Blockade of CDK7 Reverses Endocrine Therapy Resistance in Breast Cancer

 ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Results
2.1. Increased Cyclin-Dependent Kinase (CDK7) Expression Correlates with Poor Patient Survival and Tamoxifen Resistance in Estrogen Receptor + (ER+) Breast Cancer
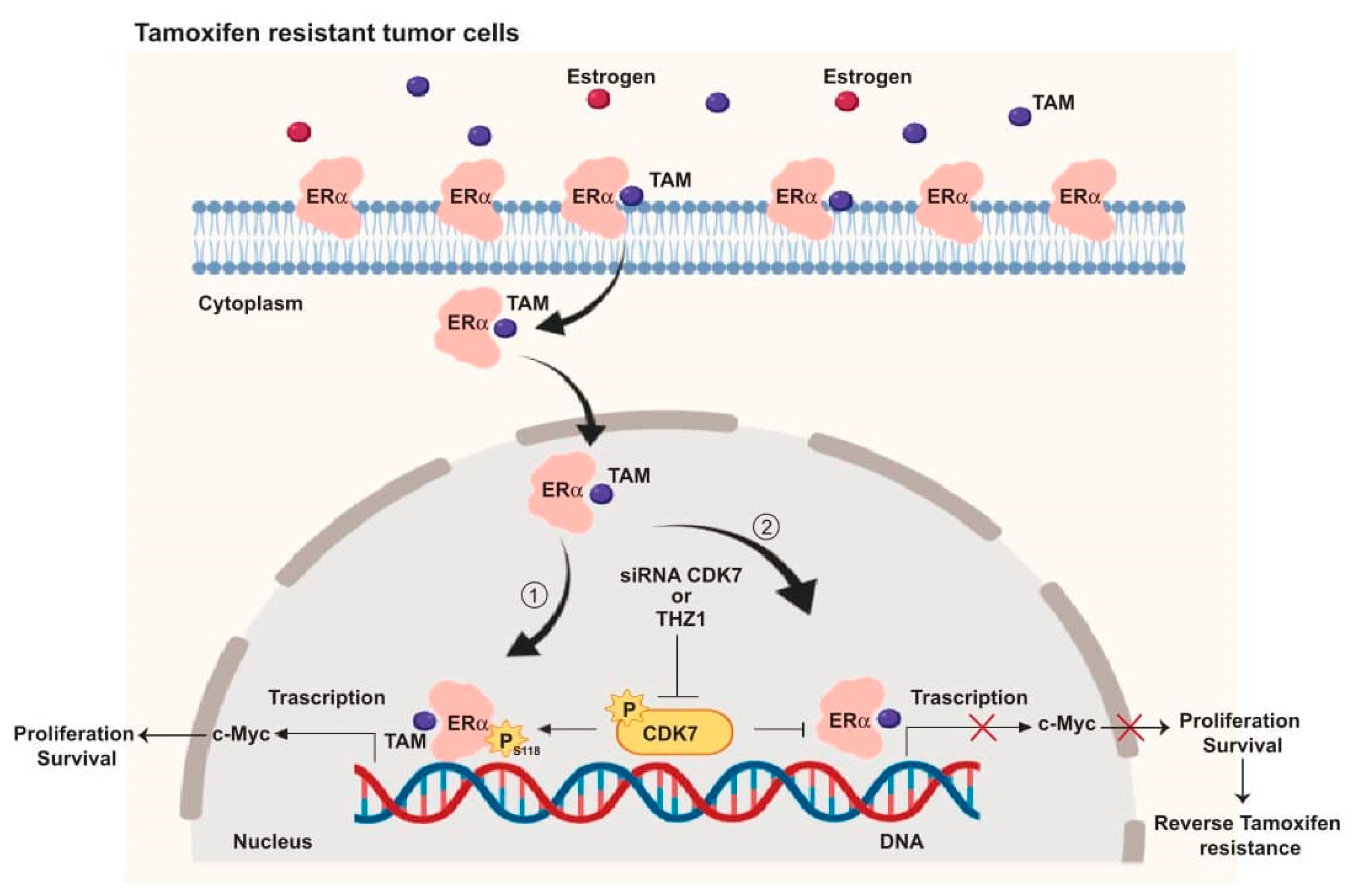
2.2. Targeting CDK7 Decreases Estrogen Receptor (ER) Activation
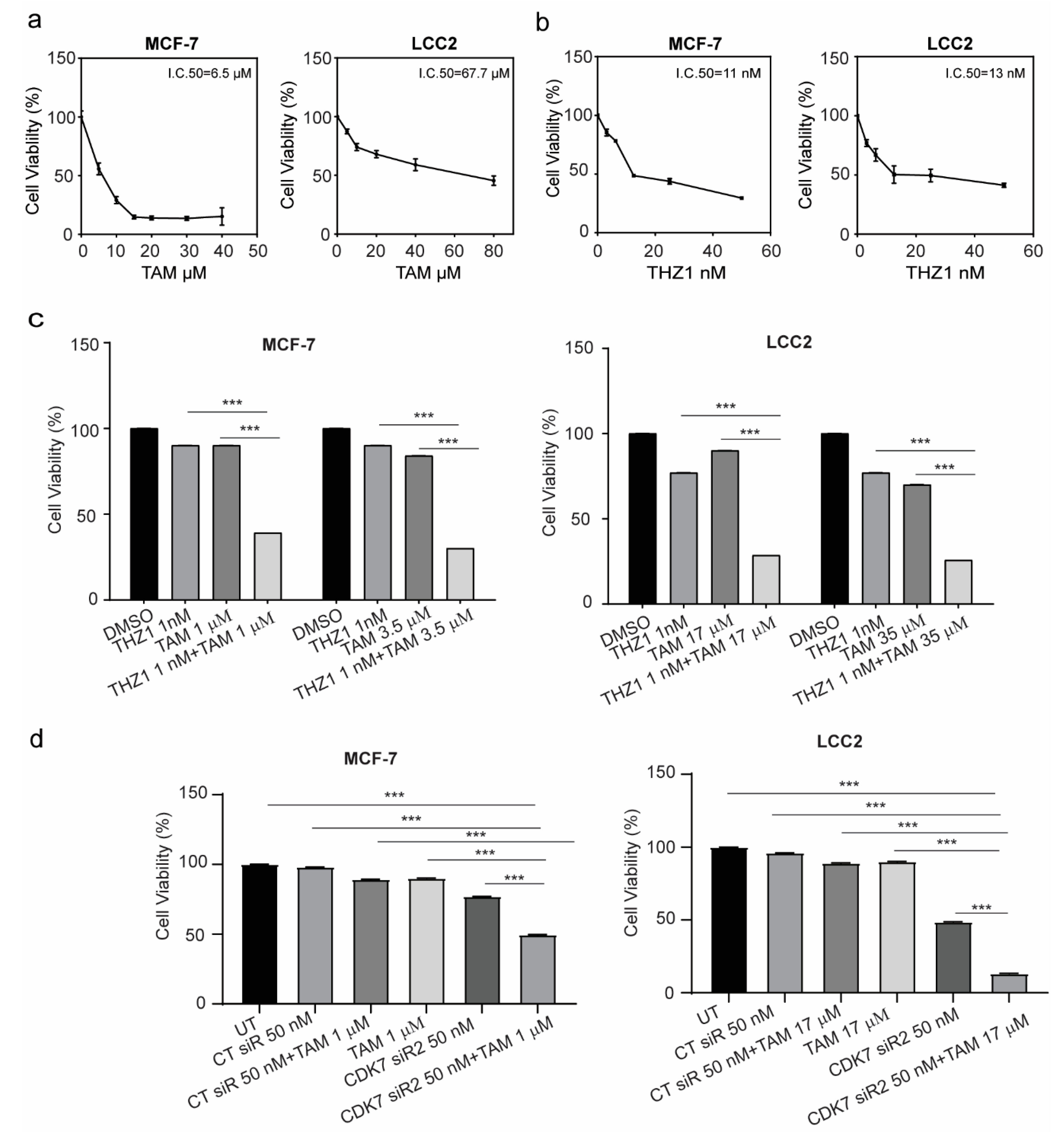
2.3. Targeting CDK7 Augmented Tamoxifen-Induced Cytotoxicity
2.4. CDK7 Inhibition Increased Tamoxifen-Induced Apoptosis and Modulated Its Effect on the Cell Cycle
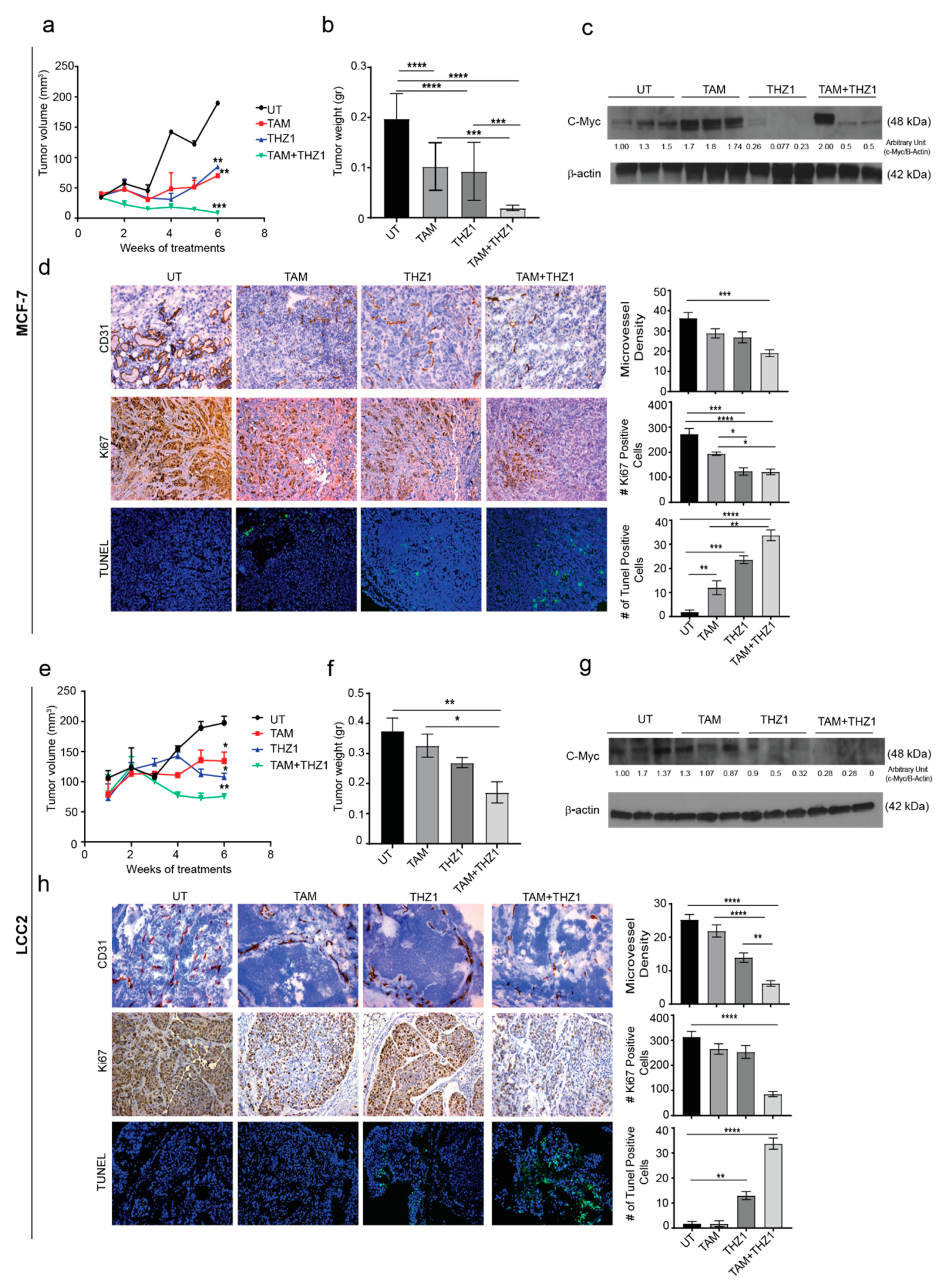
2.5. CDK7 Inhibition Enhances the Cytotoxic Effect of Tamoxifen in Murine Models of Tamoxifen-Sensitive and Tamoxifen-Resistant Breast Cancer
3. Discussion
4. Materials and Methods
4.1. TCGA Breast Cancer Cohort Data Analysis
4.2. Tamoxifen-Treated Breast Cancer Cohort Data Analysis
4.3. Cell Culture and Chemicals
4.4. siRNA Transfection
4.5. RNA Isolation and RT-PCR
4.6. Western Blotting
4.7. SRB Assay
4.8. Quantification of Cell Death
4.9. Annexin V/Propidium Iodide Staining for Apoptosis Assessment
4.10. Cell Cycle Analysis
4.11. Orthotopic Mouse Models of Breast Cancer
4.12. Immunohistochemistry
4.13. Statistical Analysis
4.14. Ethical Approval
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CDK | Cyclin-dependent kinase |
| siRNA | Small interfering RNA |
| ER | Estrogen receptor |
| Ser118 | Serine 118 |
References
- Cardoso, F.; Harbeck, N.; Fallowfield, L.; Kyriakides, S.; Senkus, E.; ESMO Guidelines Working Group. Locally recurrent or metastatic breast cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2012, 23. [Google Scholar] [CrossRef]
- Lumachi, F.; Brunello, A.; Maruzzo, M.; Basso, U.; Basso, S. Treatment of Estrogen Receptor-Positive Breast Cancer. Curr. Med. Chem. 2013, 20, 596–604. [Google Scholar] [CrossRef]
- Tao, Z.Q.; Shi, A.; Lu, C.; Song, T.; Zhang, Z.; Zhao, J. Breast Cancer: Epidemiology and Etiology. Cell Biochem. Biophys. 2015, 72, 333–338. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, B.; Liu, J.; Liu, J.; Li, C.; Dong, W.; Fang, S.; Li, M.; Song, B.; Tang, B.; et al. Mechanisms of Gefitinib-mediated reversal of tamoxifen resistance in MCF-7 breast cancer cells by inducing ERα re-expression. Sci. Rep. 2015, 5, 7835. [Google Scholar] [CrossRef]
