Update on the Neurobiology of Vascular Cognitive Impairment: From Lab to Clinic


 , , and
, , and 
Abstract
1. Introduction
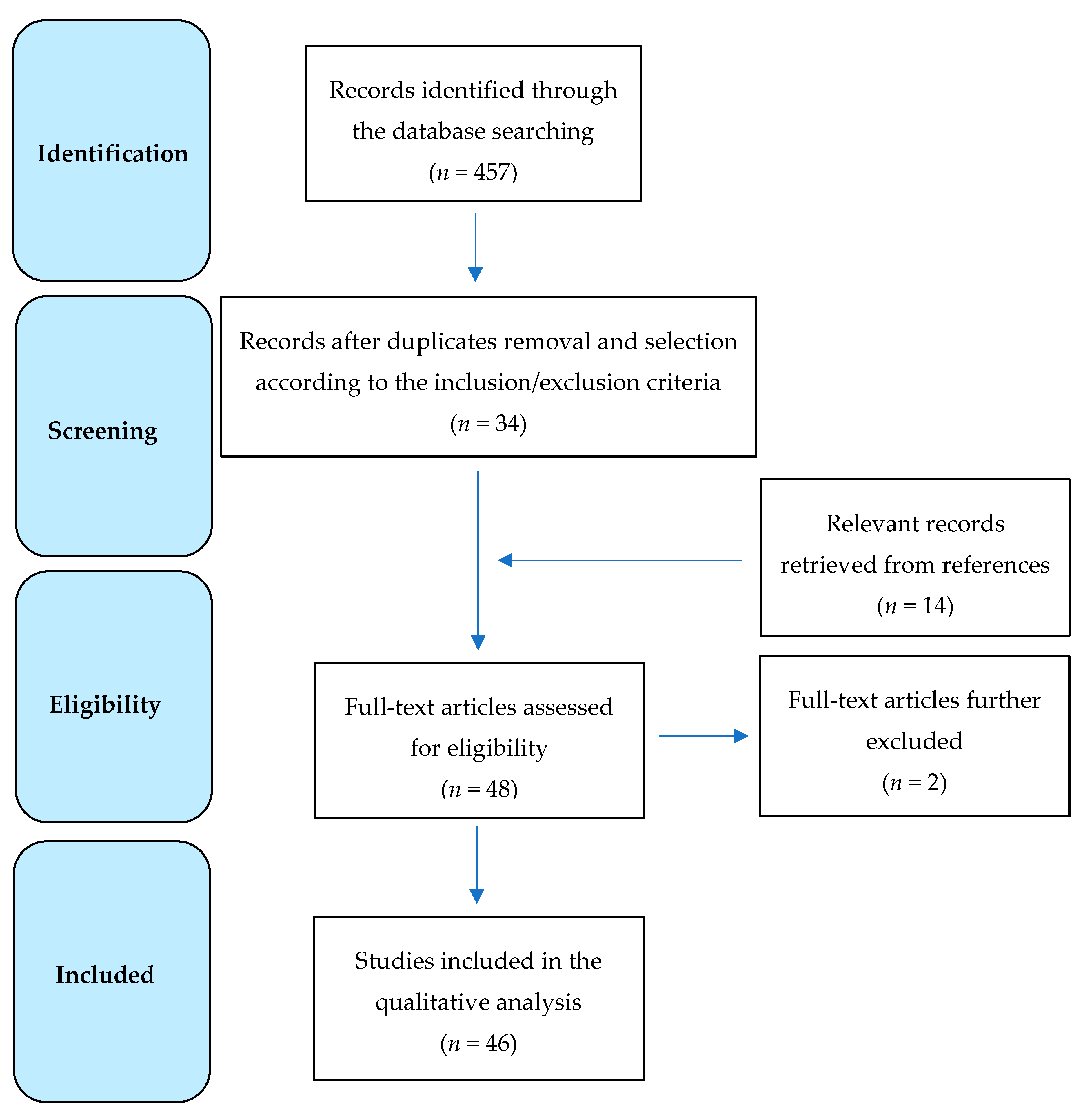
2. Data Source and Selection
- ((“biochemistries”[All Fields] OR “biochemistry”[MeSH Terms]) OR “biochemistry”[All Fields]) AND ((“dementia, vascular”[MeSH Terms] OR (“dementia”[All Fields] AND “vascular”[All Fields])) OR “vascular dementia”[All Fields]);
- ((“biochemistries”[All Fields] OR “biochemistry”[MeSH Terms]) OR “biochemistry”[All Fields]) AND “vascular”[All Fields] AND ((((“cognitive dysfunction”[MeSH Terms] OR (“cognitive”[All Fields] AND “dysfunction”[All Fields])) OR “cognitive dysfunction”[All Fields]) OR (“cognitive”[All Fields] AND “impairment”[All Fields])) OR “cognitive impairment”[All Fields]);
- “cortical excitability”[MeSH Terms] OR (“cortical”[All Fields] AND “excitability”[All Fields]) OR “cortical excitability”[All Fields] AND (“dementia, vascular”[MeSH Terms] OR (“dementia”[All Fields] AND “vascular”[All Fields]) OR “vascular dementia”[All Fields]);
- “transcranial magnetic stimulation”[MeSH Terms] OR “transcranial magnetic stimulation”[All Fields] AND “vascular”[All Fields] AND ((((“cognitive dysfunction”[MeSH Terms] OR (“cognitive”[All Fields] AND “dysfunction”[All Fields])) OR “cognitive dysfunction”[All Fields]) OR (“cognitive”[All Fields] AND “impairment”[All Fields])) OR “cognitive impairment”[All Fields]).
3. Results
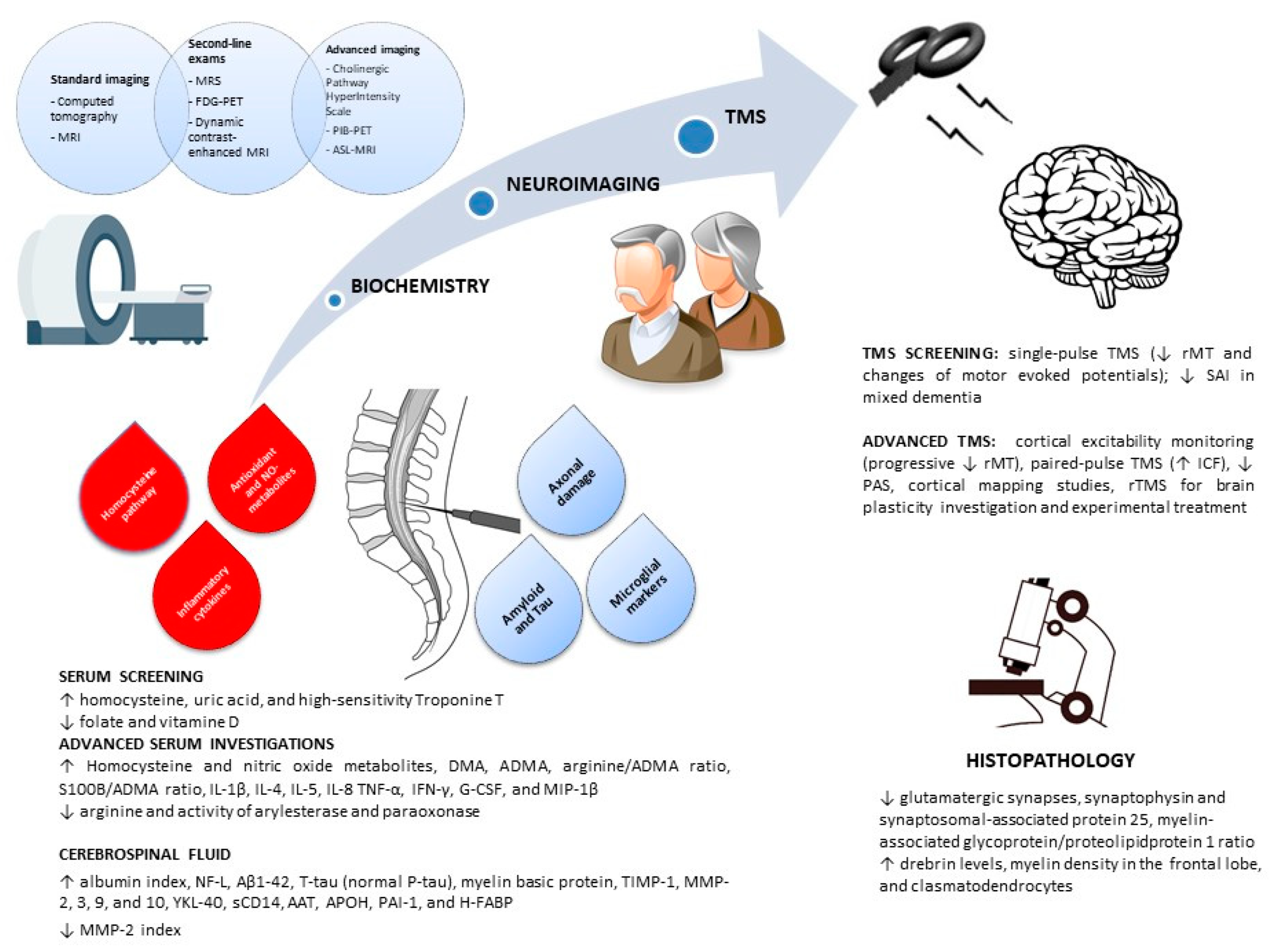
3.1. Serum
3.2. Cerebrospinal Fluid
3.3. Neuroimaging
3.4. Transcranial Magnetic Stimulation
3.5. Histopathology
4. Discussion
4.1. Summary of Findings
4.2. Limitations and Future Directions
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AAT | alpha1-antitrypsin |
| AD | Alzheimer’s disease |
| ADMA | asymmetric dimethylarginine |
| APO | apolipoprotein |
| ASL | arterial spin labelling |
| Aβ | amyloid-beta |
| Bax | BCL2 associated X protein |
| BBB | blood–brain barrier |
| Bcl-2 | B-cell lymphoma 2 |
| BDNF | brain-derived neurotrophic factor |
| CAA | cerebral amyloid angiopathy |
| CADASIL | cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy |
| CBF | cerebral blood flow |
| CNS | central nervous system |
| CSF | cerebrospinal fluid |
| DMA | dimethylarginine |
| FDG | 18-fluorodeoxyglucose |
| GABA | gamma-amino-butyric acid |
| G-CSF | granulocyte-colony stimulating factor |
| H-FABP | heart-type fatty acid binding protein |
| ICF | intracortical facilitation |
| IFN-γ | interferon-gamma |
| IL | interleukin |
| LBD | Lewy body dementia |
| L-DOPA | levo-3,4-dihydroxyphenylalanine |
| MCI | mild cognitive impairment |
| MIP-1β | macrophage inflammatory protein 1beta |
| MMP | matrix metalloproteinases |
| MMSE | Mini Mental State Examination |
| MRI | magnetic resonance imaging |
| MRS | magnetic resonance spectroscopy |
| NF-L | neurofilament-light protein |
| NO | nitric oxide |
| PAI-1 | plasminogen activator inhibitor 1 |
| PAS | paired associative stimulation |
| PET | positron emission tomography |
| PIB | Pittsburgh compound B |
| P-tau | phosphorilated tau |
| rMT | resting motor threshold |
| rTMS | repetitive transcranial magnetic stimulation |
| SAI | short-latency afferent inhibition |
| SIVD | subcortical ischemic vascular disease |
| TCD | transcranial Doppler ultrasound |
| TIMP-1 | tissue inhibitors of metalloproteinases 1 |
| TMS | transcranial magnetic stimulation |
| TNF-α | tumor necrosis factor alpha |
| T-tau | total tau |
| VaD | vascular dementia |
| VCI | vascular cognitive impairment |
| VGLUT1 | vesicular glutamate transporter 1 |
| WMLs | white matter lesions |
References
- Parfenov, V.A.; Ostroumova, O.D.; Ostroumova, T.M.; Kochetkov, A.I.; Fateeva, V.V.; Khacheva, K.K.; Khakimova, G.R.; Epstein, O.I. Vascular cognitive impairment: Pathophysiological mechanisms, insights into structural basis, and perspectives in specific treatments. Neuropsychiatr. Dis. Treat. 2019, 15, 1381–1402. [Google Scholar] [CrossRef]
- Jellinger, K.A. Pathology and pathogenesis of vascular cognitive impairment-a critical update. Front. Aging Neurosci. 2013, 5, 17. [Google Scholar] [CrossRef] [PubMed]
- Skrobot, O.A.; Black, S.E.; Chen, C.; DeCarli, C.; Erkinjuntti, T.; Ford, G.A.; Kalaria, R.N.; O’Brien, J.; Pantoni, L.; Pasquier, F.; et al. Progress toward standardized diagnosis of vascular cognitive impairment: Guidelines from the Vascular Impairment of Cognition Classification Consensus Study. Alzheimers Dement. J. Alzheimers Assoc. 2018, 14, 280–292. [Google Scholar] [CrossRef]
- O’Brien, J.T.; Thomas, A. Vascular dementia. Lancet Lond. Engl. 2015, 386, 1698–1706. [Google Scholar] [CrossRef]
- Moorhouse, P.; Rockwood, K. Vascular cognitive impairment: Current concepts and clinical developments. Lancet Neurol. 2008, 7, 246–255. [Google Scholar] [CrossRef]
- Cantone, M.; Lanza, G.; Bella, R.; Pennisi, G.; Santalucia, P.; Bramanti, P.; Pennisi, M. Fear and disgust: Case report of two uncommon emotional disturbances evoked by visual disperceptions after a right temporal-insular stroke. BMC Neurol. 2019, 19, 193. [Google Scholar] [CrossRef]
- van der Flier, W.M.; Skoog, I.; Schneider, J.A.; Pantoni, L.; Mok, V.; Chen, C.L.H.; Scheltens, P. Vascular cognitive impairment. Nat. Rev. Dis. Primer 2018, 4, 18003. [Google Scholar] [CrossRef]
- Skrobot, O.A.; Attems, J.; Esiri, M.; Hortobágyi, T.; Ironside, J.W.; Kalaria, R.N.; King, A.; Lammie, G.A.; Mann, D.; Neal, J.; et al. Vascular cognitive impairment neuropathology guidelines (VCING): The contribution of cerebrovascular pathology to cognitive impairment. Brain J. Neurol. 2016, 139, 2957–2969. [Google Scholar] [CrossRef]
- Cervellati, C.; Romani, A.; Seripa, D.; Cremonini, E.; Bosi, C.; Magon, S.; Passaro, A.; Bergamini, C.M.; Pilotto, A.; Zuliani, G. Oxidative balance, homocysteine, and uric acid levels in older patients with Late Onset Alzheimer’s Disease or Vascular Dementia. J. Neurol. Sci. 2014, 337, 156–161. [Google Scholar] [CrossRef]
- Schneider, A.L.C.; Rawlings, A.M.; Sharrett, A.R.; Alonso, A.; Mosley, T.H.; Hoogeveen, R.C.; Ballantyne, C.M.; Gottesman, R.F.; Selvin, E. High-sensitivity cardiac troponin T and cognitive function and dementia risk: The atherosclerosis risk in communities study. Eur. Heart J. 2014, 35, 1817–1824. [Google Scholar] [CrossRef]
