenzle, J.; Wolf, L.J.; Garrelfs, N.W.C.; Goeldner, J.M.; Osterberg, N.; Schindler, C.R.; Saavedra, J.E.; Weyerbrock, A. ATF3 reduces migration capacity by regulation of matrix metalloproteinases via NFκB and STAT3 inhibition in glioblastoma. Cell Death Discov. 2017, 3, 17006. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Takai, K.; Weaver, V.M.; Werb, Z. Extracellular Matrix Degradation and Remodeling in Development and Disease. Cold Spring Harb. Perspect. Biol. 2011, 3, a005058. [Google Scholar] [CrossRef] [PubMed]
- Jacob, A.; Prekeris, R. The regulation of MMP targeting to invadopodia during cancer metastasis. Front. Cell Dev. Biol. 2015, 3, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Chen, L.; Yang, Y.C.; Wang, X.M.; Wang, R.Y.; Li, L.; Wen, W.; Chang, Y.X.; Chen, C.Y.; Tang, J.; et al. CYP3A5 functions as a tumor suppressor in hepatocellular carcinoma by regulating mTORC2/Akt signaling. Cancer Res. 2015, 75, 1470–1481. [Google Scholar] [CrossRef]
- Liang, X.; Sun, R.; Zhao, X.; Zhang, Y.; Gu, Q.; Dong, X.; Zhang, D.; Sun, J.; Sun, B. Rictor regulates the vasculogenic mimicry of melanoma via the AKT-MMP-2/9 pathway. J. Cell. Mol. Med. 2017, 21, 3579–3591. [Google Scholar] [CrossRef]
- Schmidt, K.M.; Dietrich, P.; Hackl, C.; Guenzle, J.; Bronsert, P.; Wagner, C.; Fichtner-Feigl, S.; Schlitt, H.J.; Geissler, E.K.; Hellerbrand, C.; et al. Inhibition of mTORC2/RICTOR Impairs Melanoma Hepatic Metastasis. Neoplasia 2018, 20, 1198–1208. [Google Scholar] [CrossRef] [PubMed]
- Sabatini, D.M. mTOR and cancer: Insights into a complex relationship. Nat. Rev. Cancer 2006, 6, 729–734. [Google Scholar] [CrossRef]
- Kim, S.T.; Kim, S.Y.; Klempner, S.J.; Yoon, J.; Kim, N.; Ahn, S.; Bang, H.; Kim, K.; Park, W.; Park, S.H.; et al. Rapamycin-insensitive companion of mTOR (RICTOR) amplification defines a subset of advanced gastric cancer and is sensitive to AZD2014-mediated mTORC1 / 2 inhibition. Ann. Oncol. 2017, 28, 547–554. [Google Scholar] [CrossRef]
- Masri, J.; Bernath, A.; Martin, J.; Jo, O.D.; Vartanian, R.; Funk, A.; Gera, J. mTORC2 Activity Is Elevated in Gliomas and Promotes Growth and Cell Motility via Overexpression of Rictor. Cancer Res. 2007, 67, 11712–11721. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Hay, N. The Akt-mTOR tango and its relevance to cancer. Cancer Cell 2005, 8, 179–183. [Google Scholar] [CrossRef]
- Serra, V.; Kim, S.Y.; Baselga, J.; Serra, V.; Eichhorn, P.J.A.; García-garcía, C.; Ibrahim, Y.H.; Prudkin, L.; Scaltriti, M.; Pérez-garcia, J.; et al. RSK3 / 4 mediate resistance to PI3K pathway inhibitors in breast cancer Find the latest version: RSK3/4 mediate resistance to PI3K pathway inhibitors in breast cancer. J. Clin. Investig. 2014, 123, 2551–2563. [Google Scholar] [CrossRef] [PubMed]
- Lang, S.A.; Hackl, C.; Moser, C.; Fichtner-Feigl, S.; Koehl, G.E.; Schlitt, H.J.; Geissler, E.K.; Stoeltzing, O. Implication of RICTOR in the mTOR inhibitor-mediated induction of insulin-like growth factor-I receptor (IGF-IR) and human epidermal growth factor receptor-2 (Her2) expression in gastrointestinal cancer cells. Biochim. Biophys. Acta Mol. Cell Res. 2010, 1803, 435–442. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, K.E.; Rojo, F.; She, Q.B.; Solit, D.; Mills, G.B.; Smith, D.; Lane, H.; Hofmann, F.; Hicklin, D.J.; Ludwig, D.L.; et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006, 66, 1500–1508. [Google Scholar] [CrossRef] [PubMed]