- Droog, M.; Beelen, K.; Linn, S.; Zwart, W. Tamoxifen resistance: From bench to bedside. Eur. J. Pharmacol. 2013, 717, 47–57. [Google Scholar] [CrossRef]
- Johansson, H.J.; Sanchez, B.C.; Mundt, F.; Forshed, J.; Kovacs, A.; Panizza, E.; Yakhini, Z.; Helou, K.; Hultin-rosenberg, L.; Lundgren, B.; et al. Retinoic acid receptor alpha is associated with tamoxifen resistance in breast cancer. Nat. Commun. 2013, 4, 1–10. [Google Scholar] [CrossRef]
- Sutherland, R.L.; Musgrove, E. a CDK inhibitors as potential breast cancer therapeutics: New evidence for enhanced efficacy in ER+ disease. Breast Cancer Res. 2009, 11, 112. [Google Scholar] [CrossRef]
- Malumbres, M.; Barbacid, M. Cell cycle, CDKs and cancer: A changing paradigm. Nat. Rev. Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef]
- Cicenas, J.; Kalyan, K.; Sorokinas, A.; Jatulyte, A.; Valiunas, D.; Kaupinis, A.; Valius, M. Highlights of the latest advances in research on CDK inhibitors. Cancers 2014, 6, 2224–2242. [Google Scholar] [CrossRef]
- Malumbres, M.; Harlow, E.; Hunt, T.; Hunter, T.; Lahti, J.M.; Morgan, D.O.; Tsai, L.; Wolgemuth, D.J. Cyclin-dependent kinases: A family portrait. Nat Cell Biol. 2010, 11, 1275–1276. [Google Scholar] [CrossRef]
- Fisher, R.P. Cdk7: A kinase at the core of transcription and in the crosshairs of cancer drug discovery. Transcription 2019, 10, 47–56. [Google Scholar] [CrossRef]
- McDermott, M.S.J.; Sharko, A.C.; Munie, J.; Kassler, S.; Melendez, T.; Lim, C.; Broude, E.V. CDK7 Inhibition is Effective in all the Subtypes of Breast Cancer: Determinants of Response and Synergy with EGFR Inhibition. Cells 2020, 9, 638. [Google Scholar] [CrossRef]
- Patel, H.; Abduljabbar, R.; Lai, C.-F.; Periyasamy, M.; Harrod, A.; Gemma, C.; Steel, J.H.; Patel, N.; Busonero, C.; Jerjees, D.; et al. Expression of CDK7, cyclin H and MAT1 is elevated in breast cancer and is prognostic in estrogen receptor positive breast cancer Running title: CDK7 expression in breast cancer. Clin. Cancer Res. 2017, 22, 5929–5938. [Google Scholar] [CrossRef]
- Anbalagan, M.; Rowan, B.G. Estrogen receptor alpha phosphorylation and its functional impact in human breast cancer. Mol. Cell. Endocrinol. 2015, 418, 264–272. [Google Scholar] [CrossRef]
- Hur, J.Y.; Kim, H.R.; Lee, J.Y.; Park, S.; Hwang, J.A.; Kim, W.S.; Yoon, S.; Choi, C.-M.; Rho, J.K.; Lee, J.C. CDK7 inhibition as a promising therapeutic strategy for lung squamous cell carcinomas with a SOX2 amplification. Cell. Oncol. 2019, 42, 449–458. [Google Scholar] [CrossRef]
