Repurposing Tyrosine Kinase Inhibitors to Overcome Multidrug Resistance in Cancer: A Focus on Transporters and Lysosomal Sequestration

 , , ,
, , ,
Abstract
1. Introduction
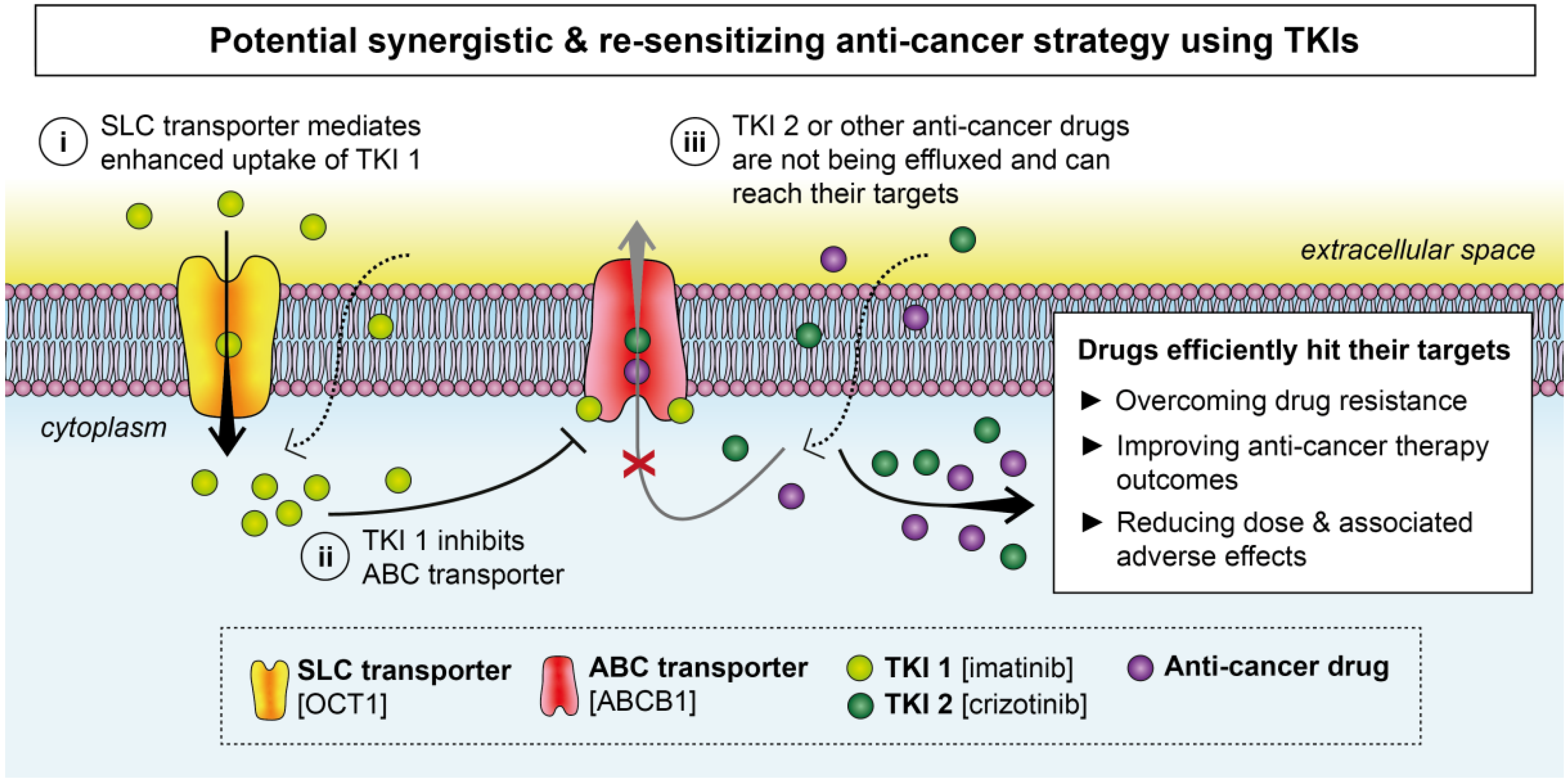
2. Effects of TKIs on Membrane Transporters
2.1. ABC Transporters
2.2. SLC Transporters
3. Lysosomal Sequestration
Overcoming Lysosomal Sequestration
4. Clinical Trials Repurposing TKIs in Combinational Strategies
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ABC | ATP-binding cassette |
| ALL | acute lymphoblastic leukemia |
| AML | acute myeloid leukemia |
| BRCP | breast cancer resistance protein |
| CML | chronic myeloid leukemia |
| EGFR | epidermal growth factor receptor |
| HER2 | human epidermal growth factor receptor 2 |
| HNSCC | head and neck squamous cell carcinoma |
| LAMP2 | lysosome-associated membrane protein 2 |
| MDR | multidrug resistance |
| MRP | multidrug resistance protein |
| NSCLC | non-small-cell lung carcinoma |
| OAT | organic anion transporter |
| OATP | organic anion-transporting polypeptide |
| OCT | organic cation transporter |
| OCTN | organic cation/carnitine transporter |
| Pgp | P-glycoprotein |
| ROS | reactive oxygen species |
| SLC | solute carrier |
| TKI | tyrosine kinase inhibitor |
| V-ATPase | vacuolar ATPase |
References
- Louvet, C.; Szot, G.L.; Lang, J.; Lee, M.R.; Martinier, N.; Bollag, G.; Zhu, S.; Weiss, A.; Bluestone, J.A. Tyrosine kinase inhibitors reverse type 1 diabetes in nonobese diabetic mice. Proc. Natl. Acad. Sci. USA 2008, 105, 18895–18900. [Google Scholar] [CrossRef]
- Weinblatt, M.E.; Kavanaugh, A.; Genovese, M.C.; Musser, T.K.; Grossbard, E.B.; Magilavy, D.B. An Oral Spleen Tyrosine Kinase (Syk) Inhibitor for Rheumatoid Arthritis. N. Engl. J. Med. 2010, 363, 1303–1312. [Google Scholar] [CrossRef]
- Emami, H.; Vucic, E.; Subramanian, S.; Abdelbaky, A.; Fayad, Z.A.; Du, S.; Roth, E.; Ballantyne, C.M.; Mohler, E.R.; Farkouh, M.E.; et al. The effect of BMS-582949, a P38 mitogen-activated protein kinase (P38 MAPK) inhibitor on arterial inflammation: A multicenter FDG-PET trial. Atherosclerosis 2015, 240, 490–496. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-P.; Hsieh, C.-H.; Wu, Y.-S. The Emergence of Drug Transporter-Mediated Multidrug Resistance to Cancer Chemotherapy. Mol. Pharm. 2011, 8, 1996–2011. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.S. Regulation of P-glycoprotein and other ABC drug transporters at the blood-brain barrier. Trends Pharmacol. Sci. 2010, 31, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Russo, A.; Saide, A.; Smaldone, S.; Faraonio, R.; Russo, G. Role of uL3 in multidrug resistance in p53-mutated lung cancer cells. Int. J. Mol. Sci. 2017, 18, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Hupfeld, T.; Chapuy, B.; Schrader, V.; Beutler, M.; Veltkamp, C.; Koch, R.; Cameron, S.; Aung, T.; Haase, D.; LaRosee, P.; et al. Tyrosinekinase inhibition facilitates cooperation of transcription factor SALL4 and ABC transporter A3 towards intrinsic CML cell drug resistance. Br. J. Haematol. 2013, 161, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Franke, R.M.; Filipski, K.K.; Hu, C.; Orwick, S.J.; de Bruijn, E.A.; Burger, H.; Baker, S.D.; Sparreboom, A. Interaction of Imatinib with Human Organic Ion Carriers. Clin. Cancer Res. 2008, 14, 3034–3038. [Google Scholar] [CrossRef]
- Li, W.; Sparidans, R.W.; Wang, Y.; Lebre, M.C.; Beijnen, J.H.; Schinkel, A.H. P-glycoprotein and breast cancer resistance protein restrict brigatinib brain accumulation and toxicity, and, alongside CYP3A, limit its oral availability. Pharmacol. Res. 2018, 137, 47–55. [Google Scholar] [CrossRef]
- Eadie, L.N.; Dang, P.; Goyne, J.M.; Hughes, T.P.; White, D.L. ABCC6 plays a significant role in the transport of nilotinib and dasatinib, and contributes to TKI resistance in vitro, in both cell lines and primary patient mononuclear cells. PLoS ONE 2018, 13, e0192180. [Google Scholar] [CrossRef]
- Zhao, H.; Huang, Y.; Shi, J.; Dai, Y.; Wu, L.; Zhou, H. ABCC10 plays a significant role in the transport of gefitinib and contributes to acquired resistance to gefitinib in NSCLC. Front. Pharmacol. 2018, 9, 1312. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.-C.; Chen, Y.-J.; Li, L.-Y.; Wei, Y.-L.; Hsu, S.-C.; Tsai, S.-L.; Chiu, P.-C.; Huang, W.-P.; Wang, Y.-N.; Chen, C.-H.; et al. Nuclear Translocation of Epidermal Growth Factor Receptor by Akt-dependent Phosphorylation Enhances Breast Cancer-resistant Protein Expression in Gefitinib-resistant Cells. J. Biol. Chem. 2011, 286, 20558–20568. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Kathawala, R.J.; Wang, Y.-J.; Zhang, Y.-K.; Patel, A.; Shukla, S.; Robey, R.W.; Talele, T.T.; Ashby, C.R.; Ambudkar, S.V.; et al. Linsitinib (OSI-906) antagonizes ATP-binding cassette subfamily G member 2 and subfamily C member 10-mediated drug resistance. Int. J. Biochem. Cell Biol. 2014, 51, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Kitazaki, T.; Oka, M.; Nakamura, Y.; Tsurutani, J.; Doi, S.; Yasunaga, M.; Takemura, M.; Yabuuchi, H.; Soda, H.; Kohno, S. Gefitinib, an EGFR tyrosine kinase inhibitor, directly inhibits the function of P-glycoprotein in multidrug resistant cancer cells. Lung Cancer 2005, 49, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.-N.; Zhang, Y.-K.; Wang, Y.-J.; Barbuti, A.M.; Zhu, X.-J.; Yu, X.-Y.; Wen, A.-W.; Wurpel, J.N.D.; Chen, Z.-S. Modulating the function of ATP-binding cassette subfamily G member 2 (ABCG2) with inhibitor cabozantinib. Pharmacol. Res. 2017, 119, 89–98. [Google Scholar] [CrossRef]