- Gao, Q.; Fan, Y.; Mu, L.-Y.; Ma, L.; Song, Z.-Q.; Zhang, Y.-N. S100B and ADMA in cerebral small vessel disease and cognitive dysfunction. J. Neurol. Sci. 2015, 354, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Zhou, K.; Li, H.; Dong, X.; Tan, G.; Chai, Y.; Wang, W.; Bi, X. Potential of serum metabolites for diagnosing post-stroke cognitive impairment. Mol. Biosyst. 2015, 11, 3287–3296. [Google Scholar] [CrossRef] [PubMed]
- Cervellati, C.; Trentini, A.; Romani, A.; Bellini, T.; Bosi, C.; Ortolani, B.; Zurlo, A.; Passaro, A.; Seripa, D.; Zuliani, G. Serum paraoxonase and arylesterase activities of paraoxonase-1 (PON-1), mild cognitive impairment, and 2-year conversion to dementia: A pilot study. J. Neurochem. 2015, 135, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Castellazzi, M.; Trentini, A.; Romani, A.; Valacchi, G.; Bellini, T.; Bonaccorsi, G.; Fainardi, E.; Cavicchio, C.; Passaro, A.; Zuliani, G.; et al. Decreased arylesterase activity of paraoxonase-1 (PON-1) might be a common denominator of neuroinflammatory and neurodegenerative diseases. Int. J. Biochem. Cell Biol. 2016, 81, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Paroni, G.; Michele, L.; Andrea, F.; Grazia, D.; Filomena, C.; Francesco, P.; Leandro, C.; Maria, U.; Carolina, G.; Massimiliano, C.; et al. Brain Atrophy, Anti-Smooth Muscle Antibody and Cognitive Impairment: An Association Study. Aging Dis. 2016, 7, 318. [Google Scholar] [CrossRef]
- Moretti, R.; Caruso, P.; Dal Ben, M.; Conti, C.; Gazzin, S.; Tiribelli, C. Vitamin D, Homocysteine, and Folate in Subcortical Vascular Dementia and Alzheimer Dementia. Front. Aging Neurosci. 2017, 9, 169. [Google Scholar] [CrossRef]
- Jonsson, M.; Zetterberg, H.; Van Straaten, E.; Lind, K.; Syversen, S.; Edman, Å.; Blennow, K.; Rosengren, L.; Pantoni, L.; Inzitari, D.; et al. Cerebrospinal fluid biomarkers of white matter lesions - cross-sectional results from the LADIS study: Biochemical markers for white-matter lesions. Eur. J. Neurol. 2010, 17, 377–382. [Google Scholar] [CrossRef]
- Formichi, P.; Parnetti, L.; Radi, E.; Cevenini, G.; Dotti, M.T.; Federico, A. CSF Biomarkers Profile in CADASIL—A Model of Pure Vascular Dementia: Usefulness in Differential Diagnosis in the Dementia Disorder. Int. J. Alzheimers Dis. 2010, 2010, 1–6. [Google Scholar] [CrossRef][Green Version]
- Spies, P.E.; Slats, D.; Sjögren, J.M.C.; Kremer, B.P.H.; Verhey, F.R.J. The Cerebrospinal Fluid Amyloid 42/40 Ratio in the Differentiation of Alzheimer’s Disease from Non-Alzheimer’s Dementia. Curr. Alzheimer Res. 2010, 7, 470–476. [Google Scholar] [CrossRef]
- Bjerke, M.; Zetterberg, H.; Edman, Å.; Blennow, K.; Wallin, A.; Andreasson, U. Cerebrospinal Fluid Matrix Metalloproteinases and Tissue Inhibitor of Metalloproteinases in Combination with Subcortical and Cortical Biomarkers in Vascular Dementia and Alzheimer’s Disease. J. Alzheimers Dis. 2011, 27, 665–676. [Google Scholar] [CrossRef]
- Candelario-Jalil, E.; Thompson, J.; Taheri, S.; Grossetete, M.; Adair, J.C.; Edmonds, E.; Prestopnik, J.; Wills, J.; Rosenberg, G.A. Matrix Metalloproteinases Are Associated With Increased Blood–Brain Barrier Opening in Vascular Cognitive Impairment. Stroke 2011, 42, 1345–1350. [Google Scholar] [CrossRef] [PubMed]
- Öhrfelt, A.; Andreasson, U.; Simon, A.; Zetterberg, H.; Edman, Å.; Potter, W.; Holder, D.; Devanarayan, V.; Seeburger, J.; Smith, A.D.; et al. Screening for New Biomarkers for Subcortical Vascular Dementia and Alzheimer’s Disease. Dement. Geriatr. Cogn. Disord. Extra 2011, 1, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Olsson, B.; Hertze, J.; Lautner, R.; Zetterberg, H.; Nägga, K.; Höglund, K.; Basun, H.; Annas, P.; Lannfelt, L.; Andreasen, N.; et al. Microglial Markers are Elevated in the Prodromal Phase of Alzheimer’s Disease and Vascular Dementia. J. Alzheimers Dis. 2012, 33, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Kaerst, L.; Kuhlmann, A.; Wedekind, D.; Stoeck, K.; Lange, P.; Zerr, I. Cerebrospinal fluid biomarkers in Alzheimer’s disease, vascular dementia and ischemic stroke patients: A critical analysis. J. Neurol. 2013, 260, 2722–2727. [Google Scholar] [CrossRef]
- Li, Y.; Ge, F.; Zhang, Y.; You, H.; Zhang, Z. Cerebrospinal Fluid Biomarkers in Dementia Patients with Cerebral Amyloid Angiopathy. Chin. Med. Sci. J. 2015, 30, 170–173. [Google Scholar] [CrossRef]
- Rosenberg, G.A.; Prestopnik, J.; Adair, J.C.; Huisa, B.N.; Knoefel, J.; Caprihan, A.; Gasparovic, C.; Thompson, J.; Erhardt, E.B.; Schrader, R. Validation of biomarkers in subcortical ischaemic vascular disease of the Binswanger type: Approach to targeted treatment trials. J. Neurol. Neurosurg. Psychiatry 2015, 86, 1324–1330. [Google Scholar] [CrossRef]
- Pascual, B.; Prieto, E.; Arbizu, J.; Marti-Climent, J.; Olier, J.; Masdeu, J.C. Brain Glucose Metabolism in Vascular White Matter Disease With Dementia: Differentiation From Alzheimer Disease. Stroke 2010, 41, 2889–2893. [Google Scholar] [CrossRef]
- Mok, V.; Leung, E.Y.L.; Chu, W.; Chen, S.; Wong, A.; Xiong, Y.; Lam, W.; Ho, C.L.; Wong, K.S. Pittsburgh compound B binding in poststroke dementia. J. Neurol. Sci. 2010, 290, 135–137. [Google Scholar] [CrossRef]
- Kim, S.H.; Kang, H.S.; Kim, H.J.; Moon, Y.; Ryu, H.J.; Kim, M.Y.; Han, S.-H. The Effect of Ischemic Cholinergic Damage on Cognition in Patients With Subcortical Vascular Cognitive Impairment. J. Geriatr. Psychiatry Neurol. 2012, 25, 122–127. [Google Scholar] [CrossRef]
- Gasparovic, C.; Prestopnik, J.; Thompson, J.; Taheri, S.; Huisa, B.; Schrader, R.; Adair, J.C.; Rosenberg, G.A. 1H-MR spectroscopy metabolite levels correlate with executive function in vascular cognitive impairment. J. Neurol. Neurosurg. Psychiatry 2013, 84, 715–721. [Google Scholar] [CrossRef]
- Kirvell, S.L.; Elliott, M.S.; Kalaria, R.N.; Hortobagyi, T.; Ballard, C.G.; Francis, P.T. Vesicular glutamate transporter and cognition in stroke: A case-control autopsy study. Neurology 2010, 75, 1803–1809. [Google Scholar] [CrossRef] [PubMed]
- Ihara, M.; Polvikoski, T.M.; Hall, R.; Slade, J.Y.; Perry, R.H.; Oakley, A.E.; Englund, E.; O’Brien, J.T.; Ince, P.G.; Kalaria, R.N. Quantification of myelin loss in frontal lobe white matter in vascular dementia, Alzheimer’s disease, and dementia with Lewy bodies. Acta Neuropathol. 2010, 119, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.; Zarow, C.; Mack, W.J.; Zheng, L.; Vinters, H.V.; Ellis, W.G.; Lyness, S.A.; Chui, H.C. Preservation of Neurons of the Nucleus Basalis in Subcortical Ischemic Vascular Disease. Arch. Neurol. 2012, 69. [Google Scholar] [CrossRef] [PubMed]