- Boyer, M.J.; Cheng, T. The CDK inhibitors: Potential targets for therapeutic stem cell manipulations? Gene Ther. 2008, 15, 117–125. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, T.; Kwiatkowski, N.; Abraham, B.J.; Lee, T.I.; Xie, S.; Yuzugullu, H.; Von, T.; Li, H.; Lin, Z.; et al. CDK7-Dependent Transcriptional Addiction in Triple-Negative Breast Cancer. Cell 2015, 163, 174–186. [Google Scholar] [CrossRef]
- Harrod, A.; Fulton, J.; Nguyen, V.T.M.; Periyasamy, M.; Ramos-Garcia, L.; Lai, C.F.; Metodieva, G.; De Giorgio, A.; Williams, R.L.; Santos, D.B.; et al. Genomic modelling of the ESR1 Y537S mutation for evaluating function and new therapeutic approaches for metastatic breast cancer. Oncogene 2017, 36, 2286–2296. [Google Scholar] [CrossRef]
- Kwiatkowski, N.; Zhang, T.; Rahl, P.B.; Abraham, B.J.; Reddy, J.; Ficarro, S.B.; Dastur, A.; Amzallag, A.; Ramaswamy, S.; Tesar, B.; et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature 2014, 511, 616–620. [Google Scholar] [CrossRef]
- Dang, C.V. MYC on the path to cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef]
- Bui, Q.T.; Im, J.H.; Jeong, S.B.; Kim, Y.-M.; Lim, S.C.; Kim, B.; Kang, K.W. Essential role of Notch4/STAT3 signaling in epithelial-mesenchymal transition of tamoxifen-resistant human breast cancer. Cancer Lett. 2017, 390, 115–125. [Google Scholar] [CrossRef]
- Ward, A.; Balwierz, A.; Zhang, J.D.; Kublbeck, M.; Pawitan, Y.; Hielscher, T.; Wiemann, S.; Sahin, O. Re-expression of microRNA-375 reverses both tamoxifen resistance and accompanying EMT-like properties in breast cancer. Oncogene 2013, 32, 1173–1182. [Google Scholar] [CrossRef]
- Martin, M.B.; Saceda, M.; Lindsey, R.K. Regulation of estrogen receptor expression in breast cancer. Adv. Exp. Med. Biol. 1993, 330, 143–153. [Google Scholar]
- Skliris, G.P.; Nugent, Z.J.; Rowan, B.G.; Penner, C.R.; Watson, P.H.; Murphy, L.C. A phosphorylation code for oestrogen receptor-alpha predicts clinical outcome to endocrine therapy in breast cancer. Endocr. Relat. Cancer 2010, 17, 589–597. [Google Scholar] [CrossRef]
- Jeselsohn, R.; Bergholz, J.S.; Pun, M.; Cornwell, M.; Liu, W.; Nardone, A.; Xiao, T.; Li, W.; Qiu, X.; Buchwalter, G.; et al. Allele-Specific Chromatin Recruitment and Therapeutic Vulnerabilities of ESR1 Activating Mutations. Cancer Cell 2018, 33, 173–186.e5. [Google Scholar] [CrossRef]