- Kuang, Y.-H.; Shen, T.; Chen, X.; Sodani, K.; Hopper-Borge, E.; Tiwari, A.K.; Lee, J.W.K.K.; Fu, L.-W.; Chen, Z.-S. Lapatinib and erlotinib are potent reversal agents for MRP7 (ABCC10)-mediated multidrug resistance. Biochem. Pharmacol. 2010, 79, 154–161. [Google Scholar] [CrossRef]
- Mi, Y.-J.; Liang, Y.-J.; Huang, H.-B.; Zhao, H.-Y.; Wu, C.-P.; Wang, F.; Tao, L.-Y.; Zhang, C.-Z.; Dai, C.-L.; Tiwari, A.K.; et al. Apatinib (YN968D1) reverses multidrug resistance by inhibiting the efflux function of multiple ATP-binding cassette transporters. Cancer Res. 2010, 70, 7981–7991. [Google Scholar] [CrossRef]
- Nakanishi, T.; Shiozawa, K.; Hassel, B.A.; Ross, D.D. Complex interaction of BCRP/ABCG2 and imatinib in BCR-ABL-expressing cells: BCRP-mediated resistance to imatinib is attenuated by imatinib-induced reduction of BCRP expression. Blood 2006, 108, 678–684. [Google Scholar] [CrossRef]
- Sen, R.; Natarajan, K.; Bhullar, J.; Shukla, S.; Fang, H.-B.; Cai, L.; Chen, Z.-S.; Ambudkar, S.V.; Baer, M.R. The novel BCR-ABL and FLT3 inhibitor ponatinib is a potent inhibitor of the MDR-associated ATP-binding cassette transporter ABCG2. Mol. Cancer Ther. 2012, 11, 2033–2044. [Google Scholar] [CrossRef]
- Tiwari, A.K.; Sodani, K.; Dai, C.-L.; Abuznait, A.H.; Singh, S.; Xiao, Z.-J.; Patel, A.; Talele, T.T.; Fu, L.; Kaddoumi, A.; et al. Nilotinib potentiates anticancer drug sensitivity in murine ABCB1-, ABCG2-, and ABCC10-multidrug resistance xenograft models. Cancer Lett. 2013, 328, 307–317. [Google Scholar] [CrossRef]
- Shukla, S.; Robey, R.W.; Bates, S.E.; Ambudkar, S.V. Sunitinib (Sutent, SU11248), a small-molecule receptor tyrosine kinase inhibitor, blocks function of the ATP-binding cassette (ABC) transporters P-glycoprotein (ABCB1) and ABCG2. Drug Metab. Dispos. 2009, 37, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Englinger, B.; Lötsch, D.; Pirker, C.; Mohr, T.; van Schoonhoven, S.; Boidol, B.; Lardeau, C.-H.; Spitzwieser, M.; Szabó, P.; Heffeter, P.; et al. Acquired nintedanib resistance in FGFR1-driven small cell lung cancer: Role of endothelin-A receptor-activated ABCB1 expression. Oncotarget 2016, 7, 50161–50179. [Google Scholar] [CrossRef] [PubMed]
- Ellegaard, A.-M.; Groth-Pedersen, L.; Oorschot, V.; Klumperman, J.; Kirkegaard, T.; Nylandsted, J.; Jaattela, M. Sunitinib and SU11652 Inhibit Acid Sphingomyelinase, Destabilize Lysosomes, and Inhibit Multidrug Resistance. Mol. Cancer Ther. 2013, 12, 2018–2020. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Chen, Z.; Franke, R.; Orwick, S.; Zhao, M.; Rudek, M.A.; Sparreboom, A.; Baker, S.D. Interaction of the Multikinase Inhibitors Sorafenib and Sunitinib with Solute Carriers and ATP-Binding Cassette Transporters. Clin. Cancer Res. 2009, 15, 6062–6069. [Google Scholar] [CrossRef] [PubMed]
- Sodani, K.; Patel, A.; Anreddy, N.; Singh, S.; Yang, D.-H.; Kathawala, R.J.; Kumar, P.; Talele, T.T.; Chen, Z.-S. Telatinib reverses chemotherapeutic multidrug resistance mediated by ABCG2 efflux transporter in vitro and in vivo. Biochem. Pharmacol. 2014, 89, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Vispute, S.G.; Chen, J.-J.; Sun, Y.-L.; Sodani, K.S.; Singh, S.; Pan, Y.; Talele, T.; Ashby, C.R.; Chen, Z.-S. Vemurafenib (PLX4032, Zelboraf®), a BRAF Inhibitor, Modulates ABCB1-, ABCG2-, and ABCC10-Mediated Multidrug Resistance. J. Can. Res. Updates 2013, 2, 306–317. [Google Scholar]
- Zhang, H.; Patel, A.; Wang, Y.-J.; Zhang, Y.-K.; Kathawala, R.J.; Qiu, L.-H.; Patel, B.A.; Huang, L.-H.; Shukla, S.; Yang, D.-H.; et al. The BTK Inhibitor Ibrutinib (PCI-32765) Overcomes Paclitaxel Resistance in ABCB1- and ABCC10-Overexpressing Cells and Tumors. Mol. Cancer Ther. 2017, 16, 1021–1030. [Google Scholar] [CrossRef]
- Hiwase, D.K.; White, D.; Zrim, S.; Saunders, V.; Melo, J.V.; Hughes, T.P. Nilotinib-mediated inhibition of ABCB1 increases intracellular concentration of dasatinib in CML cells: Implications for combination TKI therapy. Leukemia 2010, 24, 658–660. [Google Scholar] [CrossRef]
- Zhao, X.; Xie, J.; Chen, X.; Sim, H.M.; Zhang, X.; Liang, Y.; Singh, S.; Talele, T.T.; Sun, Y.; Ambudkar, S.V.; et al. Neratinib Reverses ATP-Binding Cassette B1-Mediated Chemotherapeutic Drug Resistance In Vitro, In Vivo, and Ex Vivo. Mol. Pharmacol. 2012, 82, 47–58. [Google Scholar] [CrossRef]
- Kathawala, R.J.; Sodani, K.; Chen, K.; Patel, A.; Abuznait, A.H.; Anreddy, N.; Sun, Y.-L.; Kaddoumi, A.; Ashby, C.R.; Chen, Z.-S. Masitinib Antagonizes ATP-Binding Cassette Subfamily C Member 10-Mediated Paclitaxel Resistance: A Preclinical Study. Mol. Cancer Ther. 2014, 13, 714–723. [Google Scholar] [CrossRef]
- Minocha, M.; Khurana, V.; Qin, B.; Pal, D.; Mitra, A.K. Enhanced brain accumulation of pazopanib by modulating P-gp and Bcrp1 mediated efflux with canertinib or erlotinib. Int. J. Pharm. 2012, 436, 127–134. [Google Scholar] [CrossRef] [PubMed]
- van Hoppe, S.; Sparidans, R.W.; Wagenaar, E.; Beijnen, J.H.; Schinkel, A.H. Breast cancer resistance protein (BCRP/ABCG2) and P-glycoprotein (P-gp/ABCB1) transport afatinib and restrict its oral availability and brain accumulation. Pharmacol. Res. 2017, 120, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Chen, Y.; To, K.K.W.; Wang, F.; Li, D.; Chen, L.; Fu, L. Alectinib (CH5424802) antagonizes ABCB1- and ABCG2-mediated multidrug resistance in vitro, in vivo and ex vivo. Exp. Mol. Med. 2017, 49, e303. [Google Scholar] [CrossRef] [PubMed]