- Bendell, J.C.; Kelley, R.K.; Shih, K.C.; Grabowsky, J.A.; Bergsland, E.; Jones, S.; Martin, T.; Infante, J.R.; Mischel, P.S.; Matsutani, T.; et al. A phase I dose-escalation study to assess safety, tolerability, pharmacokinetics, and preliminary efficacy of the dual mTORC1/mTORC2 kinase inhibitor CC-223 in patients with advanced solid tumors or multiple myeloma. Cancer 2015, 121, 3481–3490. [Google Scholar] [CrossRef]
- Mateo, J.; Olmos, D.; Dumez, H.; Poondru, S.; Samberg, N.L.; Barr, S.; Van Tornout, J.M.; Jie, F.; Sandhu, S.; Tan, D.S.; et al. A first in man, dose-finding study of the mTORC1/mTORC2 inhibitor OSI-027 in patients with advanced solid malignancies. Br. J. Cancer 2016, 114, 889–896. [Google Scholar] [CrossRef]
- Statz, C.M.; Patterson, S.E.; Mockus, S.M. mTOR Inhibitors in Castration-Resistant Prostate Cancer: A Systematic Review. Target. Oncol. 2017, 12, 47–59. [Google Scholar] [CrossRef]
- Sun, B.; Chen, L.; Fu, H.; Guo, L.; Guo, H.; Zhang, N. Upregulation of RICTOR gene transcription by the proinflammatory cytokines through NF-κB pathway contributes to the metastasis of renal cell carcinoma. Tumor Biol. 2016, 37, 4457–4466. [Google Scholar] [CrossRef]
- Schmidt, K.M.; Hellerbrand, C.; Ruemmele, P.; Michalski, C.W.; Kong, B.; Kroemer, A.; Hackl, C.; Schlitt, H.J.; Geissler, E.K.; Lang, S.A. Inhibition of mTORC2 component RICTOR impairs tumor growth in pancreatic cancer models. Oncotarget 2017, 8, 24491–24505. [Google Scholar] [CrossRef]
- Benavides-Serrato, A.; Lee, J.; Holmes, B.; Landon, K.A.; Bashir, T.; Jung, M.E.; Lichtenstein, A.; Gera, J. Specific blockade of Rictor-mTOR association inhibits mTORC2 activity and is cytotoxic in glioblastoma. PLoS ONE 2017, e0176599. [Google Scholar] [CrossRef]
- Barrandon, Y.; Green, H. Three clonal types of keratinocyte with different capacities for multiplication. Proc. Nati. Acad. Sci. USA 1987, 84, 2302–2306. [Google Scholar] [CrossRef]
- Harries, M.; Malvehy, J.; Lebbe, C.; Heron, L.; Amelio, J.; Szabo, Z.; Schadendorf, D. Treatment patterns of advanced malignant melanoma (stage III–IV)—A review of current standards in Europe. Eur. J. Cancer 2016, 60, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Forschner, A.; Eichner, F.; Amaral, T.; Keim, U.; Garbe, C.; Eigentler, T.K. Improvement of overall survival in stage IV melanoma patients during 2011–2014: Analysis of real-world data in 441 patients of the German Central Malignant Melanoma Registry (CMMR). J. Cancer Res. Clin. Oncol. 2017, 143, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Murugan, A.K. mTOR: Role in cancer, metastasis and drug resistance. Semin. Cancer Biol. 2019, 59, 92–111. [Google Scholar] [CrossRef] [PubMed]
- Hua, H.; Kong, Q.; Zhang, H.; Wang, J.; Luo, T.; Jiang, Y. Targeting mTOR for cancer therapy. J. Hematol. Oncol. 2019, 12, 1–19. [Google Scholar] [CrossRef]
- Zhang, S.; Qian, G.; Zhang, Q.Q.; Yao, Y.; Wang, D.; Chen, Z.G.; Wang, L.J.; Chen, M.; Sun, S.Y. MTORC2 suppresses GSK3-dependent snail degradation to positively regulate cancer cell invasion and metastasis. Cancer Res. 2019, 79, 3725–3736. [Google Scholar] [CrossRef]