- Sarwar, N.; Kim, J.-S.; Jiang, J.; Peston, D.; Sinnett, H.D.; Madden, P.; Gee, J.M.; Nicholson, R.I.; Lykkesfeldt, A.E.; Shousha, S.; et al. Phosphorylation of ERalpha at serine 118 in primary breast cancer and in tamoxifen-resistant tumours is indicative of a complex role for ERalpha phosphorylation in breast cancer progression. Endocr. Relat. Cancer 2006, 13, 851–861. [Google Scholar] [CrossRef]
- Chen, M.; Cui, Y.K.; Huang, W.H.; Man, K.; Zhang, G.J. Phosphorylation of estrogen receptor α at serine 118 is correlated with breast cancer resistance to tamoxifen. Oncol. Lett. 2013, 6, 118–124. [Google Scholar] [CrossRef]
- Schachter, M.M.; Merrick, K.A.; Larochelle, S.; Hirschi, A.; Zhang, C.; Shokat, K.M.; Rubin, S.M.; Fisher, R.P. A Cdk7-Cdk4 T-loop phosphorylation cascade promotes G1 progression. Mol. Cell 2013, 50, 250–260. [Google Scholar] [CrossRef]
- Schacter, M.; Fisher, R.P. The CDK-activating kinase Cdk7. Taking yes for an answer. Cell Cycle 2012, 12, 3239–3240. [Google Scholar] [CrossRef]
- Finn, R.S.; Crown, J.P.; Lang, I.; Boer, K.; Bondarenko, I.M.; Kulyk, S.O.; Ettl, J.; Patel, R.; Pinter, T.; Schmidt, M.; et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): A randomised phase 2 study. Lancet. Oncol. 2015, 16, 25–35. [Google Scholar] [CrossRef]
- Turner, N.C.; Huang Bartlett, C.; Cristofanilli, M. Palbociclib in Hormone-Receptor-Positive Advanced Breast Cancer. N. Engl. J. Med. 2015, 373, 1672–1673. [Google Scholar] [CrossRef]
- Cheng, Z.-J.; Miao, D.-L.; Su, Q.-Y.; Tang, X.-L.; Wang, X.-L.; Deng, L.-B.; Shi, H.-D.; Xin, H.-B. THZ1 suppresses human non-small-cell lung cancer cells in vitro through interference with cancer metabolism. Acta Pharmacol. Sin. 2019, 40, 814–822. [Google Scholar] [CrossRef] [PubMed]
- Mbonye, U.; Wang, B.; Gokulrangan, G.; Shi, W.; Yang, S.; Karn, J. Cyclin-dependent kinase 7 (CDK7)-mediated phosphorylation of the CDK9 activation loop promotes P-TEFb assembly with Tat and proviral HIV reactivation. J. Biol. Chem. 2018, 293, 10009–10025. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.L.; Daley, B.J.; Enderson, B.L.; Zhou, J.-R.; Karlstad, M.D. Genistein inhibits tamoxifen effects on cell proliferation and cell cycle arrest in T47D breast cancer cells. Am. Surg. 2002, 68, 575–578. [Google Scholar]
- Kilker, R.L.; Hartl, M.W.; Rutherford, T.M.; Planas-Silva, M.D. Cyclin D1 expression is dependent on estrogen receptor function in tamoxifen-resistant breast cancer cells. J. Steroid Biochem. Mol. Biol. 2004, 92, 63–71. [Google Scholar] [CrossRef]