- D’Cunha, R.; Bae, S.; Murry, D.J.; An, G. TKI combination therapy: Strategy to enhance dasatinib uptake by inhibiting Pgp- and BCRP-mediated efflux. Biopharm. Drug Dispos. 2016, 37, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Chuan Tang, S.; Nguyen, L.N.; Sparidans, R.W.; Wagenaar, E.; Beijnen, J.H.; Schinkel, A.H. Increased oral availability and brain accumulation of the ALK inhibitor crizotinib by coadministration of the P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) inhibitor elacridar. Int. J. Cancer 2014, 134, 1484–1494. [Google Scholar] [CrossRef]
- Xiang, Q.; Zhang, D.; Wang, J.; Zhang, H.; Zheng, Z.; Yu, D.; Li, Y.; Xu, J.; Chen, Y.; Shang, C. Cabozantinib reverses multidrug resistance of human hepatoma HepG2/adr cells by modulating the function of P-glycoprotein. Liver Int. 2015, 35, 1010–1023. [Google Scholar] [CrossRef]
- Tao, L.; Liang, Y.; Wang, F.; Chen, L.; Yan, Y.; Dai, C.; Fu, L. Cediranib (recentin, AZD2171) reverses ABCB1- and ABCC1-mediated multidrug resistance by inhibition of their transport function. Cancer Chemother. Pharmacol. 2009, 64, 961–969. [Google Scholar] [CrossRef]
- Hu, J.; Zhang, X.; Wang, F.; Wang, X.; Yang, K.; Xu, M.; To, K.K.W.; Li, Q.; Fu, L. Effect of ceritinib (LDK378) on enhancement of chemotherapeutic agents in ABCB1 and ABCG2 overexpressing cells in vitro and in vivo. Oncotarget 2015, 6, 44643–44659. [Google Scholar] [CrossRef]
- Wang, Y.-J.; Kathawala, R.J.; Zhang, Y.-K.; Patel, A.; Kumar, P.; Shukla, S.; Fung, K.L.; Ambudkar, S.V.; Talele, T.T.; Chen, Z.-S. Motesanib (AMG706), a potent multikinase inhibitor, antagonizes multidrug resistance by inhibiting the efflux activity of the ABCB1. Biochem. Pharmacol. 2014, 90, 367–378. [Google Scholar] [CrossRef]
- Chen, Z.; Chen, Y.; Xu, M.; Chen, L.; Zhang, X.; To, K.K.W.; Zhao, H.; Wang, F.; Xia, Z.; Chen, X.; et al. Osimertinib (AZD9291) Enhanced the Efficacy of Chemotherapeutic Agents in ABCB1- and ABCG2-Overexpressing Cells In Vitro, In Vivo, and Ex Vivo. Mol. Cancer Ther. 2016, 15, 1845–1858. [Google Scholar] [CrossRef]
- Liu, K.-J.; He, J.-H.; Su, X.-D.; Sim, H.-M.; Xie, J.-D.; Chen, X.-G.; Wang, F.; Liang, Y.-J.; Singh, S.; Sodani, K.; et al. Saracatinib (AZD0530) is a potent modulator of ABCB1-mediated multidrug resistance in vitro and in vivo. Int. J. Cancer 2013, 132, 224–235. [Google Scholar] [CrossRef]
- Zheng, L.; Wang, F.; Li, Y.; Zhang, X.; Chen, L.; Liang, Y.; Dai, C.; Yan, Y.; Tao, L.; Mi, Y.; et al. Vandetanib (Zactima, ZD6474) Antagonizes ABCC1- and ABCG2-Mediated Multidrug Resistance by Inhibition of Their Transport Function. PLoS ONE 2009, 4, e5172. [Google Scholar] [CrossRef] [PubMed]
- To, K.K.W.; Poon, D.C.; Wei, Y.; Wang, F.; Lin, G.; Fu, L. Vatalanib sensitizes ABCB1 and ABCG2-overexpressing multidrug resistant colon cancer cells to chemotherapy under hypoxia. Biochem. Pharmacol. 2015, 97, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, C.; Duan, Y.; Huo, X.; Meng, Q.; Liu, Z.; Sun, H.; Ma, X.; Liu, K. Afatinib Decreases P-Glycoprotein Expression to Promote Adriamycin Toxicity of A549T Cells. J. Cell. Biochem. 2018, 119, 414–423. [Google Scholar] [CrossRef] [PubMed]
- Hegedűs, C.; Özvegy-Laczka, C.; Apáti, Á.; Magócsi, M.; Német, K.; Őrfi, L.; Kéri, G.; Katona, M.; Takáts, Z.; Váradi, A.; et al. Interaction of nilotinib, dasatinib and bosutinib with ABCB1 and ABCG2: Implications for altered anti-cancer effects and pharmacological properties. Br. J. Pharmacol. 2009, 158, 1153–1164. [Google Scholar] [CrossRef]
- Shukla, S.; Sauna, Z.E.; Ambudkar, S.V. Evidence for the interaction of imatinib at the transport-substrate site(s) of the multidrug-resistance-linked ABC drug transporters ABCB1 (P-glycoprotein) and ABCG2. Leukemia 2008, 22, 445–447. [Google Scholar] [CrossRef]
- Dai, C.-L.; Tiwari, A.K.; Wu, C.-P.; Su, X.; Wang, S.-R.; Liu, D.; Ashby, C.R.; Huang, Y.; Robey, R.W.; Liang, Y.-J.; et al. Lapatinib (Tykerb, GW572016) Reverses Multidrug Resistance in Cancer Cells by Inhibiting the Activity of ATP-Binding Cassette Subfamily B Member 1 and G Member 2. Cancer Res. 2008, 68, 7905–7914. [Google Scholar] [CrossRef]
- Radic-Sarikas, B.; Halasz, M.; Huber, K.V.M.; Winter, G.E.; Tsafou, K.P.; Papamarkou, T.; Brunak, S.; Kolch, W.; Superti-Furga, G. Lapatinib potentiates cytotoxicity of YM155 in neuroblastoma via inhibition of the ABCB1 efflux transporter. Sci. Rep. 2017, 7, 1–8. [Google Scholar] [CrossRef]
- Zhang, H.; Patel, A.; Ma, S.L.; Li, X.J.; Zhang, Y.K.; Yang, P.Q.; Kathawala, R.J.; Wang, Y.J.; Anreddy, N.; Fu, L.W.; et al. In vitro, in vivo and ex vivo characterization of ibrutinib: A potent inhibitor of the efflux function of the transporter MRP1. Br. J. Pharmacol. 2014, 171, 5845–5857. [Google Scholar] [CrossRef]
- Shibayama, Y.; Nakano, K.; Maeda, H.; Taguchi, M.; Ikeda, R.; Sugawara, M.; Iseki, K.; Takeda, Y.; Yamada, K. Multidrug resistance protein 2 implicates anticancer drug-resistance to sorafenib. Biol. Pharm. Bull. 2011, 34, 433–435. [Google Scholar] [CrossRef]
- Gay, C.; Toulet, D.; Le Corre, P. Pharmacokinetic drug-drug interactions of tyrosine kinase inhibitors: A focus on cytochrome P450, transporters, and acid suppression therapy. Hematol. Oncol. 2017, 35, 259–280. [Google Scholar] [CrossRef] [PubMed]
- Radich, J.P.; Dai, H.; Mao, M.; Oehler, V.; Schelter, J.; Druker, B.; Sawyers, C.; Shah, N.; Stock, W.; Willman, C.L.; et al. Gene expression changes associated with progression and response in chronic myeloid leukemia. Proc. Natl. Acad. Sci. USA 2006, 103, 2794–2799. [Google Scholar] [CrossRef] [PubMed]
- Tomonari, T.; Takeishi, S.; Taniguchi, T.; Tanaka, T.; Tanaka, H.; Fujimoto, S.; Kimura, T.; Okamoto, K.; Miyamoto, H.; Muguruma, N.; et al. MRP3 as a novel resistance factor for sorafenib in hepatocellular carcinoma. Oncotarget 2016, 7, 7207–7215. [Google Scholar] [CrossRef] [PubMed]