- Thomas, T.; Miners, S.; Love, S. Post-mortem assessment of hypoperfusion of cerebral cortex in Alzheimer’s disease and vascular dementia. Brain 2015, 138, 1059–1069. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, L.I.; Tayler, H.M.; Love, S. Synaptic protein levels altered in vascular dementia: Synaptic proteins in vascular dementia. Neuropathol. Appl. Neurobiol. 2015, 41, 533–543. [Google Scholar] [CrossRef]
- Chen, A.; Akinyemi, R.O.; Hase, Y.; Firbank, M.J.; Ndung’u, M.N.; Foster, V.; Craggs, L.J.L.; Washida, K.; Okamoto, Y.; Thomas, A.J.; et al. Frontal white matter hyperintensities, clasmatodendrosis and gliovascular abnormalities in ageing and post-stroke dementia. Brain 2016, 139, 242–258. [Google Scholar] [CrossRef] [PubMed]
- Pennisi, G.; Ferri, R.; Alagona, G.; Pennisi, M.; Malaguarnera, G.; Motta, M.; Bella, R. Motor cortex hyperexcitability in subcortical ischemic vascular dementia. Arch. Gerontol. Geriatr. 2011, 53, e111–e113. [Google Scholar] [CrossRef]
- Nardone, R.; De Blasi, P.; Seidl, M.; Höller, Y.; Caleri, F.; Tezzon, F.; Ladurner, G.; Golaszewski, S.; Trinka, E. Cognitive function and cholinergic transmission in patients with subcortical vascular dementia and microbleeds: A TMS study. J. Neural Transm. 2011, 118, 1349–1358. [Google Scholar] [CrossRef][Green Version]
- Bella, R.; Ferri, R.; Cantone, M.; Pennisi, M.; Lanza, G.; Malaguarnera, G.; Spampinato, C.; Giordano, D.; Raggi, A.; Pennisi, G. Motor cortex excitability in vascular depression. Int. J. Psychophysiol. 2011, 82, 248–253. [Google Scholar] [CrossRef]
- Bella, R.; Ferri, R.; Pennisi, M.; Cantone, M.; Lanza, G.; Malaguarnera, G.; Spampinato, C.; Giordano, D.; Alagona, G.; Pennisi, G. Enhanced motor cortex facilitation in patients with vascular cognitive impairment-no dementia. Neurosci. Lett. 2011, 503, 171–175. [Google Scholar] [CrossRef]
- Bella, R.; Ferri, R.; Lanza, G.; Cantone, M.; Pennisi, M.; Puglisi, V.; Vinciguerra, L.; Spampinato, C.; Mazza, T.; Malaguarnera, G.; et al. TMS follow-up study in patients with vascular cognitive impairment-no dementia. Neurosci. Lett. 2013, 534, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Lanza, G.; Bella, R.; Giuffrida, S.; Cantone, M.; Pennisi, G.; Spampinato, C.; Giordano, D.; Malaguarnera, G.; Raggi, A.; Pennisi, M. Preserved transcallosal inhibition to transcranial magnetic stimulation in nondemented elderly patients with leukoaraiosis. BioMed Res. Int. 2013, 2013, 351680. [Google Scholar] [CrossRef]
- List, J.; Duning, T.; Kürten, J.; Deppe, M.; Wilbers, E.; Flöel, A. Cortical plasticity is preserved in nondemented older individuals with severe ischemic small vessel disease. Hum. Brain Mapp. 2013, 34, 1464–1476. [Google Scholar] [CrossRef]
- Palomar, F.J.; Suárez, A.; Franco, E.; Carrillo, F.; Gil-Néciga, E.; Mir, P. Abnormal sensorimotor plasticity in CADASIL correlates with neuropsychological impairment. J. Neurol. Neurosurg. Psychiatry 2013, 84, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Concerto, C.; Lanza, G.; Cantone, M.; Pennisi, M.; Giordano, D.; Spampinato, C.; Ricceri, R.; Pennisi, G.; Aguglia, E.; Bella, R. Different patterns of cortical excitability in major depression and vascular depression: A transcranial magnetic stimulation study. BMC Psychiatry 2013, 13, 300. [Google Scholar] [CrossRef] [PubMed]
- Nardone, R.; Höller, Y.; Thomschewski, A.; Kunz, A.B.; Lochner, P.; Golaszewski, S.; Trinka, E.; Brigo, F. Dopamine differently modulates central cholinergic circuits in patients with Alzheimer disease and CADASIL. J. Neural Transm. Vienna Austria 1996 2014, 121, 1313–1320. [Google Scholar] [CrossRef] [PubMed]
- List, J.; Hertel-Zens, S.; Kübke, J.C.; Lesemann, A.; Schreiber, S.J.; Flöel, A. Cortical reorganization due to impaired cerebral autoregulation in individuals with occlusive processes of the internal carotid artery. Brain Stimulat. 2014, 7, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Guerra, A.; Petrichella, S.; Vollero, L.; Ponzo, D.; Pasqualetti, P.; Määttä, S.; Mervaala, E.; Könönen, M.; Bressi, F.; Iannello, G.; et al. Neurophysiological features of motor cortex excitability and plasticity in Subcortical Ischemic Vascular Dementia: A TMS mapping study. Clin. Neurophysiol. 2015, 126, 906–913. [Google Scholar] [CrossRef] [PubMed]
- Bella, R.; Cantone, M.; Lanza, G.; Ferri, R.; Vinciguerra, L.; Puglisi, V.; Pennisi, M.; Ricceri, R.; Di Lazzaro, V.; Pennisi, G. Cholinergic circuitry functioning in patients with vascular cognitive impairment--no dementia. Brain Stimulat. 2016, 9, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Pennisi, M.; Lanza, G.; Cantone, M.; Ricceri, R.; Spampinato, C.; Pennisi, G.; Di Lazzaro, V.; Bella, R. Correlation between Motor Cortex Excitability Changes and Cognitive Impairment in Vascular Depression: Pathophysiological Insights from a Longitudinal TMS Study. Neural Plast. 2016, 2016, 8154969. [Google Scholar] [CrossRef]
- Schmitz, M.; Hermann, P.; Oikonomou, P.; Stoeck, K.; Ebert, E.; Poliakova, T.; Schmidt, C.; Llorens, F.; Zafar, S.; Zerr, I. Cytokine profiles and the role of cellular prion protein in patients with vascular dementia and vascular encephalopathy. Neurobiol. Aging 2015, 36, 2597–2606. [Google Scholar] [CrossRef] [PubMed]
- Miwa, K.; Tanaka, M.; Okazaki, S.; Yagita, Y.; Sakaguchi, M.; Mochizuki, H.; Kitagawa, K. Increased Total Homocysteine Levels Predict the Risk of Incident Dementia Independent of Cerebral Small-Vessel Diseases and Vascular Risk Factors. J. Alzheimers Dis. 2015, 49, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Fleszar, M.G.; Wiśniewski, J.; Zboch, M.; Diakowska, D.; Gamian, A.; Krzystek-Korpacka, M. Targeted metabolomic analysis of nitric oxide/L-arginine pathway metabolites in dementia: Association with pathology, severity, and structural brain changes. Sci. Rep. 2019, 9, 13764. [Google Scholar] [CrossRef] [PubMed]
- Taheri, S.; Gasparovic, C.; Huisa, B.N.; Adair, J.C.; Edmonds, E.; Prestopnik, J.; Grossetete, M.; Shah, N.J.; Wills, J.; Qualls, C.; et al. Blood–Brain Barrier Permeability Abnormalities in Vascular Cognitive Impairment. Stroke 2011, 42, 2158–2163. [Google Scholar] [CrossRef]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G. Preferred Reporting Items for Systematic Reviews and Meta-Analyses: The PRISMA Statement. PLoS Med. 2009, 6. [Google Scholar] [CrossRef]