- Span, P.N.; Tjan-Heijnen, V.C.G.; Manders, P.; Beex, L.V.A.M.; Sweep, C.G.J. Cyclin-E is a strong predictor of endocrine therapy failure in human breast cancer. Oncogene 2003, 22, 4898–4904. [Google Scholar] [CrossRef]
- Sun, B.; Mason, S.; Wilson, R.C.; Hazard, S.E.; Wang, Y.; Fang, R.; Wang, Q.; Yeh, E.S.; Yang, M.; Roberts, T.M.; et al. Inhibition of the transcriptional kinase CDK7 overcomes therapeutic resistance in HER2-positive breast cancers. Oncogene 2020, 39, 50–63. [Google Scholar] [CrossRef]
- Huang, T.; Ding, X.; Xu, G.; Chen, G.; Cao, Y.; Peng, C.; Shen, S.; Lv, Y.; Wang, L.; Zou, X. CDK7 inhibitor THZ1 inhibits MCL1 synthesis and drives cholangiocarcinoma apoptosis in combination with BCL2/BCL-XL inhibitor ABT-263. Cell Death Dis. 2019, 10. [Google Scholar] [CrossRef]
- Chipumuro, E.; Marco, E.; Christensen, C.L.; Kwiatkowski, N.; Zhang, T.; Hatheway, C.M.; Abraham, B.J.; Sharma, B.; Yeung, C.; Altabef, A.; et al. CDK7 inhibition suppresses super-enhancer-linked oncogenic transcription in MYCN-driven cancer. Cell 2014, 159, 1126–1139. [Google Scholar] [CrossRef]
- Zeng, M.; Kwiatkowski, N.P.; Zhang, T.; Nabet, B.; Xu, M.; Liang, Y.; Quan, C.; Wang, J.; Hao, M.; Palakurthi, S.; et al. Targeting MYC dependency in ovarian cancer through inhibition of CDK7 and CDK12/13. Elife 2018, 7, 1–20. [Google Scholar] [CrossRef]
- Liang, Y.C.; Tsai, S.H.; Chen, L.; Lin-Shiau, S.Y.; Lin, J.K. Resveratrol-induced G 2 arrest through the inhibition of CDK7 and p34 CDC2 kinases in colon carcinoma HT29 cells. Biochem. Pharmacol. 2003, 65, 1053–1060. [Google Scholar] [CrossRef]
- Larochelle, S.; Merrick, K.A.; Terret, M.-E.; Wohlbold, L.; Barboza, N.M.; Zhang, C.; Shokat, K.M.; Jallepalli, P.V.; Fisher, R.P. Requirements for Cdk7 in the assembly of Cdk1/cyclin B and activation of Cdk2 revealed by chemical genetics in human cells. Mol. Cell 2007, 25, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, M.S.; Heidemann, M.; Tietjen, J.R.; Zhang, D.W.; Chapman, R.D.; Eick, D.; Ansari, A.Z. TFIIH kinase places bivalent marks on the carboxy-terminal domain of RNA polymerase II. Mol. Cell 2009, 34, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Pan, C.; Sun, J.; Gilbert, C.; Drews-Elger, K.; Azzam, D.J.; Picon-Ruiz, M.; Kim, M.; Ullmer, W.; El-Ashry, D.; et al. VEGF drives cancer-initiating stem cells through VEGFR-2/Stat3 signaling to upregulate Myc and Sox2. Oncogene 2015, 34, 3107–3119. [Google Scholar] [CrossRef] [PubMed]