- Cheung, L.; Yu, D.M.T.; Neiron, Z.; Failes, T.W.; Arndt, G.M.; Fletcher, J.I. Identification of new MRP4 inhibitors from a library of FDA approved drugs using a high-throughput bioluminescence screen. Biochem. Pharmacol. 2015, 93, 380–388. [Google Scholar] [CrossRef]
- Macias, R.I.R.; Sánchez-Martín, A.; Rodríguez-Macías, G.; Sánchez-Abarca, L.I.; Lozano, E.; Herraez, E.; Odero, M.D.; Díez-Martín, J.L.; Marin, J.J.G.; Briz, O. Role of drug transporters in the sensitivity of acute myeloid leukemia to sorafenib. Oncotarget 2018, 9, 28474–28485. [Google Scholar] [CrossRef]
- Shen, T.; Kuang, Y.-H.; Ashby, C.R.; Lei, Y.; Chen, A.; Zhou, Y.; Chen, X.; Tiwari, A.K.; Hopper-Borge, E.; Ouyang, J.; et al. Imatinib and Nilotinib Reverse Multidrug Resistance in Cancer Cells by Inhibiting the Efflux Activity of the MRP7 (ABCC10). PLoS ONE 2009, 4, e7520. [Google Scholar] [CrossRef]
- Sun, Y.-L.; Kumar, P.; Sodani, K.; Patel, A.; Pan, Y.; Baer, M.R.; Chen, Z.-S.; Jiang, W.-Q.; Pan, Y.; Pan, Y.; et al. Ponatinib enhances anticancer drug sensitivity in MRP7-overexpressing cells. Oncol. Rep. 2014, 31, 1605–1612. [Google Scholar] [CrossRef]
- Chen, Y.-J.; Huang, W.-C.; Wei, Y.-L.; Hsu, S.-C.; Yuan, P.; Lin, H.Y.; Wistuba, I.I.; Lee, J.J.; Yen, C.-J.; Su, W.-C.; et al. Elevated BCRP/ABCG2 Expression Confers Acquired Resistance to Gefitinib in Wild-Type EGFR-Expressing Cells. PLoS ONE 2011, 6, e21428. [Google Scholar] [CrossRef]
- Wang, D.-S.; Patel, A.; Shukla, S.; Zhang, Y.-K.; Wang, Y.-J.; Kathawala, R.J.; Robey, R.W.; Zhang, L.; Yang, D.-H.; Talele, T.T.; et al. Icotinib antagonizes ABCG2-mediated multidrug resistance, but not the pemetrexed resistance mediated by thymidylate synthase and ABCG2. Oncotarget 2014, 5, 4529–4542. [Google Scholar] [CrossRef]
- Kathawala, R.J.; Chen, J.-J.; Zhang, Y.-K.; Wang, Y.-J.; Patel, A.; Wang, D.-S.; Talele, T.T.; Ashby, C.R.; Chen, Z.-S. Masitinib antagonizes ATP-binding cassette subfamily G member 2-mediated multidrug resistance. Int. J. Oncol. 2014, 44, 1634–1642. [Google Scholar] [CrossRef]
- Li, J.; Kumar, P.; Anreddy, N.; Zhang, Y.-K.; Wang, Y.-J.; Chen, Y.; Talele, T.T.; Gupta, K.; Trombetta, L.D.; Chen, Z.-S. Quizartinib (AC220) reverses ABCG2-mediated multidrug resistance: In vitro and in vivo studies. Oncotarget 2017, 8, 93785–93799. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Yee, S.W.; Kim, R.B.; Giacomini, K.M. SLC transporters as therapeutic targets: Emerging opportunities. Nat. Rev. Drug Discov. 2015, 14, 543–560. [Google Scholar] [CrossRef] [PubMed]
- Engler, J.R.; Frede, A.; Saunders, V.A.; Zannettino, A.C.W.; Hughes, T.P.; White, D.L. Chronic Myeloid Leukemia CD34 cells have reduced uptake of imatinib due to low OCT-1 Activity. Leukemia 2010, 24, 765–770. [Google Scholar] [CrossRef] [PubMed]
- White, D.L.; Dang, P.; Engler, J.; Frede, A.; Zrim, S.; Osborn, M.; Saunders, V.A.; Manley, P.W.; Hughes, T.P. Functional activity of the OCT-1 protein is predictive of long-term outcome in patients with chronic-phase chronic myeloid leukemia treated with imatinib. J. Clin. Oncol. 2010, 28, 2761–2767. [Google Scholar] [CrossRef]
- Minematsu, T.; Giacomini, K.M. Interactions of Tyrosine Kinase Inhibitors with Organic Cation Transporters and Multidrug and Toxic Compound Extrusion Proteins. Mol. Cancer Ther. 2011, 10, 531–539. [Google Scholar] [CrossRef]
- Davies, A.; Jordanides, N.E.; Giannoudis, A.; Lucas, C.M.; Hatziieremia, S.; Harris, R.J.; Jørgensen, H.G.; Holyoake, T.L.; Pirmohamed, M.; Clark, R.E.; et al. Nilotinib concentration in cell lines and primary CD34+ chronic myeloid leukemia cells is not mediated by active uptake or efflux by major drug transporters. Leukemia 2009, 23, 1999–2006. [Google Scholar] [CrossRef]
- Elmeliegy, M.A.; Carcaboso, A.M.; Tagen, M.; Bai, F.; Stewart, C.F. Role of ATP-Binding Cassette and Solute Carrier Transporters in Erlotinib CNS Penetration and Intracellular Accumulation. Clin. Cancer Res. 2011, 17, 89–99. [Google Scholar] [CrossRef]
- Arakawa, H.; Omote, S.; Tamai, I. Inhibitory Effect of Crizotinib on Creatinine Uptake by Renal Secretory Transporter OCT2. J. Pharm. Sci. 2017, 106, 2899–2903. [Google Scholar] [CrossRef]
- Morrow, C.J.; Ghattas, M.; Smith, C.; Bönisch, H.; Bryce, R.A.; Hickinson, D.M.; Green, T.P.; Dive, C. Src family kinase inhibitor Saracatinib (AZD0530) impairs oxaliplatin uptake in colorectal cancer cells and blocks organic cation transporters. Cancer Res. 2010, 70, 5931–5941. [Google Scholar] [CrossRef]
- Zimmerman, E.I.; Gibson, A.A.; Hu, S.; Vasilyeva, A.; Orwick, S.J.; Du, G.; Mascara, G.P.; Ong, S.S.; Chen, T.; Vogel, P.; et al. Therapeutics, Targets, and Chemical Biology Multikinase Inhibitors Induce Cutaneous Toxicity through OAT6-Mediated Uptake and MAP3K7-Driven Cell Death. Cancer Res. 2016, 76, 117–126. [Google Scholar] [CrossRef]
- Hu, S.; Mathijssen, R.H.J.; de Bruijn, P.; Baker, S.D.; Sparreboom, A. Inhibition of OATP1B1 by tyrosine kinase inhibitors: In vitro–in vivo correlations. Br. J. Cancer 2014, 110, 894–898. [Google Scholar] [CrossRef] [PubMed]
- Bauer, M.; Matsuda, A.; Wulkersdorfer, B.; Philippe, C.; Traxl, A.; Özvegy-Laczka, C.; Stanek, J.; Nics, L.; Klebermass, E.-M.; Poschner, S.; et al. Influence of OATPs on Hepatic Disposition of Erlotinib Measured With Positron Emission Tomography. Clin. Pharmacol. Ther. 2018, 104, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.; Wang, L.; Clark, R.E.; Pirmohamed, M.; Reiffers, J.; Goldman, J.M.; Melo, J.V. Active transport of imatinib into and out of cells: Implications for drug resistance. Blood 2004, 104, 3739–3745. [Google Scholar] [CrossRef] [PubMed]