- Lang, S.A.; Moser, C.; Fichnter-Feigl, S.; Schachtschneider, P.; Hellerbrand, C.; Schmitz, V.; Schlitt, H.J.; Geissler, E.K.; Stoeltzing, O. Targeting heat-shock protein 90 improves efficacy of rapamycin in a model of hepatocellular carcinoma in mice. Hepatology 2009, 49, 523–532. [Google Scholar] [CrossRef]
- Sarbassov, D.; Guertin, D.; Ali, S.; Sabatini, D. Phosphorylation and regulation of Akt//PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef]
- Chen, B.W.; Chen, W.; Liang, H.; Liu, H.; Liang, C.; Zhi, X.; Hu, L.-Q.; Yu, X.-Z.; Wei, T.; Ma, T.; et al. Inhibition of mTORC2 Induces Cell-Cycle Arrest and Enhances the Cytotoxicity of Doxorubicin by Suppressing MDR1 Expression in HCC Cells. Mol. Cancer Ther. 2015, 14, 1805–1815. [Google Scholar] [CrossRef]
- Geng, L.; Donnelly, E.; McMahon, G.; Lin, P.C.; Sierra-Rivera, E.; Oshinka, H.; Hallahan, D. Inhibition of vascular endothelial growth factor receptor signaling leads to reversal of tumor resistance to radiotherapy. Cancer Res. 2001, 61, 2413–2419. [Google Scholar]
- Bhagwat, S.V.; Gokhale, P.C.; Crew, A.P.; Cooke, A.; Yao, Y.; Mantis, C.; Kahler, J.; Workman, J.; Bittner, M.; Dudkin, L.; et al. Preclinical Characterization of OSI-027, a Potent and Selective Inhibitor of mTORC1 and mTORC2: Distinct from Rapamycin. Mol. Cancer Ther. 2011, 10, 1394–1406. [Google Scholar] [CrossRef]
- Romano, M.F.; Avellino, R.; Petrella, A.; Bisogni, R.; Romano, S.; Venuta, S. Rapamycin inhibits doxorubicin-induced NF-κB/Rel nuclear activity and enhances the apoptosis of melanoma cells. Eur. J. Cancer 2004, 40, 2829–2836. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Lei, Z.; Li, B.; Zhou, Y.; Zhang, G.M.; Feng, Z.H.; Zhang, B.; Shen, G.X.; Huang, B. Rapamycin inhibits lung metastasis of B16 melanoma cells through down-regulating alphav integrin expression and up-regulating apoptosis signaling. Cancer Sci. 2010, 101, 494–500. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, A.; Cornu, M.; Cybulski, N.; Polak, P.; Betz, C.; Trapani, F.; Terracciano, L.; Heim, M.H.; Rüegg, M.A.; Hall, M.N. Hepatic mTORC2 Activates Glycolysis and Lipogenesis through Akt, Glucokinase, and SREBP1c. Cell Metab. 2012, 15, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Driscoll, D.R.; Karim, S.A.; Sano, M.; Gay, D.M.; Jacob, W.; Yu, J.; Mizukami, Y.; Gopinathan, A.; Jodrell, D.I.; Evans, T.R.J.; et al. mTORC2 signaling drives the development and progression of pancreatic cancer. Cancer Res. 2017, 76, 6911–6923. [Google Scholar] [CrossRef]
- Weiler, M.; Blaes, J.; Pusch, S.; Sahm, F.; Czabanka, M.; Luger, S.; Bunse, L.; Solecki, G.; Eichwald, V.; Jugold, M.; et al. MTOR target NDRG1 confers MGMT-dependent resistance to alkylating chemotherapy. Proc. Natl. Acad. Sci. USA 2014, 111, 409–414. [Google Scholar] [CrossRef]
- Sun, J.; Zhang, D.; Bae, D.H.; Sahni, S.; Jansson, P.; Zheng, Y.; Zhao, Q.; Yue, F.; Zheng, M.; Kovacevic, Z.; et al. Metastasis suppressor, NDRG1, mediates its activity through signaling pathways and molecular motors. Carcinogenesis 2013, 34, 1943–1954. [Google Scholar] [CrossRef]
- Cheng, J.; Xie, H.Y.; Xu, X.; Wu, J.; Wei, X.; Su, R.; Zhang, W.; Lv, Z.; Zheng Shusen, S.; Zhou, L. NDRG1 as a biomarker for metastasis, recurrence and of poor prognosis in hepatocellular carcinoma. Cancer Lett. 2011, 310, 35–45. [Google Scholar] [CrossRef]