- Liao, D.J.; Dickson, R.B. c-Myc in breast cancer. Endocr. Relat. Cancer 2000, 7, 143–164. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.S.L.; Jin, V.X.; Fan, M.; Smith, L.T.; Liyanarachchi, S.; Yan, P.S.; Leu, Y.-W.; Chan, M.W.Y.; Plass, C.; Nephew, K.P.; et al. Combinatorial analysis of transcription factor partners reveals recruitment of c-MYC to estrogen receptor-alpha responsive promoters. Mol. Cell 2006, 21, 393–404. [Google Scholar] [CrossRef]
- Shang, Y.; Brown, M. Molecular determinants for the tissue specificity of SERMs. Science 2002, 295, 2465–2468. [Google Scholar] [CrossRef]
- Baudino, T.A.; Mckay, C.; Pendeville-samain, H.; Nilsson, J.A.; Maclean, K.H.; White, E.L.; Davis, A.C.; Ihle, J.N.; Cleveland, J.L. c-Myc is essential for vasculogenesis and angiogenesis during development and tumor progression. Genes Dev. 2002, 16, 2530–2543. [Google Scholar] [CrossRef]
- Mezquita, P.; Parghi, S.S.; Brandvold, K.A.; Ruddell, A. Myc regulates VEGF production in B cells by stimulating initiation of VEGF mRNA translation. Oncogene 2005, 24, 889–901. [Google Scholar] [CrossRef]
- Michel, T.; Kankura, A.; Salinas Medina, M.L.; Kurz, J.; Behring, A.; Avci-Adali, M.; Nolte, A.; Schlensak, C.; Wendel, H.P.; Krajewski, S. In Vitro Evaluation of a Novel mRNA-Based Therapeutic Strategy for the Treatment of Patients Suffering from Alpha-1-Antitrypsin Deficiency. Nucleic Acid Ther. 2015, 25, 235–244. [Google Scholar] [CrossRef]
- Sears, R.; Ohtani, K.; Nevins, J.R. Identification of positively and negatively acting elements regulating expression of the E2F2 gene in response to cell growth signals. Mol. Cell. Biol. 1997, 17, 5227–5235. [Google Scholar] [CrossRef]
- Cheng, C.-C.; Shi, L.-H.; Wang, X.-J.; Wang, S.-X.; Wan, X.-Q.; Liu, S.-R.; Wang, Y.-F.; Lu, Z.; Wang, L.-H.; Ding, Y. Stat3/Oct-4/c-Myc signal circuit for regulating stemness-mediated doxorubicin resistance of triple-negative breast cancer cells and inhibitory effects of WP1066. Int. J. Oncol. 2018, 53, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; Li, T.G.; Qi, X.; Zhao, D.F.; Zhao, G.Q. WNT/β-catenin pathway up-regulates Stat3 and converges on LIF to prevent differentiation of mouse embryonic stem cells. Dev. Biol. 2006, 290, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.r-project.org/,R-software (accessed on 20 May 2019).