- White, D.L.; Saunders, V.A.; Dang, P.; Engler, J.; Zannettino, A.C.W.; Cambareri, A.C.; Quinn, S.R.; Manley, P.W.; Hughes, T.P. OCT-1–mediated influx is a key determinant of the intracellular uptake of imatinib but not nilotinib (AMN107): Reduced OCT-1 activity is the cause of low in vitro sensitivity to imatinib. Blood 2006, 108, 697–704. [Google Scholar] [CrossRef] [PubMed]
- De Duve, C.; De Barsy, T.; Poole, B.; Trouet, A.; Tulkens, P.; van Hoof, F. Lysosomotropic agents. Biochem. Pharmacol. 1974, 23, 2495–2531. [Google Scholar] [CrossRef]
- Kazmi, F.; Hensley, T.; Pope, C.; Funk, R.S.; Loewen, G.J.; Buckley, D.B.; Parkinson, A. Lysosomal Sequestration (Trapping) of Lipophilic Amine (Cationic Amphiphilic) Drugs in Immortalized Human Hepatocytes (Fa2N-4 Cells). Drug Metab. Dispos. 2013, 41, 897–905. [Google Scholar] [CrossRef]
- Gotink, K.J.; Broxterman, H.J.; Labots, M.; De Haas, R.R.; Dekker, H.; Honeywell, R.J.; Rudek, M.A.; Beerepoot, L.V.; Musters, R.J.; Jansen, G.; et al. Lysosomal sequestration of sunitinib: A novel mechanism of drug resistance. Clin. Cancer Res. 2011, 17, 7337–7346. [Google Scholar] [CrossRef]
- Wilson, J.N.; Liu, W.; Brown, A.S.; Landgraf, R. Binding-induced, turn-on fluorescence of the EGFR/ERBB kinase inhibitor, lapatinib. Org. Biomol. Chem. 2015, 13, 5006–5011. [Google Scholar] [CrossRef]
- Burger, H.; den Dekker, A.T.; Segeletz, S.; Boersma, A.W.M.; de Bruijn, P.; Debiec-Rychter, M.; Taguchi, T.; Sleijfer, S.; Sparreboom, A.; Mathijssen, R.H.J.; et al. Lysosomal Sequestration Determines Intracellular Imatinib Levels. Mol. Pharmacol. 2015, 88, 477–487. [Google Scholar] [CrossRef]
- Fu, D.; Zhou, J.; Zhu, W.S.; Manley, P.W.; Wang, Y.K.; Hood, T.; Wylie, A.; Xie, X.S. Imaging the intracellular distribution of tyrosine kinase inhibitors in living cells with quantitative hyperspectral stimulated Raman scattering. Nat. Chem. 2014, 6, 614–622. [Google Scholar] [CrossRef]
- Englinger, B.; Kallus, S.; Senkiv, J.; Heilos, D.; Gabler, L.; van Schoonhoven, S.; Terenzi, A.; Moser, P.; Pirker, C.; Timelthaler, G.; et al. Intrinsic fluorescence of the clinically approved multikinase inhibitor nintedanib reveals lysosomal sequestration as resistance mechanism in FGFR-driven lung cancer. J. Exp. Clin. Cancer Res. 2017, 36, 122. [Google Scholar] [CrossRef] [PubMed]
- Nadanaciva, S.; Lu, S.; Gebhard, D.F.; Jessen, B.A.; Pennie, W.D.; Will, Y. A high content screening assay for identifying lysosomotropic compounds. Toxicol. Vitr. 2011, 25, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Colombo, F.; Trombetta, E.; Cetrangolo, P.; Maggioni, M.; Razini, P.; De Santis, F.; Torrente, Y.; Prati, D.; Torresani, E.; Porretti, L. Giant Lysosomes as a Chemotherapy Resistance Mechanism in Hepatocellular Carcinoma Cells. PLoS ONE 2014, 9, e114787. [Google Scholar] [CrossRef] [PubMed]
- Gotink, K.J.; Rovithi, M.; de Haas, R.R.; Honeywell, R.J.; Dekker, H.; Poel, D.; Azijli, K.; Peters, G.J.; Broxterman, H.J.; Verheul, H.M.W. Cross-resistance to clinically used tyrosine kinase inhibitors sunitinib, sorafenib and pazopanib. Cell. Oncol. 2015, 38, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Ferrao, P.; Sincock, P.; Cole, S.; Ashman, L. Intracellular P-gp contributes to functional drug efflux and resistance in acute myeloid leukaemia. Leuk. Res. 2001, 25, 395–405. [Google Scholar] [CrossRef]
- Molinari, A.; Calcabrini, A.; Meschini, S.; Stringaro, A.; Crateri, P.; Toccacieli, L.; Marra, M.; Colone, M.; Cianfriglia, M.; Arancia, G. Subcellular Detection and Localization of the Drug Transporter P-Glycoprotein in Cultured Tumor Cells. Curr. Protein Pept. Sci. 2002, 3, 653–670. [Google Scholar] [CrossRef]
- Chapuy, B.; Panse, M.; Radunski, U.; Koch, R.; Wenzel, D.; Inagaki, N.; Haase, D.; Truemper, L.; Wulf, G.G. ABC transporter A3 facilitates lysosomal sequestration of imatinib and modulates susceptibility of chronic myeloid leukemia cell lines to this drug. Haematologica 2009, 94, 1528–1536. [Google Scholar] [CrossRef]
- Al-Akra, L.; Bae, D.-H.; Sahni, S.; Huang, M.L.H.; Park, K.C.; Lane, D.J.R.; Jansson, P.J.; Richardson, D.R. Tumor stressors induce two mechanisms of intracellular P-glycoprotein-mediated resistance that are overcome by lysosomal-targeted thiosemicarbazones. J. Biol. Chem. 2018, 293, 3562–3587. [Google Scholar] [CrossRef]
- Yamagishi, T.; Sahni, S.; Sharp, D.M.; Arvind, A.; Jansson, P.J.; Richardson, D.R. P-glycoprotein mediates drug resistance via a novel mechanism involving lysosomal sequestration. J. Biol. Chem. 2013, 288, 31761–31771. [Google Scholar] [CrossRef]
- Zama, I.N.; Hutson, T.E.; Elson, P.; Cleary, J.M.; Choueiri, T.K.; Heng, D.Y.C.; Ramaiya, N.; Michaelson, M.D.; Garcia, J.A.; Knox, J.J.; et al. Sunitinib rechallenge in metastatic renal cell carcinoma patients. Cancer 2010, 116, 5400–5406. [Google Scholar] [CrossRef]
- Gotink, K.J.; Broxterman, H.J.; Honeywell, R.J.; Dekker, H.; de Haas, R.R.; Miles, K.M.; Adelaiye, R.; Griffioen, A.W.; Peters, G.J.; Pili, R.; et al. Acquired tumor cell resistance to sunitinib causes resistance in a HT-29 human colon cancer xenograft mouse model without affecting sunitinib biodistribution or the tumor microvasculature. Oncoscience 2014, 1, 844–853. [Google Scholar] [CrossRef]
- McAfee, Q.; Zhang, Z.; Samanta, A.; Levi, S.M.; Ma, X.-H.; Piao, S.; Lynch, J.P.; Uehara, T.; Sepulveda, A.R.; Davis, L.E.; et al. Autophagy inhibitor Lys05 has single-agent antitumor activity and reproduces the phenotype of a genetic autophagy deficiency. Proc. Natl. Acad. Sci. USA 2012, 109, 8253–8258. [Google Scholar] [CrossRef]
