Calciprotein Particles Cause Endothelial Dysfunction under Flow

 ,
,
Abstract
1. Introduction
2. Results
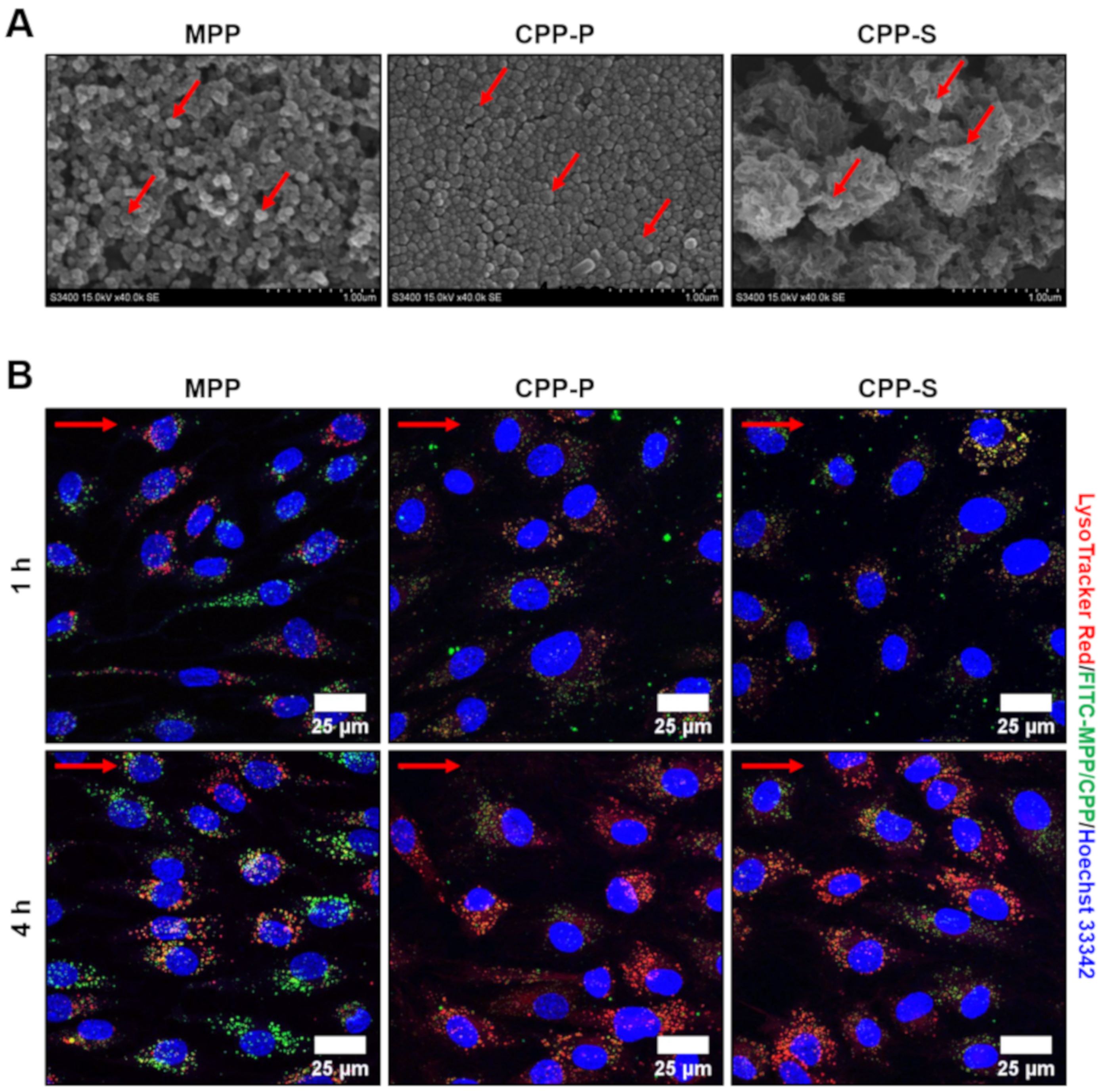