- Liu, J.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018, 173, 400–416.e11. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://portal.gdc.cancer.gov/ (accessed on 20 May 2019).
- Chanrion, M.; Negre, V.; Fontaine, H.; Salvetat, N.; Bibeau, F.; Mac Grogan, G.; Mauriac, L.; Katsaros, D.; Molina, F.; Theillet, C.; et al. A gene expression signature that can predict the recurrence of tamoxifen-treated primary breast cancer. Clin. Cancer Res. 2008, 14, 1744–1752. [Google Scholar] [CrossRef]
- Brünner, N.; Frandsen, T.L.; Holst-hansen, C.; Brãnner, N.; Lippman, M.E.; Clarke, R.; Bei, M.; Thompson, E.W.; Wakeling, A.E. MCF7/LCC2: A 4-Hydroxytamoxifen Resistant Human Breast Cancer Variant That Retains Sensitivity to the Steroidal Advances in Brief MCF7/LCC2: A 4-Hydroxytamoxifen Resistant Human Breast Cancer Variant That Retains Sensitivity to the Steroidal Antiest. Cancer Res. 1993, 3229–3232. [Google Scholar]
- Brünner, N.; Boysen, B.; Jirus, S.; Skaar, T.C.; Holst-Hansen, C.; Lippman, J.; Frandsen, T.; Spang-Thomsen, M.; Fuqua, S.A.W.; Clarke, R. MCF7/LCC9: An antiestrogen-resistant MCF-7 variant in which acquired resistance to the steroidal antiestrogen ICI 182,780 confers an early cross- resistance to the nonsteroidal antiestrogen tamoxifen. Cancer Res. 1997, 57, 3486–3493. [Google Scholar]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; & Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2020, 9, 671–675. [Google Scholar] [CrossRef]
- Whitfield, J.; Littlewood, T.; Soucek, L. Tamoxifen administration to mice. Cold Spring Harb. Protoc. 2015, 2015, 269–271. [Google Scholar] [CrossRef]
- Cayrol, F.; Praditsuktavorn, P.; Fernando, T.M.; Kwiatkowski, N.; Marullo, R.; Calvo-Vidal, M.N.; Phillip, J.; Pera, B.; Yang, S.N.; Takpradit, K.; et al. THZ1 targeting CDK7 suppresses STAT transcriptional activity and sensitizes T-cell lymphomas to BCL2 inhibitors. Nat. Commun. 2017, 8, 14290. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Forward Sequence | Reverse Sequence |
|---|---|---|
| CDK7 | 5′-AGGATGTATGGTGTAGGTGTGGA-3′ | 5′-AAGATGTGATGCAAAGGTATTCC-3′ |
| GAPDH | 5′-TGAAGGTCGGAGTCAACGGATTT-3′ | 5′-GCCATGGAATTTGCCATGGGTGG-3′ |
| Primary Antibody | Purchasing Company | Dilution Factor | Secondary Company | Purchasing Company | Dilution Factor |
|---|---|---|---|---|---|
| Anti-estrogen receptor alpha | Santa-Cruz biotechnology, Dallas, TX, USA | 1:100 | Horseradish peroxidase (HRP) linked secondary antibody Anti-mouse | Sigma–Aldrich, St. Louis, MO, USA | 1:2000 |
| Anti-Ser118 | Cell signal Technology, Danvers, MA, USA | 1:1000 | HRP linked secondary antibody Anti-rabbit | Sigma–Aldrich, St. Louis, MO, USA | 1:2000 |
| Anti-Stat3 | Cell signal Technology, Danvers, MA, USA | 1:1000 | HRP linked secondary antibody Anti-rabbit | Sigma–Aldrich, St. Louis, MO, USA | 1:2000 |
| Anti-β-catenin | Santa-Cruz biotechnology, Dallas, TX, USA | 1:100 | HRP linked secondary antibody Anti-mouse | Sigma–Aldrich, St. Louis, MO, USA | 1:2000 |
| Anti-CDK 2 | Cell signal Technology, Danvers, MA, USA | 1:1000 | HRP linked secondary antibody Anti-mouse | Sigma–Aldrich, St. Louis, MO, USA | 1:2000 |
| Anti-CDK 4 | Cell signal Technology, Danvers, MA, USA | 1:1000 | HRP linked secondary antibody Anti-rabbit | Sigma–Aldrich, St. Louis, MO, USA | 1:2000 |