- Rosenfeld, M.R.; Ye, X.; Supko, J.G.; Desideri, S.; Grossman, S.A.; Brem, S.; Mikkelson, T.; Wang, D.; Chang, Y.C.; Hu, J.; et al. A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy 2014, 10, 1359–1368. [Google Scholar] [CrossRef]
- Rangwala, R.; Chang, Y.C.; Hu, J.; Algazy, K.M.; Evans, T.L.; Fecher, L.A.; Schuchter, L.M.; Torigian, D.A.; Panosian, J.T.; Troxel, A.B.; et al. Combined MTOR and autophagy inhibition: Phase I trial of hydroxychloroquine and temsirolimus in patients with advanced solid tumors and melanoma. Autophagy 2014, 10, 1391–1402. [Google Scholar] [CrossRef]
- Nowak-Sliwinska, P.; Weiss, A.; van Beijnum, J.R.; Wong, T.J.; Kilarski, W.W.; Szewczyk, G.; Verheul, H.M.W.; Sarna, T.; van den Bergh, H.; Griffioen, A.W. Photoactivation of lysosomally sequestered sunitinib after angiostatic treatment causes vascular occlusion and enhances tumor growth inhibition. Cell Death Dis. 2015, 6, e1641. [Google Scholar] [CrossRef]
- Jansson, P.J.; Yamagishi, T.; Arvind, A.; Seebacher, N.; Gutierrez, E.; Stacy, A.; Maleki, S.; Sharp, D.; Sahni, S.; Richardson, D.R. Di-2-pyridylketone 4,4-dimethyl-3-thiosemicarbazone (Dp44mT) overcomes multidrug resistance by a novel mechanism involving the hijacking of lysosomal P-glycoprotein (Pgp). J. Biol. Chem. 2015, 290, 9588–9603. [Google Scholar] [CrossRef]
- Lan, C.-Y.; Wang, Y.; Xiong, Y.; Li, J.-D.; Shen, J.-X.; Li, Y.-F.; Zheng, M.; Zhang, Y.-N.; Feng, Y.-L.; Liu, Q.; et al. Apatinib combined with oral etoposide in patients with platinum-resistant or platinum-refractory ovarian cancer (AEROC): A phase 2, single-arm, prospective study. Lancet Oncol. 2018, 19, 1239–1246. [Google Scholar] [CrossRef]
- Wang, L.; Liang, L.; Yang, T.; Qiao, Y.; Xia, Y.; Liu, L.; Li, C.; Lu, P.; Jiang, X. A pilot clinical study of apatinib plus irinotecan in patients with recurrent high-grade glioma: Clinical Trial/Experimental Study. Medicine (Baltimore) 2017, 96, e9053. [Google Scholar] [CrossRef]
- Symonds, R.P.; Gourley, C.; Davidson, S.; Carty, K.; McCartney, E.; Rai, D.; Banerjee, S.; Jackson, D.; Lord, R.; McCormack, M.; et al. Cediranib combined with carboplatin and paclitaxel in patients with metastatic or recurrent cervical cancer (CIRCCa): A randomised, double-blind, placebo-controlled phase 2 trial. Lancet Oncol. 2015, 16, 1515–1524. [Google Scholar] [CrossRef]
- Valle, J.W.; Wasan, H.; Lopes, A.; Backen, A.C.; Palmer, D.H.; Morris, K.; Duggan, M.; Cunningham, D.; Anthoney, D.A.; Corrie, P.; et al. Cediranib or placebo in combination with cisplatin and gemcitabine chemotherapy for patients with advanced biliary tract cancer (ABC-03): A randomised phase 2 trial. Lancet Oncol. 2015, 16, 967–978. [Google Scholar] [CrossRef]
- Ahn, H.K.; Han, B.; Lee, S.J.; Lim, T.; Sun, J.-M.; Ahn, J.S.; Ahn, M.-J.; Park, K. ALK inhibitor crizotinib combined with intrathecal methotrexate treatment for non-small cell lung cancer with leptomeningeal carcinomatosis. Lung Cancer 2012, 76, 253–254. [Google Scholar] [CrossRef]
- Neal, J.W.; Dahlberg, S.E.; Wakelee, H.A.; Aisner, S.C.; Bowden, M.; Huang, Y.; Carbone, D.P.; Gerstner, G.J.; Lerner, R.E.; Rubin, J.L.; et al. Erlotinib, cabozantinib, or erlotinib plus cabozantinib as second-line or third-line treatment of patients with EGFR wild-type advanced non-small-cell lung cancer (ECOG-ACRIN 1512): A randomised, controlled, open-label, multicentre, phase 2 trial. Lancet Oncol. 2016, 17, 1661–1671. [Google Scholar] [CrossRef]
- Hirte, H.; Oza, A.; Swenerton, K.; Ellard, S.L.; Grimshaw, R.; Fisher, B.; Tsao, M.; Seymour, L. A phase II study of erlotinib (OSI-774) given in combination with carboplatin in patients with recurrent epithelial ovarian cancer (NCIC CTG IND.149). Gynecol. Oncol. 2010, 118, 308–312. [Google Scholar] [CrossRef]
- Massarelli, E.; Lin, H.; Ginsberg, L.E.; Tran, H.T.; Lee, J.J.; Canales, J.R.; Williams, M.D.; Blumenschein, G.R.; Lu, C.; Heymach, J.V.; et al. Phase II trial of everolimus and erlotinib in patients with platinum-resistant recurrent and/or metastatic head and neck squamous cell carcinoma. Ann. Oncol. 2015, 26, 1476–1480. [Google Scholar] [CrossRef]
- Yang, Z.Y.; Yuan, J.Q.; Di, M.Y.; Zheng, D.Y.; Chen, J.Z.; Ding, H.; Wu, X.Y.; Huang, Y.F.; Mao, C.; Tang, J.L. Gemcitabine Plus Erlotinib for Advanced Pancreatic Cancer: A Systematic Review with Meta-Analysis. PLoS ONE 2013, 8, e57528. [Google Scholar] [CrossRef]
- Lim, S.H.; Yun, J.; Lee, M.-Y.; Kim, H.J.; Kim, K.H.; Kim, S.H.; Lee, S.-C.; Bae, S.B.; Kim, C.K.; Lee, N.; et al. A randomized phase II clinical trial of gemcitabine, oxaliplatin, erlotinib combination chemotherapy versus gemcitabine and erlotinib in previously untreated patients with locally advanced or metastatic pancreatic cancer. J. Clin. Oncol. 2018, 36, 344. [Google Scholar] [CrossRef]
- Stewart, C.F.; Tagen, M.; Schwartzberg, L.S.; Blakely, L.J.; Tauer, K.W.; Smiley, L.M. Phase I dosage finding and pharmacokinetic study of intravenous topotecan and oral erlotinib in adults with refractory solid tumors. Cancer Chemother. Pharmacol. 2014, 73, 561–568. [Google Scholar] [CrossRef]
- Hosomi, Y.; Morita, S.; Sugawara, S.; Kato, T.; Fukuhara, T.; Gemma, A.; Takahashi, K.; Fujita, Y.; Harada, T.; Minato, K.; et al. Gefitinib Alone Versus Gefitinib Plus Chemotherapy for Non-Small-Cell Lung Cancer With Mutated Epidermal Growth Factor Receptor: NEJ009 Study. J. Clin. Oncol. 2020, 38, 115–123. [Google Scholar] [CrossRef]
- Cetin, B.; Benekli, M.; Turker, I.; Koral, L.; Ulas, A.; Dane, F.; Oksuzoglu, B.; Kaplan, M.A.; Koca, D.; Boruban, C.; et al. Lapatinib plus capecitabine for HER2-positive advanced breast cancer: A multicentre study of Anatolian Society of Medical Oncology (ASMO). J. Chemother. 2014, 26, 300–305. [Google Scholar] [CrossRef]