- Pennisi, M.; Malaguarnera, G.; Di Bartolo, G.; Lanza, G.; Bella, R.; Chisari, E.M.; Cauli, O.; Vicari, E.; Malaguarnera, M. Decrease in Serum Vitamin D Level of Older Patients with Fatigue. Nutrients 2019, 11, 2531. [Google Scholar] [CrossRef]
- Zelante, G.; Ricceri, R.; Lanza, G.; Fiumanò, G.; Pennisi, G.; Bella, R. Retroclival subdural hematoma after a lumbar puncture: An uncommon complication for a common procedure. Neurol. India 2017, 65, 1400–1401. [Google Scholar] [CrossRef]
- Vinciguerra, L.; Cantone, M.; Lanza, G.; Bramanti, A.; Santalucia, P.; Puglisi, V.; Pennisi, G.; Bella, R. Migrainous Infarction And Cerebral Vasospasm: Case Report And Literature Review. J. Pain Res. 2019, 12, 2941–2950. [Google Scholar] [CrossRef]
- Iemolo, F.; Duro, G.; Rizzo, C.; Castiglia, L.; Hachinski, V.; Caruso, C. Pathophysiology of vascular dementia. Immun. Ageing A 2009, 6, 13. [Google Scholar] [CrossRef]
- Liu, W.; Wong, A.; Law, A.C.K.; Mok, V.C.T. Cerebrovascular Disease, Amyloid Plaques, and Dementia. Stroke 2015, 46, 1402–1407. [Google Scholar] [CrossRef]
- Schneider, J.A.; Arvanitakis, Z.; Bang, W.; Bennett, D.A. Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology 2007, 69, 2197–2204. [Google Scholar] [CrossRef]
- Iadecola, C. The Pathobiology of Vascular Dementia. Neuron 2013, 80, 844–866. [Google Scholar] [CrossRef] [PubMed]
- Heiss, W.-D.; Rosenberg, G.A.; Thiel, A.; Berlot, R.; de Reuck, J. Neuroimaging in vascular cognitive impairment: A state-of-the-art review. BMC Med. 2016, 14, 174. [Google Scholar] [CrossRef] [PubMed]
- Malojcic, B.; Giannakopoulos, P.; Sorond, F.A.; Azevedo, E.; Diomedi, M.; Oblak, J.P.; Carraro, N.; Boban, M.; Olah, L.; Schreiber, S.J.; et al. Ultrasound and dynamic functional imaging in vascular cognitive impairment and Alzheimer’s disease. BMC Med. 2017, 15, 27. [Google Scholar] [CrossRef] [PubMed]
- Lanza, G.; Papotto, M.; Pennisi, G.; Bella, R.; Ferri, R. Epileptic seizure as a precipitating factor of vascular progressive supranuclear palsy: A case report. J. Stroke Cerebrovasc. Dis. 2014, 23, e379–e381. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Liu, C.Y.; Smith, R.X.; Jog, M.; Langham, M.; Krasileva, K.; Chen, Y.; Ringman, J.M.; Wang, D.J.J. Assessing intracranial vascular compliance using dynamic arterial spin labeling. NeuroImage 2016, 124, 433–441. [Google Scholar] [CrossRef]
- Gao, Y.-Z.; Zhang, J.-J.; Liu, H.; Wu, G.-Y.; Xiong, L.; Shu, M. Regional cerebral blood flow and cerebrovascular reactivity in Alzheimer’s disease and vascular dementia assessed by arterial spinlabeling magnetic resonance imaging. Curr. Neurovasc. Res. 2013, 10, 49–53. [Google Scholar] [CrossRef]
- Firbank, M.J.; He, J.; Blamire, A.M.; Singh, B.; Danson, P.; Kalaria, R.N.; O’Brien, J.T. Cerebral blood flow by arterial spin labeling in poststroke dementia. Neurology 2011, 76, 1478–1484. [Google Scholar] [CrossRef]
- Wu, W.-C.; Lin, S.-C.; Wang, D.J.; Chen, K.-L.; Li, Y.-D. Measurement of cerebral white matter perfusion using pseudocontinuous arterial spin labeling 3T magnetic resonance imaging--an experimental and theoretical investigation of feasibility. PLoS ONE 2013, 8, e82679. [Google Scholar] [CrossRef]
- Rossini, P.M.; Burke, D.; Chen, R.; Cohen, L.G.; Daskalakis, Z.; Di Iorio, R.; Di Lazzaro, V.; Ferreri, F.; Fitzgerald, P.B.; George, M.S.; et al. Non-invasive electrical and magnetic stimulation of the brain, spinal cord, roots and peripheral nerves: Basic principles and procedures for routine clinical and research application. An updated report from an I.F.C.N. Committee. Clin. Neurophysiol. 2015, 126, 1071–1107. [Google Scholar] [CrossRef]
- Cantone, M.; Lanza, G.; Vinciguerra, L.; Puglisi, V.; Ricceri, R.; Fisicaro, F.; Vagli, C.; Bella, R.; Ferri, R.; Pennisi, G.; et al. Age, Height, and Sex on Motor Evoked Potentials: Translational Data From a Large Italian Cohort in a Clinical Environment. Front. Hum. Neurosci. 2019, 13. [Google Scholar] [CrossRef] [PubMed]
- Lanza, G.; Cantone, M.; Puglisi, V.; Vinciguerra, L.; Fisicaro, F.; Vagli, C.; Bella, R.; Pennisi, G.; Di Lazzaro, V.; Pennisi, M. “Mute” plantar response: Does the cortico-spinal tract “speak”? Brain Stimulat. 2019, S1935861X1930292X. [Google Scholar] [CrossRef] [PubMed]
- Pennisi, M.; Lanza, G.; Cantone, M.; Ricceri, R.; Ferri, R.; D’Agate, C.C.; Pennisi, G.; Di Lazzaro, V.; Bella, R. Cortical involvement in celiac disease before and after long-term gluten-free diet: A Transcranial Magnetic Stimulation study. PLoS ONE 2017, 12, e0177560. [Google Scholar] [CrossRef] [PubMed]
- Lanza, G.; Lanuzza, B.; Aricò, D.; Cantone, M.; Cosentino, F.I.I.; Bella, R.; Pennisi, G.; Ferri, R.; Pennisi, M. Impaired short-term plasticity in restless legs syndrome: A pilot rTMS study. Sleep Med. 2018, 46, 1–4. [Google Scholar] [CrossRef]
- Cantone, M.; Lanza, G.; Le Pira, A.; Barone, R.; Pennisi, G.; Bella, R.; Pennisi, M.; Fiumara, A. Adjunct Diagnostic Value of Transcranial Magnetic Stimulation in Mucopolysaccharidosis-Related Cervical Myelopathy: A Pilot Study. Brain Sci. 2019, 9, 200. [Google Scholar] [CrossRef]
- Lanza, G.; Cantone, M.; Lanuzza, B.; Pennisi, M.; Bella, R.; Pennisi, G.; Ferri, R. Distinctive patterns of cortical excitability to transcranial magnetic stimulation in obstructive sleep apnea syndrome, restless legs syndrome, insomnia, and sleep deprivation. Sleep Med. Rev. 2015, 19, 39–50. [Google Scholar] [CrossRef]
- Lanza, G.; Lanuzza, B.; Aricò, D.; Cantone, M.; Cosentino, F.I.I.; Pennisi, M.; Bella, R.; Pennisi, G.; Ferri, R. Direct comparison of cortical excitability to transcranial magnetic stimulation in obstructive sleep apnea syndrome and restless legs syndrome. Sleep Med. 2015, 16, 138–142. [Google Scholar] [CrossRef]
- Lanza, G.; Aricò, D.; Lanuzza, B.; Cosentino, F.I.I.; Tripodi, M.; Giardina, F.; Bella, R.; Puligheddu, M.; Pennisi, G.; Ferri, R.; et al. Facilitatory/inhibitory intracortical imbalance in REM sleep behavior disorder: Early electrophysiological marker of neurodegeneration? Sleep 2020, 43. [Google Scholar] [CrossRef]
- Lanza, G.; Bella, R.; Cantone, M.; Pennisi, G.; Ferri, R.; Pennisi, M. Cognitive Impairment and Celiac Disease: Is Transcranial Magnetic Stimulation a Trait d’Union between Gut and Brain? Int. J. Mol. Sci. 2018, 19, 2243. [Google Scholar] [CrossRef]
- Cantone, M.; Bramanti, A.; Lanza, G.; Pennisi, M.; Bramanti, P.; Pennisi, G.; Bella, R. Cortical Plasticity in Depression. ASN NEURO 2017, 9. [Google Scholar] [CrossRef]