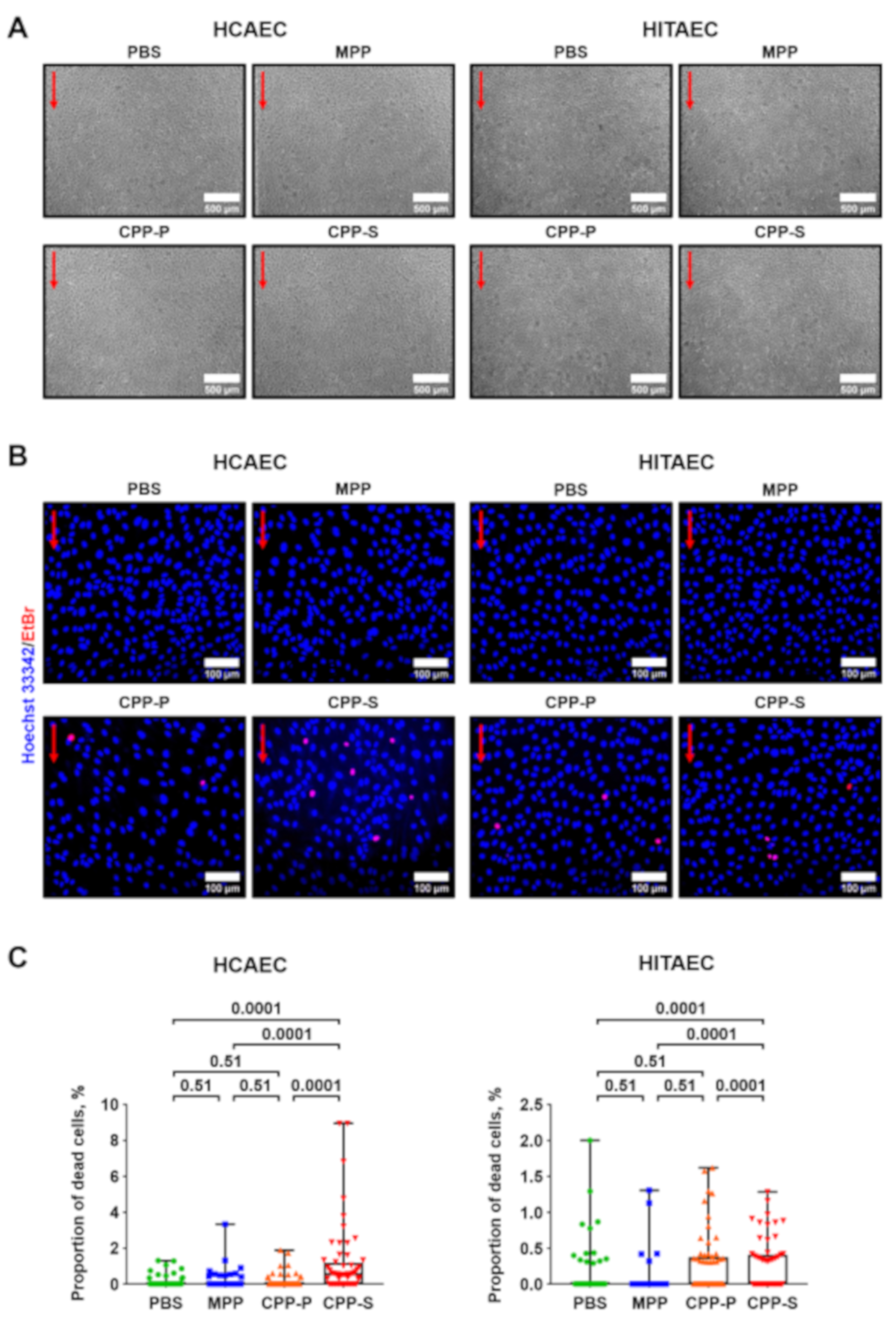
2.1. Calciprotein Particles (CPPs) Are Rapidly Internalised by ECs and Promote Endothelial