- Di Leo, A.; Gomez, H.L.; Aziz, Z.; Zvirbule, Z.; Bines, J.; Arbushites, M.C.; Guerrera, S.F.; Koehler, M.; Oliva, C.; Stein, S.H.; et al. Phase III, double-blind, randomized study comparing lapatinib plus paclitaxel with placebo plus paclitaxel as first-line treatment for metastatic breast cancer. J. Clin. Oncol. 2008, 26, 5544–5552. [Google Scholar] [CrossRef]
- Saura, C.; Garcia-saenz, J.A.; Xu, B.; Harb, W.; Moroose, R.; Pluard, T.; Cortés, J.; Kiger, C.; Germa, C.; Wang, K.; et al. Safety and Efficacy of Neratinib in Combination With Capecitabine in Patients With Metastatic Human Epidermal Growth Factor Receptor 2–Positive Breast Cancer. J. Clin. Oncol. 2014, 32, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Chow, L.; Xu, B.; Gupta, S.; Freyman, A.; Zhao, Y.; Abbas, R.; Van, M.V.; Bondarenko, I. Combination neratinib ( HKI-272 ) and paclitaxel therapy in patients with HER2-positive metastatic breast cancer. Br. J. Cancer 2013, 108, 1985–1993. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.-Y.; Joo, Y.-D.; Lim, S.-N.; Kim, S.-D.; Lee, J.-H.; Lee, J.-H.; Kim, D.H.; Kim, K.; Jung, C.W.; Kim, I.; et al. Nilotinib combined with multiagent chemotherapy for newly diagnosed Philadelphia-positive acute lymphoblastic leukemia. Blood 2015, 126, 746–756. [Google Scholar] [CrossRef]
- Reck, M.; Kaiser, R.; Mellemgaard, A.; Douillard, J.-Y.; Orlov, S.; Krzakowski, M.; von Pawel, J.; Gottfried, M.; Bondarenko, I.; Liao, M.; et al. Docetaxel plus nintedanib versus docetaxel plus placebo in patients with previously treated non-small-cell lung cancer (LUME-Lung 1): A phase 3, double-blind, randomised controlled trial. Lancet Oncol. 2014, 15, 143–155. [Google Scholar] [CrossRef]
- Serve, H.; Brunnberg, U.; Ottmann, O.; Brandts, C.; Steffen, B.; Krug, U.; Wagner, R.; Müller-Tidow, C.; Berdel, W.E.; Cristina Sauerland, M.; et al. Sorafenib in combination with intensive chemotherapy in elderly patients with acute myeloid leukemia: Results from a randomized, placebo-controlled trial. J. Clin. Oncol. 2013, 31, 3110–3118. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Shi, Q.; Knox, J.J.; Kaubisch, A.; Niedzwiecki, D.; Posey, J.; Tan, B.R.; Kavan, P.; Goel, R.; Lammers, P.E.; et al. Assessment of Treatment With Sorafenib Plus Doxorubicin vs Sorafenib Alone in Patients With Advanced Hepatocellular Carcinoma. JAMA Oncol. 2019, 5, 1582. [Google Scholar] [CrossRef]
- Sheng, X.; Cao, D.; Yuan, J.; Zhou, F.; Wei, Q.; Xie, X.; Cui, C.; Chi, Z.; Si, L.; Li, S.; et al. Sorafenib in combination with gemcitabine plus cisplatin chemotherapy in metastatic renal collecting duct carcinoma: A prospective, multicentre, single-arm, phase 2 study. Eur. J. Cancer 2018, 100, 1–7. [Google Scholar] [CrossRef]
- Crown, J.P.; Diéras, V.; Staroslawska, E.; Yardley, D.A.; Bachelot, T.; Davidson, N.; Wildiers, H.; Fasching, P.A.; Capitain, O.; Ramos, M.; et al. Phase III trial of sunitinib in combination with capecitabine versus capecitabine monotherapy for the treatment of patients with pretreated metastatic breast cancer. J. Clin. Oncol. 2013, 31, 2870–2878. [Google Scholar] [CrossRef]
- Bergh, J.; Bondarenko, I.M.; Lichinitser, M.R.; Liljegren, A.; Greil, R.; Voytko, N.L.; Makhson, A.N.; Cortes, J.; Lortholary, A.; Bischoff, J.; et al. First-line treatment of advanced breast cancer with sunitinib in combination with docetaxel versus docetaxel alone: Results of a prospective, randomized phase III study. J. Clin. Oncol. 2012, 30, 921–929. [Google Scholar] [CrossRef]
- Yi, J.H.; Lee, J.; Lee, J.; Park, S.H.; Park, J.O.; Yim, D.-S.; Park, Y.S.; Lim, H.Y.; Kang, W.K. Randomised phase II trial of docetaxel and sunitinib in patients with metastatic gastric cancer who were previously treated with fluoropyrimidine and platinum. Br. J. Cancer 2012, 106, 1469–1474. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Ross, R.W.; Jacobus, S.; Vaishampayan, U.; Yu, E.Y.; Quinn, D.I.; Hahn, N.M.; Hutson, T.E.; Sonpavde, G.; Morrissey, S.C.; et al. Double-blind, randomized trial of docetaxel plus vandetanib versus docetaxel plus placebo in platinum-pretreated metastatic urothelial cancer. J. Clin. Oncol. 2012, 30, 507–512. [Google Scholar] [CrossRef]
- Yang, C.; Gottfried, M.; Chan, V.; Raats, J.; De Marinis, F.; Abratt, R.P.; Read, J.; Vansteenkiste, J.F. Vandetanib Plus Pemetrexed for the Second-Line Treatment of Advanced Non-Small-Cell Lung Cancer: A Randomized, Double-Blind Phase III Trial. J. Clin. Oncol. 2011, 29, 1067–1074. [Google Scholar]
- Beretta, G.L.; Benedetti, V.; Cossa, G.; Assaraf, Y.G.A.; Bram, E.; Gatti, L.; Corna, E.; Carenini, N.; Colangelo, D.; Howell, S.B.; et al. Increased levels and defective glycosylation of MRPs in ovarian carcinoma cells resistant to oxaliplatin. Biochem. Pharmacol. 2010, 79, 1108–1117. [Google Scholar]
- Rudin, D.; Li, L.; Niu, N.; Kalari, K.R.; Gilbert, J.A.; Ames, M.M.; Wang, L. Gemcitabine Cytotoxicity: Interaction of Efflux and Deamination. J. Drug Metab. Toxicol. 2011, 2, 1–10. [Google Scholar] [CrossRef]
- Adamska, A.; Falasca, M. ATP-binding cassette transporters in progression and clinical outcome of pancreatic cancer: What is the way forward? World J. Gastroenterol. 2018, 24, 3222–3238. [Google Scholar] [CrossRef]
- Kemp, J.A.; Shim, M.S.; Heo, C.Y.; Kwon, Y.J. “Combo” nanomedicine: Co-delivery of multi-modal therapeutics for efficient, targeted, and safe cancer therapy. Adv. Drug Deliv. Rev. 2016, 98, 3–18. [Google Scholar]