- Pennisi, G.; Lanza, G.; Giuffrida, S.; Vinciguerra, L.; Puglisi, V.; Cantone, M.; Pennisi, M.; D’Agate, C.C.; Naso, P.; Aprile, G.; et al. Excitability of the Motor Cortex in De Novo Patients with Celiac Disease. PLoS ONE 2014, 9, e102790. [Google Scholar] [CrossRef] [PubMed]
- Bella, R.; Lanza, G.; Cantone, M.; Giuffrida, S.; Puglisi, V.; Vinciguerra, L.; Pennisi, M.; Ricceri, R.; D’Agate, C.C.; Malaguarnera, G.; et al. Effect of a Gluten-Free Diet on Cortical Excitability in Adults with Celiac Disease. PLoS ONE 2015, 10, e0129218. [Google Scholar] [CrossRef] [PubMed]
- Pennisi, M.; Bramanti, A.; Cantone, M.; Pennisi, G.; Bella, R.; Lanza, G. Neurophysiology of the “Celiac Brain”: Disentangling Gut-Brain Connections. Front. Neurosci. 2017, 11, 498. [Google Scholar] [CrossRef] [PubMed]
- Pennisi, G.; Ferri, R.; Cantone, M.; Lanza, G.; Pennisi, M.; Vinciguerra, L.; Malaguarnera, G.; Bella, R. A review of transcranial magnetic stimulation in vascular dementia. Dement. Geriatr. Cogn. Disord. 2011, 31, 71–80. [Google Scholar] [CrossRef]
- Lanza, G.; Bramanti, P.; Cantone, M.; Pennisi, M.; Pennisi, G.; Bella, R. Vascular Cognitive Impairment through the Looking Glass of Transcranial Magnetic Stimulation. Behav. Neurol. 2017, 2017. [Google Scholar] [CrossRef]
- Pennisi, G.; Ferri, R.; Lanza, G.; Cantone, M.; Pennisi, M.; Puglisi, V.; Malaguarnera, G.; Bella, R. Transcranial magnetic stimulation in Alzheimer’s disease: A neurophysiological marker of cortical hyperexcitability. J. Neural Transm. 2011, 118, 587–598. [Google Scholar] [CrossRef]
- Cantone, M.; Di Pino, G.; Capone, F.; Piombo, M.; Chiarello, D.; Cheeran, B.; Pennisi, G.; Di Lazzaro, V. The contribution of transcranial magnetic stimulation in the diagnosis and in the management of dementia. Clin. Neurophysiol. 2014, 125, 1509–1532. [Google Scholar] [CrossRef]
- Rossini, P.M.; Rossi, S.; Babiloni, C.; Polich, J. Clinical neurophysiology of aging brain: From normal aging to neurodegeneration. Prog. Neurobiol. 2007, 83, 375–400. [Google Scholar] [CrossRef]
- Di Lazzaro, V.; Oliviero, A.; Pilato, F.; Saturno, E.; Dileone, M.; Tonali, P.A. Motor cortex hyperexcitability to transcranial magnetic stimulation in Alzheimer’s disease: Evidence of impaired glutamatergic neurotransmission? Ann. Neurol. 2003, 53, 824–825. [Google Scholar] [CrossRef]
- Di Lazzaro, V.; Oliviero, A.; Pilato, F.; Saturno, E.; Dileone, M.; Marra, C.; Daniele, A.; Ghirlanda, S.; Gainotti, G.; Tonali, P.A. Motor cortex hyperexcitability to transcranial magnetic stimulation in Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 2004, 75, 555–559. [Google Scholar] [CrossRef]
- Paula-Lima, A.C.; Brito-Moreira, J.; Ferreira, S.T. Deregulation of excitatory neurotransmission underlying synapse failure in Alzheimer’s disease. J. Neurochem. 2013, 126, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Nardone, R.; Bergmann, J.; Tezzon, F.; Ladurner, G.; Golaszewski, S. Cholinergic dysfunction in subcortical ischaemic vascular dementia: A transcranial magnetic stimulation study. J. Neural Transm. 2008, 115, 737–743. [Google Scholar] [CrossRef] [PubMed]
- Di Lazzaro, V.; Pilato, F.; Dileone, M.; Profice, P.; Marra, C.; Ranieri, F.; Quaranta, D.; Gainotti, G.; Tonali, P.A. In vivo functional evaluation of central cholinergic circuits in vascular dementia. Clin. Neurophysiol. 2008, 119, 2494–2500. [Google Scholar] [CrossRef] [PubMed]
- Nardone, R.; Bergmann, J.; Christova, M.; Caleri, F.; Tezzon, F.; Ladurner, G.; Trinka, E.; Golaszewski, S. Short latency afferent inhibition differs among the subtypes of mild cognitive impairment. J. Neural Transm. 2012, 119, 463–471. [Google Scholar] [CrossRef]
- Nardone, R.; Golaszewski, S.; Ladurner, G.; Tezzon, F.; Trinka, E. A review of transcranial magnetic stimulation in the in vivo functional evaluation of central cholinergic circuits in dementia. Dement. Geriatr. Cogn. Disord. 2011, 32, 18–25. [Google Scholar] [CrossRef]
- Spampinato, C.; Aguglia, E.; Concerto, C.; Pennisi, M.; Lanza, G.; Bella, R.; Cantone, M.; Pennisi, G.; Kavasidis, I.; Giordano, D. Transcranial magnetic stimulation in the assessment of motor cortex excitability and treatment of drug-resistant major depression. IEEE Trans. Neural Syst. Rehabil. Eng. 2013, 21, 391–403. [Google Scholar] [CrossRef]
- Lanza, G.; Cantone, M.; Aricò, D.; Lanuzza, B.; Cosentino, F.I.I.; Paci, D.; Papotto, M.; Pennisi, M.; Bella, R.; Pennisi, G.; et al. Clinical and electrophysiological impact of repetitive low-frequency transcranial magnetic stimulation on the sensory–motor network in patients with restless legs syndrome. Ther. Adv. Neurol. Disord. 2018, 11, 175628641875997. [Google Scholar] [CrossRef]
- Fisicaro, F.; Lanza, G.; Bella, R.; Pennisi, M. “Self-Neuroenhancement”: The Last Frontier of Noninvasive Brain Stimulation? J. Clin. Neurol. 2020, 16, 158–159. [Google Scholar] [CrossRef]
- Concerto, C.; Lanza, G.; Cantone, M.; Ferri, R.; Pennisi, G.; Bella, R.; Aguglia, E. Repetitive transcranial magnetic stimulation in patients with drug-resistant major depression: A six-month clinical follow-up study. Int. J. Psychiatry Clin. Pract. 2015, 19, 252–258. [Google Scholar] [CrossRef]
- Fisicaro, F.; Lanza, G.; Grasso, A.A.; Pennisi, G.; Bella, R.; Paulus, W.; Pennisi, M. Repetitive transcranial magnetic stimulation in stroke rehabilitation: Review of the current evidence and pitfalls. Ther. Adv. Neurol. Disord. 2019, 12, 175628641987831. [Google Scholar] [CrossRef]
- Rektorova, I.; Megova, S.; Bares, M.; Rektor, I. Cognitive functioning after repetitive transcranial magnetic stimulation in patients with cerebrovascular disease without dementia: A pilot study of seven patients. J. Neurol. Sci. 2005, 229–230, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Swartz, R.H.; Sahlas, D.J.; Black, S.E. Strategic involvement of cholinergic pathways and executive dysfunction: Does location of white matter signal hyperintensities matter? J. Stroke Cerebrovasc. Dis. Off. J. Natl. Stroke Assoc. 2003, 12, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Manganelli, F.; Ragno, M.; Cacchiò, G.; Iodice, V.; Trojano, L.; Silvaggio, F.; Scarcella, M.; Grazioli, M.; Santoro, L.; Perretti, A. Motor cortex cholinergic dysfunction in CADASIL: A transcranial magnetic demonstration. Clin. Neurophysiol. 2008, 119, 351–355. [Google Scholar] [CrossRef] [PubMed]