- Ridley, A.J. Cell Migration: Integrating Signals from Front to Back. Science 2003, 302, 1704–1710. [Google Scholar] [CrossRef]
- Sun, D.; Zhang, Y.; Qi, Y.; Zhou, X.; Lv, G. Prognostic significance of MMP-7 expression in colorectal cancer: A meta-analysis. Cancer Epidemiol. 2015, 39, 135–142. [Google Scholar] [CrossRef]
- Zhou, R.; Xu, L.; Ye, M.; Liao, M.; Du, H.; Chen, H. Formononetin inhibits migration and invasion of MDA-MB-231 and 4T1 breast cancer cells by suppressing MMP-2 and MMP-9 through PI3K/AKT signaling pathways. Horm. Metab. Res. 2014, 46, 753–760. [Google Scholar] [CrossRef]
- Oh, B.Y.; Hong, H.K.; Lee, W.Y.; Cho, Y.B. Animal models of colorectal cancer with liver metastasis. Cancer Lett. 2017, 387, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Koch, A.; Lang, S.A.; Wild, P.J.; Gantner, S.; Mahli, A.; Spanier, G.; Berneburg, M.; Müller, M.; Bosserhoff, A.K.; Hellerbrand, C. Glucose transporter isoform 1 expression enhances metastasis of malignant melanoma cells. Oncotarget 2015, 6, 32748–32760. [Google Scholar] [CrossRef] [PubMed]
- Guenzle, J.; Garrelfs, N.W.C.; Goeldner, J.M.; Weyerbrock, A. Cyclooxygenase (COX) Inhibition by Acetyl Salicylic Acid (ASA) Enhances Antitumor Effects of Nitric Oxide in Glioblastoma In Vitro. Mol. Neurobiol. 2019, 56, 6046–6055. [Google Scholar] [CrossRef] [PubMed]
- Beaver, C.M.; Ahmed, A.; Masters, J.R. Clonogenicity: Holoclones and Meroclones Contain Stem Cells. PLoS ONE 2014, 9, e89834. [Google Scholar] [CrossRef] [PubMed]
- Contag, C.H.; Bachmann, M.H. Advances in In Vivo Bioluminescence Imaging of Gene Expression. Annu. Rev. Biomed. Eng. 2002, 4, 235–260. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.T.; Schonig, K.; Berger, S.; Graewe, W.; Bujard, H. Long-term, noninvasive imaging of regulated gene expression in living mice. Genesis 2001, 29, 116–122. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guenzle, J.; Akasaka, H.; Joechle, K.; Reichardt, W.; Venkatasamy, A.; Hoeppner, J.; Hellerbrand, C.; Fichtner-Feigl, S.; Lang, S.A. Pharmacological Inhibition of mTORC2 Reduces Migration and Metastasis in Melanoma. Int. J. Mol. Sci. 2021, 22, 30. https://doi.org/10.3390/ijms22010030
Guenzle J, Akasaka H, Joechle K, Reichardt W, Venkatasamy A, Hoeppner J, Hellerbrand C, Fichtner-Feigl S, Lang SA. Pharmacological Inhibition of mTORC2 Reduces Migration and Metastasis in Melanoma. International Journal of Molecular Sciences. 2021; 22(1):30. https://doi.org/10.3390/ijms22010030
Chicago/Turabian StyleGuenzle, Jessica, Harue Akasaka, Katharina Joechle, Wilfried Reichardt, Aina Venkatasamy, Jens Hoeppner, Claus Hellerbrand, Stefan Fichtner-Feigl, and Sven A. Lang. 2021. "Pharmacological Inhibition of mTORC2 Reduces Migration and Metastasis in Melanoma" International Journal of Molecular Sciences 22, no. 1: 30. https://doi.org/10.3390/ijms22010030
APA StyleGuenzle, J., Akasaka, H., Joechle, K., Reichardt, W., Venkatasamy, A., Hoeppner, J., Hellerbrand, C., Fichtner-Feigl, S., & Lang, S. A. (2021). Pharmacological Inhibition of mTORC2 Reduces Migration and Metastasis in Melanoma. International Journal of Molecular Sciences, 22(1), 30. https://doi.org/10.3390/ijms22010030