| Anti-CDK 6 | Cell signal Technology, Danvers, MA, USA | 1:1000 | HRP linked secondary antibody Anti-rabbit | Sigma–Aldrich, St. Louis, MO, USA | 1:2000 |
| Anti-CDK 7 | Cell signal Technology, Danvers, MA, USA | 1:1000 | HRP linked secondary antibody Anti-mouse | Sigma–Aldrich, St. Louis, MO, USA | 1:2000 |
| Anti-cyclin A1 | Santa-Cruz biotechnology, Dallas, TX, USA | 1:100 | HRP linked secondary antibody Anti-mouse | Sigma–Aldrich, St. Louis, MO, USA | 1:2000 |
| Anti-cyclin B1 | Cell signal Technology, Danvers, MA, USA | 1:1000 | HRP linked secondary antibody Anti-rabbit | Sigma–Aldrich, St. Louis, MO, USA | 1:2000 |
| Anti-cyclin H | Cell signal Technology, Danvers, MA, USA | 1:1000 | HRP linked secondary antibody Anti-rabbit | Sigma–Aldrich, St. Louis, MO, USA | 1:2000 |
| Anti-cyclin D1 | Cell signal Technology, Danvers, MA, USA | 1:1000 | HRP linked secondary antibody Anti-rabbit | Sigma–Aldrich, St. Louis, MO, USA | 1:2000 |
| Anti-cyclin E1 | Cell signal Technology, Danvers, MA, USA | 1:1000 | HRP linked secondary antibody Anti-rabbit | Sigma–Aldrich, St. Louis, MO, USA | 1:2000 |
| Anti-p21 | Santa-Cruz biotechnology, Dallas, TX, USA | 1:100 | HRP linked secondary antibody Anti-mouse | Sigma–Aldrich, St. Louis, MO, USA | 1:2000 |
| Anti-p27 | Santa-Cruz biotechnology, Dallas, TX, USA | 1:100 | HRP linked secondary antibody Anti-mouse | 1:2000 | |
| Caspase-3 | Sigma–Aldrich, St. Louis, MO, USA | 1:1000 | HRP linked secondary antibody Anti-rabbit | Sigma–Aldrich, St. Louis, MO, USA | 1:2000 |
| Cleaved PARP | Cell signal Technology, Danvers, MA, USA | 1:1000 | HRP linked secondary antibody Anti-rabbit | Sigma–Aldrich, St. Louis, MO, USA | 1:2000 |
| PARP | Cell signal Technology, Danvers, MA, USA | 1:1000 | HRP linked secondary antibody Anti-rabbit | Sigma–Aldrich, St. Louis, MO, USA | 1:2000 |
| C-Myc | Cell signal Technology, Danvers, MA, USA | 1:1000 | HRP linked secondary antibody Anti-rabbit | Sigma–Aldrich, St. Louis, MO, USA | 1:2000 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Attia, Y.M.; Shouman, S.A.; Salama, S.A.; Ivan, C.; Elsayed, A.M.; Amero, P.; Rodriguez-Aguayo, C.; Lopez-Berestein, G. Blockade of CDK7 Reverses Endocrine Therapy Resistance in Breast Cancer. Int. J. Mol. Sci. 2020, 21, 2974. https://doi.org/10.3390/ijms21082974
Attia YM, Shouman SA, Salama SA, Ivan C, Elsayed AM, Amero P, Rodriguez-Aguayo C, Lopez-Berestein G. Blockade of CDK7 Reverses Endocrine Therapy Resistance in Breast Cancer. International Journal of Molecular Sciences. 2020; 21(8):2974. https://doi.org/10.3390/ijms21082974
Chicago/Turabian StyleAttia, Yasmin M., Samia A. Shouman, Salama A. Salama, Cristina Ivan, Abdelrahman M. Elsayed, Paola Amero, Cristian Rodriguez-Aguayo, and Gabriel Lopez-Berestein. 2020. "Blockade of CDK7 Reverses Endocrine Therapy Resistance in Breast Cancer" International Journal of Molecular Sciences 21, no. 8: 2974. https://doi.org/10.3390/ijms21082974
APA StyleAttia, Y. M., Shouman, S. A., Salama, S. A., Ivan, C., Elsayed, A. M., Amero, P., Rodriguez-Aguayo, C., & Lopez-Berestein, G. (2020). Blockade of CDK7 Reverses Endocrine Therapy Resistance in Breast Cancer. International Journal of Molecular Sciences, 21(8), 2974. https://doi.org/10.3390/ijms21082974