- Thiel, G. Synapsin I, Synapsin II, and Synaptophysin: Marker Proteins of Synaptic Vesicles. Brain Pathol. 1993, 3, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Lang, B.; Kindy, M.S.; Kozel, F.A.; Schultz, S.K.; Taheri, S. Multi-Parametric Classification of Vascular Cognitive Impairment and Dementia: The Impact of Diverse Cerebrovascular Injury Biomarkers. J. Alzheimers Dis. 2018, 62, 39–60. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood–brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef]
- Farrall, A.J.; Wardlaw, J.M. Blood–brain barrier: Ageing and microvascular disease--systematic review and meta-analysis. Neurobiol. Aging 2009, 30, 337–352. [Google Scholar] [CrossRef]
- Wallin, A.; Sjögren, M.; Edman, A.; Blennow, K.; Regland, B. Symptoms, vascular risk factors and blood–brain barrier function in relation to CT white-matter changes in dementia. Eur. Neurol. 2000, 44, 229–235. [Google Scholar] [CrossRef]
- Kalaria, R.N. Neuropathological diagnosis of vascular cognitive impairment and vascular dementia with implications for Alzheimer’s disease. Acta Neuropathol. 2016, 131, 659–685. [Google Scholar] [CrossRef]
- Bella, R.; Pennisi, G.; Cantone, M.; Palermo, F.; Pennisi, M.; Lanza, G.; Zappia, M.; Paolucci, S. Clinical presentation and outcome of geriatric depression in subcortical ischemic vascular disease. Gerontology 2010, 56, 298–302. [Google Scholar] [CrossRef]
- Pennisi, G.; Bella, R.; Lanza, G. Motor cortex plasticity in subcortical ischemic vascular dementia: What can TMS say? Clin. Neurophysiol. 2015, 126, 851–852. [Google Scholar] [CrossRef] [PubMed]
- Kaminsky, Y.G.; Tikhonova, L.A.; Kosenko, E.A. Critical analysis of Alzheimer’s amyloid-beta toxicity to mitochondria. Front. Biosci. 2015, 20, 173–197. [Google Scholar] [CrossRef] [PubMed]
- Lanza, G.; Cantone, M.; Musso, S.; Borgione, E.; Scuderi, C.; Ferri, R. Early-onset subcortical ischemic vascular dementia in an adult with mtDNA mutation 3316G>A. J. Neurol. 2018, 265, 968–969. [Google Scholar] [CrossRef] [PubMed]
- Grazina, M.; Silva, F.; Santana, I.; Santiago, B.; Mendes, C.; Simões, M.; Oliveira, M.; Cunha, L.; Oliveira, C. Frontotemporal dementia and mitochondrial DNA transitions. Neurobiol. Dis. 2004, 15, 306–311. [Google Scholar] [CrossRef] [PubMed]
- Ballard, C.; Rowan, E.; Stephens, S.; Kalaria, R.; Kenny, R.A. Prospective follow-up study between 3 and 15 months after stroke: Improvements and decline in cognitive function among dementia-free stroke survivors >75 years of age. Stroke 2003, 34, 2440–2444. [Google Scholar] [CrossRef] [PubMed]
- Ferreri, F.; Pauri, F.; Pasqualetti, P.; Fini, R.; Dal Forno, G.; Rossini, P.M. Motor cortex excitability in Alzheimer’s disease: A transcranial magnetic stimulation study. Ann. Neurol. 2003, 53, 102–108. [Google Scholar] [CrossRef]
- Ferreri, F.; Pasqualetti, P.; Määttä, S.; Ponzo, D.; Guerra, A.; Bressi, F.; Chiovenda, P.; Del Duca, M.; Giambattistelli, F.; Ursini, F.; et al. Motor cortex excitability in Alzheimer’s disease: A transcranial magnetic stimulation follow-up study. Neurosci. Lett. 2011, 492, 94–98. [Google Scholar] [CrossRef]
- Román, G.C.; Sachdev, P.; Royall, D.R.; Bullock, R.A.; Orgogozo, J.-M.; López-Pousa, S.; Arizaga, R.; Wallin, A. Vascular cognitive disorder: A new diagnostic category updating vascular cognitive impairment and vascular dementia. J. Neurol. Sci. 2004, 226, 81–87. [Google Scholar] [CrossRef]
- Bordet, R.; Ihl, R.; Korczyn, A.D.; Lanza, G.; Jansa, J.; Hoerr, R.; Guekht, A. Towards the concept of disease-modifier in post-stroke or vascular cognitive impairment: A consensus report. BMC Med. 2017, 15, 107. [Google Scholar] [CrossRef]
- Yang, H.; Shi, O.; Jin, Y.; Henrich-Noack, P.; Qiao, H.; Cai, C.; Tao, H.; Tian, X. Functional protection of learning and memory abilities in rats with vascular dementia. Restor. Neurol. Neurosci. 2014, 32, 689–700. [Google Scholar] [CrossRef]
- Wang, F.; Chang, G.; Yu, Q.; Geng, X. The neuroprotection of repetitive transcranial magnetic stimulation pre-treatment in vascular dementia rats. J. Mol. Neurosci. MN 2015, 56, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Yukimasa, T.; Yoshimura, R.; Tamagawa, A.; Uozumi, T.; Shinkai, K.; Ueda, N.; Tsuji, S.; Nakamura, J. High-frequency repetitive transcranial magnetic stimulation improves refractory depression by influencing catecholamine and brain-derived neurotrophic factors. Pharmacopsychiatry 2006, 39, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.-Y.; Liu, Y.; Xie, J.-C.; Liu, N.-N.; Tian, X. Effects of repetitive transcranial magnetic stimulation on synaptic plasticity and apoptosis in vascular dementia rats. Behav. Brain Res. 2015, 281, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-Q.; Li, L.; Huo, J.-T.; Cheng, M.; Li, L.-H. Effects of repetitive transcranial magnetic stimulation on cognitive function and cholinergic activity in the rat hippocampus after vascular dementia. Neural Regen. Res. 2018, 13, 1384–1389. [Google Scholar] [CrossRef]
- Wang, F.; Zhang, C.; Hou, S.; Geng, X. Synergistic Effects of Mesenchymal Stem Cell Transplantation and Repetitive Transcranial Magnetic Stimulation on Promoting Autophagy and Synaptic Plasticity in Vascular Dementia. J. Gerontol. A. Biol. Sci. Med. Sci. 2019, 74, 1341–1350. [Google Scholar] [CrossRef]
- Román, G.C. Lacunar dementia. In Senile Dementia of the Alzheimer Type; Alan, R. Liss Inc.: New York, NY, USA, 1985; pp. 131–151. [Google Scholar]
- Hamel, E. Cholinergic modulation of the cortical microvascular bed. Prog. Brain Res. 2004, 145, 171–178. [Google Scholar] [CrossRef]
- Sato, A.; Sato, Y.; Uchida, S. Activation of the intracerebral cholinergic nerve fibers originating in the basal forebrain increases regional cerebral blood flow in the rat’s cortex and hippocampus. Neurosci. Lett. 2004, 361, 90–93. [Google Scholar] [CrossRef]
- Román, G.C. Brain hypoperfusion: A critical factor in vascular dementia. Neurol. Res. 2004, 26, 454–458. [Google Scholar] [CrossRef]
- Mok, V.; Ding, D.; Fu, J.; Xiong, Y.; Chu, W.W.C.; Wang, D.; Abrigo, J.M.; Yang, J.; Wong, A.; Zhao, Q.; et al. Transcranial Doppler ultrasound for screening cerebral small vessel disease: A community study. Stroke 2012, 43, 2791–2793. [Google Scholar] [CrossRef]
- Heliopoulos, I.; Artemis, D.; Vadikolias, K.; Tripsianis, G.; Piperidou, C.; Tsivgoulis, G. Association of ultrasonographic parameters with subclinical white-matter hyperintensities in hypertensive patients. Cardiovasc. Psychiatry Neurol. 2012, 2012, 616572. [Google Scholar] [CrossRef]