- Zhou, Z.; Kennell, C.; Jafari, M.; Lee, J.Y.; Ruiz-Torres, S.J.; Waltz, S.E.; Lee, J.H. Sequential delivery of erlotinib and doxorubicin for enhanced triple negative Breast cancer treatment using polymeric nanoparticle. Int. J. Pharm. 2017, 530, 300–307. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| ABC Transporter | Substrate | Inhibitor | Substrate/Inhibitor |
|---|---|---|---|
| ABCA3 | dasatinib [7]; imatinib [7]; nilotinib [7] | – | – |
| ABCB1 (P-glycoprotein, MDR1) | brigatinib [9]; crizotinib [35] | cabozantinib [36]; canertinib * [31]; cediranib * [37]; ceritinib [38]; erlotinib [34]; gefitinib [14]; motesanib * [39]; neratinib [29]; osimertinib [40]; regorafenib [34]; saracatinib * [41]; sorafenib [34]; sunitinib [21]; vandetanib [42]; vatalanib * [43] | afatinib [44]; alectinib [33]; apatinib * [17]; bosutinib [45]; dasatinib [45]; ibrutinib [27]; imatinib [46]; lapatinib [47,48]; nilotinib [45]; nintedanib [22]; pazopanib [31,34]; ponatinib [19] |
| ABCC1 (MRP1) | – | cediranib * [37]; ibrutinib [49]; sunitinib [21]; vandetanib [42] | – |
| ABCC2 (MRP2) | sorafenib [50] | sunitinib [51] | – |
| ABCC3 (MRP3) | imatinib [52]; sorafenib [53] | – | – |
| ABCC4 (MRP4) | imatinib [8] | erlotinib [54]; gefitinib [54]; sorafenib [55]; sunitinib [51] | – |
| ABCC6 (MRP6) | dasatinib [10]; nilotinib [10] | – | – |
| ABCC10 (MRP7) | gefitinib [11] | erlotinib [16]; ibrutinib [27]; imatinib [56]; lapatinib [16]; linsitinib * [13]; masitinib * [30]; nilotinib [20]; ponatinib [57]; sorafenib [24] | – |
| ABCC11 (MRP8) | – | sorafenib [24] | – |
| ABCG2 (BRCP) | brigatinib [9]; gefitinib [58] | axitinib [34]; cabozantinib [15]; canertinib * [31]; ceritinib [38]; erlotinib [34]; icotinib * [59]; linsitinib * [13]; masitinib * [60]; osimertinib [40]; quizartinib * [61]; regorafenib [34]; sorafenib [24]; sunitinib [21]; tandutinib * [15]; vandetanib [42]; vatalanib * [43] | afatinib [32]; alectinib [33]; apatinib * [17]; bosutinib [45]; dasatinib [45]; imatinib [46]; lapatinib [47]; nilotinib [45]; pazopanib [31,34]; ponatinib [19]; telatinib * [25] |
| SLC Transporter | Substrate | Inhibitor |
|---|---|---|
| OCT1 (SLC22A1) | imatinib [63,64] sorafenib [55] | crizotinib [51] erlotinib [65] gefitinib [65] nilotinib [66] sunitinib [65] |
| OCT2 (SLC22A2) | erlotinib [67] | crizotinib [68] gefitinib [65] nilotinib [65] saracatinib [69] sunitinib [65] vandetanib [68] |
| OCT3 (SLC22A3) | – | gefitinib [65] nilotinib [65] sunitinib [65] |
| OCTN2 (SLC22A5) | imatinib [8] | – |
| OAT3 (SLC22A8) | erlotinib [67] | – |
| OAT6 (SLC22A20) | sorafenib [70] | – |
| OATP1A2 (SLCO1A2) | imatinib [8] | – |
| OATP1B1 (SLCO1B1) | – | axitinib [71] lapatinib [51] nilotinib [71] pazopanib [71] sorafenib [71] |
| OATP1B3 (SLCO1B3) | imatinib [8] | – |
| OATP2B1 (SLCO2B1) | erlotinib [72] | – |
| TKI | pKa 1 | LogP 2 | Reference |
|---|---|---|---|
| dasatinib | 8.49 | 3.82 | [82] |
| gefitinib | 6.85 | 3.75 | [76] |
| imatinib | 8.10 | 4.50 | [79] |
| lapatinib | 7.20 | 4.64 | [76] |
| nilotinib | 6.30 | 5.36 | [80] |
| nintedanib | 7.90 | 3.60 | [81] |
| pazopanib | 5.07 | 3.60 | [84] |
| sorafenib | 4.34 | 2.03 | [83] |
| sunitinib | 9.04 | 5.20 | [77] |
| Combination of Drugs | Malignancy | Reference | |
|---|---|---|---|
| apatinib * | + etoposide + irinotecan | ovarian cancer high-grade glioma | [97] [98] |
| cediranib * | + carboplatin, paclitaxel + cisplatin, gemcitabine | cervical cancer biliary tract cancer | [99] [100] |
| crizotinib | + methotrexate | NSCLC | [101] |
| erlotinib | + cabozantinib + carboplatin + everolimus + gemcitabine + gemcitabine, oxaliplatin + topotecan | NSCLC ovarian carcinoma HNSCC pancreatic cancer pancreatic cancer solid tumors | [102] [103] [104] [105] [106] [107] |
| gefitinib | + carboplatin, pemetrexed | NSCLC | [108] |
| lapatinib | + capecitabine + paclitaxel | breast cancer breast cancer | [109] [110] |
| neratinib | + capecitabine + paclitaxel | breast cancer breast cancer | [111] [112] |
| nilotinib | + vincristine, daunorubucin | ALL | [113] |
| nintedanib | + docetaxel | NSCLC | [114] |
| sorafenib | + cytarabine, daunorubicin + doxorubicin + gemcitabine, cisplatin | AML hepatocellular carcinoma collecting duct carcinoma | [115] [116] [117] |
| sunitinib | + capecitabine + docetaxel | breast cancer breast cancer, gastric cancer | [118] [119,120] |
| vandetanib | + docetaxel + pemetrexed | urothelial cancer NSCLC | [121] [122] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krchniakova, M.; Skoda, J.; Neradil, J.; Chlapek, P.; Veselska, R. Repurposing Tyrosine Kinase Inhibitors to Overcome Multidrug Resistance in Cancer: A Focus on Transporters and Lysosomal Sequestration. Int. J. Mol. Sci. 2020, 21, 3157. https://doi.org/10.3390/ijms21093157
Krchniakova M, Skoda J, Neradil J, Chlapek P, Veselska R. Repurposing Tyrosine Kinase Inhibitors to Overcome Multidrug Resistance in Cancer: A Focus on Transporters and Lysosomal Sequestration. International Journal of Molecular Sciences. 2020; 21(9):3157. https://doi.org/10.3390/ijms21093157
Chicago/Turabian StyleKrchniakova, Maria, Jan Skoda, Jakub Neradil, Petr Chlapek, and Renata Veselska. 2020. "Repurposing Tyrosine Kinase Inhibitors to Overcome Multidrug Resistance in Cancer: A Focus on Transporters and Lysosomal Sequestration" International Journal of Molecular Sciences 21, no. 9: 3157. https://doi.org/10.3390/ijms21093157
APA StyleKrchniakova, M., Skoda, J., Neradil, J., Chlapek, P., & Veselska, R. (2020). Repurposing Tyrosine Kinase Inhibitors to Overcome Multidrug Resistance in Cancer: A Focus on Transporters and Lysosomal Sequestration. International Journal of Molecular Sciences, 21(9), 3157. https://doi.org/10.3390/ijms21093157