- Bakker, S.L.; de Leeuw, F.E.; de Groot, J.C.; Hofman, A.; Koudstaal, P.J.; Breteler, M.M. Cerebral vasomotor reactivity and cerebral white matter lesions in the elderly. Neurology 1999, 52, 578–583. [Google Scholar] [CrossRef] [PubMed]
- Naqvi, J.; Yap, K.H.; Ahmad, G.; Ghosh, J. Transcranial Doppler ultrasound: A review of the physical principles and major applications in critical care. Int. J. Vasc. Med. 2013, 2013, 629378. [Google Scholar] [CrossRef]
- Vinciguerra, L.; Bösel, J. Noninvasive Neuromonitoring: Current Utility in Subarachnoid Hemorrhage, Traumatic Brain Injury, and Stroke. Neurocrit. Care 2017, 27, 122–140. [Google Scholar] [CrossRef] [PubMed]
- Sabayan, B.; Jansen, S.; Oleksik, A.M.; van Osch, M.J.P.; van Buchem, M.A.; van Vliet, P.; de Craen, A.J.M.; Westendorp, R.G.J. Cerebrovascular hemodynamics in Alzheimer’s disease and vascular dementia: A meta-analysis of transcranial Doppler studies. Ageing Res. Rev. 2012, 11, 271–277. [Google Scholar] [CrossRef]
- Vicenzini, E.; Ricciardi, M.C.; Altieri, M.; Puccinelli, F.; Bonaffini, N.; Di Piero, V.; Lenzi, G.L. Cerebrovascular reactivity in degenerative and vascular dementia: A transcranial Doppler study. Eur. Neurol. 2007, 58, 84–89. [Google Scholar] [CrossRef]
- Vinciguerra, L.; Lanza, G.; Puglisi, V.; Pennisi, M.; Cantone, M.; Bramanti, A.; Pennisi, G.; Bella, R. Transcranial Doppler ultrasound in vascular cognitive impairment-no dementia. PLoS ONE 2019, 14. [Google Scholar] [CrossRef] [PubMed]
- Puglisi, V.; Bramanti, A.; Lanza, G.; Cantone, M.; Vinciguerra, L.; Pennisi, M.; Bonanno, L.; Pennisi, G.; Bella, R. Impaired Cerebral Haemodynamics in Vascular Depression: Insights From Transcranial Doppler Ultrasonography. Front. Psychiatry 2018, 9, 316. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Authors, Year | Study Design | Patients (n). | VCI Subtype | Main Findings |
|---|---|---|---|---|
| Serum | ||||
| Cervellati, et al. 2014 [9] | Cross-sectional | 54 | Not reported | ↑ homocysteine and uric acid ↓ residual antioxidant power ↑ slightly of hydroperoxide level |
| Schneider, et al. 2014 [10] | Prospective | 34 | Not reported | High-sensitivity cardiac troponin T associated with increased risk of vascular dementia |
| Gao, et al. 2015 [11] | Cross-sectional | 210 | Isolated or multiple lacunar infarcts; leukoaraiosis | ↑ plasma levels of S100 Calcium-Binding protein B and asymmetric dimethylarginine associated with small vessel disease in patients with cognitive decline |
| Liu, et al. 2015 [12] | Cross-sectional | 30 | Not reported | ↑ carnitine, glutamine, uric acid, tyrosine, kynurenine, and phenylalanine ↓ 3-indolepropionic acid, stearoyl-carnitine, valine, isoleucine, tryptophan, lysophosphatidylcholines, and palmitoylcarnitine |
| Cervellati, et al. 2015 [13] | Case-control | 21 | Not reported | ↓ paraoxonase and arylesterase activity, which was associated with risk of conversion of mild cognitive impairment into vascular dementia |
| Castellazzi, et al. 2016 [14] | Cross-sectional | 65 | Not reported | ↓ serum arylesterase level = paraoxonase level |
| Paroni, et al. 2016 [15] | Cross-sectional | 31 | Cortical/subcortical or strategic infarcts | Anti-smooth muscle antibody associated with brain atrophy |
| Moretti, et al. 2017 [16] | Cross-sectional | 456 | White matter lesions | ↓ folate and vitamin D ↑ homocysteine |
| Cerebrospinal fluid | ||||
| Jonsson, et al. 2010 [17] | Cross-sectional | 53 | White matter lesions | ↑ neurofilament light protein associated with white matter lesion severity; less strong evidence for sulfatide |
| Formichi, et al. 2010 [18] | Cross-sectional | 10 | CADASIL | ↓ amyloid-beta 42, overlapping to Alzheimer’s disease = total tau and phosphorilated tau, which differed from Alzheimer’s disease |
| Spies, et al. 2010 [19] | Cross-sectional | 26 | Not reported | ↑ amyloid-beta 42 and amyloid-beta 42/amyloid-beta 40 ratio in vascular dementia than Alzheimer’s disease |
| Bjerke, et al. 2011 [20] | Case-control | 26 | SIVD | ↑ major basic protein, neurofilament light protein, heart-fatty acid binding protein, total tau, tissue inhibitor of metalloproteinases-1, and matrix metalloproteinase-10 |
| Candelario-Jalil, et al. 2011 [21] | Cross-sectional | 60 | SIVD; multiple strokes; leukoaraiosis | ↓ matrix metalloproteinase-2 index, with a negative correlation with albumin ratio ↑ matrix metalloproteinase-3 activity |
| Öhrfelt, et al. 2011 [22] | Cross-sectional | 8 | White matter lesions; lacunar infarcts | ↑ alpha1-antitrypsin, apolipoprotein H, plasminogen activator inhibitor-1, heart-fatty acid binding protein, and tissue inhibitor of metalloproteinases-1 |
| Olsson, et al. 2012 [23] | Prospective | 19 | SIVD | ↑ chitinase-3-like protein 1 and soluble cluster of differentation 14; chitinase-3-like protein 1 differentiated between stable mild cognitive impairmennt and those converting into Alzheimer’s disease and vascular dementia ↑ cerebrospinal fluid/serum albumin ratio |
| Kaerst, et al. 2013 [24] | Retrospective case-control | 44 | Not reported | ↓ amyloid beta 42 |
| Li, et al. 2015 [25] | Cross-sectional | 5 | Cerebral amyloid angiopathy | = amyloid-beta 42, amyloid-beta 40, phosphorylated tau, and amyloid-beta 42/amyloid-beta 40 ratio between cerebral amyloid angiopathy and Alzheimer’s disease |
| Rosenberg, et al. 2015 [26] | Prospective | 62 | SIVD | ↑ albumin index ↓ matrix metalloproteinase-2 index |
| Neuroimaging | ||||
| Pascual, et al. 2010 [27] | Cross-sectional | 12 | Confluent white matter lesions | ↓ metabolism in both frontal lobes and right supramarginal gyrus at the 18-fluoro-deoxyglucose-positron emission tomography |
| Mok, et al. 2010 [28] | Cross-sectional | 10 | SIVD; large post-stroke lesions | Pittsburgh compound B-positron emission tomography binding was commonly observed |
| Kim, et al. 2012 [29] | Cross-sectional | 48 | White matter lesions | Cholinergic pathway deficit |
| Gasparovic, et al. 2013 [30] | Cross-sectional | 60 | Multiple stroke; SIVD; hypoxic hypoperfusion | Correlation between total creatine, N-acetyl-aspartyl compounds and test scores of executive function; metabolite levels correlated with total white matter lesion volume |
| Combined serum and cerebrospinal fluid | ||||
| Schmitz, et al. 2015 [51] | Cross-sectional | 42 | SIVD | Interleukin-8 and macrophage inflammatory protein-1β correlated with dementia severity; association between cellular prion protein and cytokine levels and between cellular prion protein and degenerative marker proteins |
| Combined serum and neuroimaging | ||||
| Miwa, et al. 2015 [52] | Case-control | 18 | SIVD | Total homocysteine level contributed to the increased risk susceptibility of dementia |
| Fleszar, et al. 2019 [53] | Cross-sectional | 40 | Strategic infarcts; white matter lesions | ↑ dimethyarginine, L-citrulline, asymmetric dimethylarginine, and symmetric; dimethyarginine, L-arginine/ asymmetric dimethylarginine, and dimethyarginine independently predicted Hachinski Ischemic score |
| Combined cerebrospinal fluid and neuroimaging | ||||
| Taheri, et al. 2011 [54] | Cross-sectional | 60 | SIVD; multiple and lacunar infarcts; leukoaraiosis | ↑ albumin index and blood–brain barrier permeability |
| Transcranial magnetic stimulation | ||||
| Pennisi et al. 2011 [37] | Cross-sectional | 20 vascular dementia 20 mild VCI | SIVD dementia | ↑ cortical excitability in demented patients only |
| Nardone, et al. 2011 [38] | Cross-sectional | 28 | SIVD dementia | Microbleeds on cholinergic function are independent of white matter lesion extent and ischemic stroke |
| Bella, et al. 2011 [39] | Cross-sectional | 15 major depressive disorder 10 non-depressed | SIVD mild VCI | Neurophysiology of vascular depression differs from major depressive disorder and seems to be similar to that of SIVD |
| Bella, et al. 2011 [40] | Cross-sectional | 10 | SIVD mild VCI | ↑ intracortical excitatory neuronal circuits |
| Bella, et al. 2013 [41] | Case-control | 9 | SIVD mild VCI | ↑ excitability during the progression of VCI |
| Lanza, et al. 2013 [42] | Cross-sectional | 15 | Leukoaraiosis mild VCI | = transcallosal inhibitory functioning, unlike Alzheimer’s disease and mild cognitive impairment |
| List, et al. 2013 [43] | Cross-sectional | 20 | Leukoaraiosis mild VCI | ↑ cortical plasticity as a compensatory mechanism |
| Palomar, et al. 2013 [44] | Cross-sectional | 10 | CADASIL | Acetylcholine and glutamate involved; abnormal sensory-motor plasticity correlated with cognition |
| Concerto, et al. 2013 [45] | Cross-sectional | 11 depressed 11 major depressive disorder | SIVD mild VCI | Distinctive patterns of cortical excitability between late-onset vascular depression and early-onset non-vascular major depressive disorder |
| Nardone, et al. 2014 [46] | Cross-sectional | 8 VCI 8 Alzheimer’s disease | CADASIL | ↓ cholinergic functioning, with restoration by L-3,4-dihydroxyphenylalanine in Alzheimer’s disease only |
| List, et al. 2014 [47] | Cross-sectional | 12 | Mild VCI (post-stroke in 3 of them) | ↓ long-term potentiation-like plasticity in the affected hemisphere = motor learning between hemispheres, maybe due to γ-amino-butyric acid B-effect in the affected side |
| Guerra, et al. 2015 [48] | Cross-sectional | 7 VCI 9 Alzheimer’s disease | SIVD dementia | ↑ excitability and plasticity in Alzheimer’s disease and vascular dementia; the hyperexcitability promoted plasticity |
| Bella, et al. 2016 [49] | Cross-sectional | 25 | SIVD mild VCI | Central cholinergic pathway not clearly affected |
| Pennisi, et al. 2016 [50] | Case-control | 16 major depressive disorder 11 non-depressed | SIVD mild VCI | ↑ risk of dementia in vascular depression, probably due to subcortical vascular lesions or to the lack of compensatory functional cortical changes |
| Histopathology | ||||
| Kirvell, et al. 2010 [31] | Case-control | 18 | Not reported | ↓ glutamatergic synapses |
| Ihara, et al. 2010 [32] | Case-control | 20 | White matter lesions | ↓ myelin density in the frontal lobe |
| Jung, et al. 2012 [33] | Case-control | 16 | SIVD | No neuron loss in the nucleus basalis of Meynert; no evidence of retrograde degeneration |
| Thomas, et al. 2015 [34] | Case-control | 17 | Not reported | ↓ myelin-associated glycoprotein/proteolipidprotein 1 ratio |
| Sinclair, et al. 2015 [35] | Case-control | 11 | Multiple infarcts | ↓ synaptophysin and synaptosomal-associated protein 25 ↑ drebrin levels |
| Chen, et al. 2016 [36] | Case-control | 27 | Multiple infarcts; lacunae; SIVD | ↑ clasmatodendrocytes |
| Technique | Potential Applications and Expected Findings |
|---|---|
| Serum | Screening and advanced protocols that correlate with cognitive performance and progression |
| Search for genetic susceptibility for mood disorders associated with vascular pathology | |
| CSF | Screening for early identification of post-stroke cognitive impairment and other VCI subtypes |
| Markers of blood–brain barrier involvement or neuroinflammation specifically related to VCI | |
| Neuro- imaging | Implementation and diffusion of advanced structural and functional neuroimaging technique |
| Neurosonological markers of early hemodynamic changes in VCI or vascular depression | |
| TMS | Detailed analysis of neurotransmission pathways and other “pharmaco-TMS” studies in VCI |
| Advanced brain stimulation protocols to screen and follow mild VCI at risk for dementia | |
| All/Mix | Proposal and application of a multidimensional diagnostic panel to screen population at risk |
| Design and optimization of customized cognitive rehabilitation strategies or drug trials |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vinciguerra, L.; Lanza, G.; Puglisi, V.; Fisicaro, F.; Pennisi, M.; Bella, R.; Cantone, M. Update on the Neurobiology of Vascular Cognitive Impairment: From Lab to Clinic. Int. J. Mol. Sci. 2020, 21, 2977. https://doi.org/10.3390/ijms21082977
Vinciguerra L, Lanza G, Puglisi V, Fisicaro F, Pennisi M, Bella R, Cantone M. Update on the Neurobiology of Vascular Cognitive Impairment: From Lab to Clinic. International Journal of Molecular Sciences. 2020; 21(8):2977. https://doi.org/10.3390/ijms21082977
Chicago/Turabian StyleVinciguerra, Luisa, Giuseppe Lanza, Valentina Puglisi, Francesco Fisicaro, Manuela Pennisi, Rita Bella, and Mariagiovanna Cantone. 2020. "Update on the Neurobiology of Vascular Cognitive Impairment: From Lab to Clinic" International Journal of Molecular Sciences 21, no. 8: 2977. https://doi.org/10.3390/ijms21082977
APA StyleVinciguerra, L., Lanza, G., Puglisi, V., Fisicaro, F., Pennisi, M., Bella, R., & Cantone, M. (2020). Update on the Neurobiology of Vascular Cognitive Impairment: From Lab to Clinic. International Journal of Molecular Sciences, 21(8), 2977. https://doi.org/10.3390/ijms21082977