Dysfunction Rather than Endothelial Injury under Flow
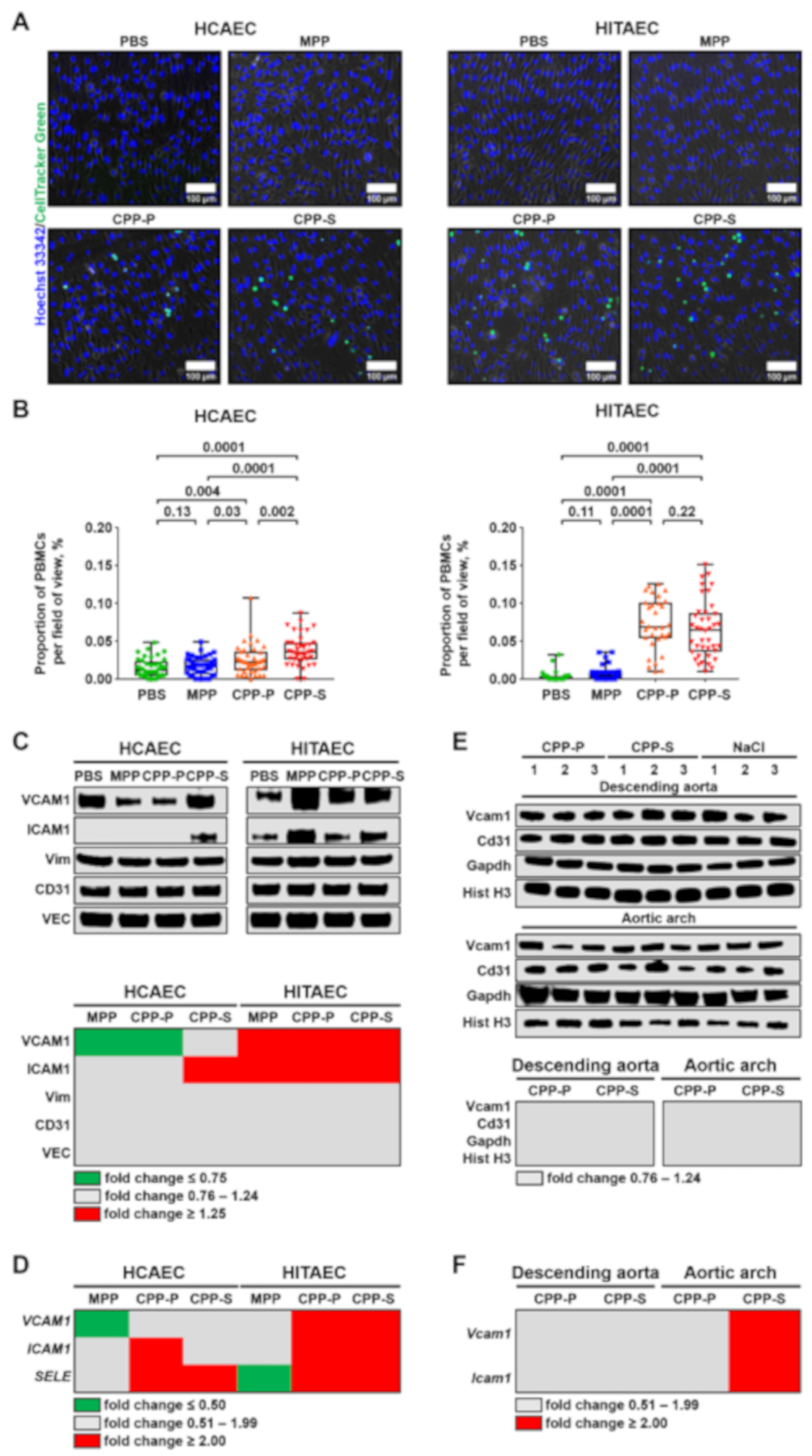
2.2. CPPs Instigate Attachment of PBMCs to the ECs
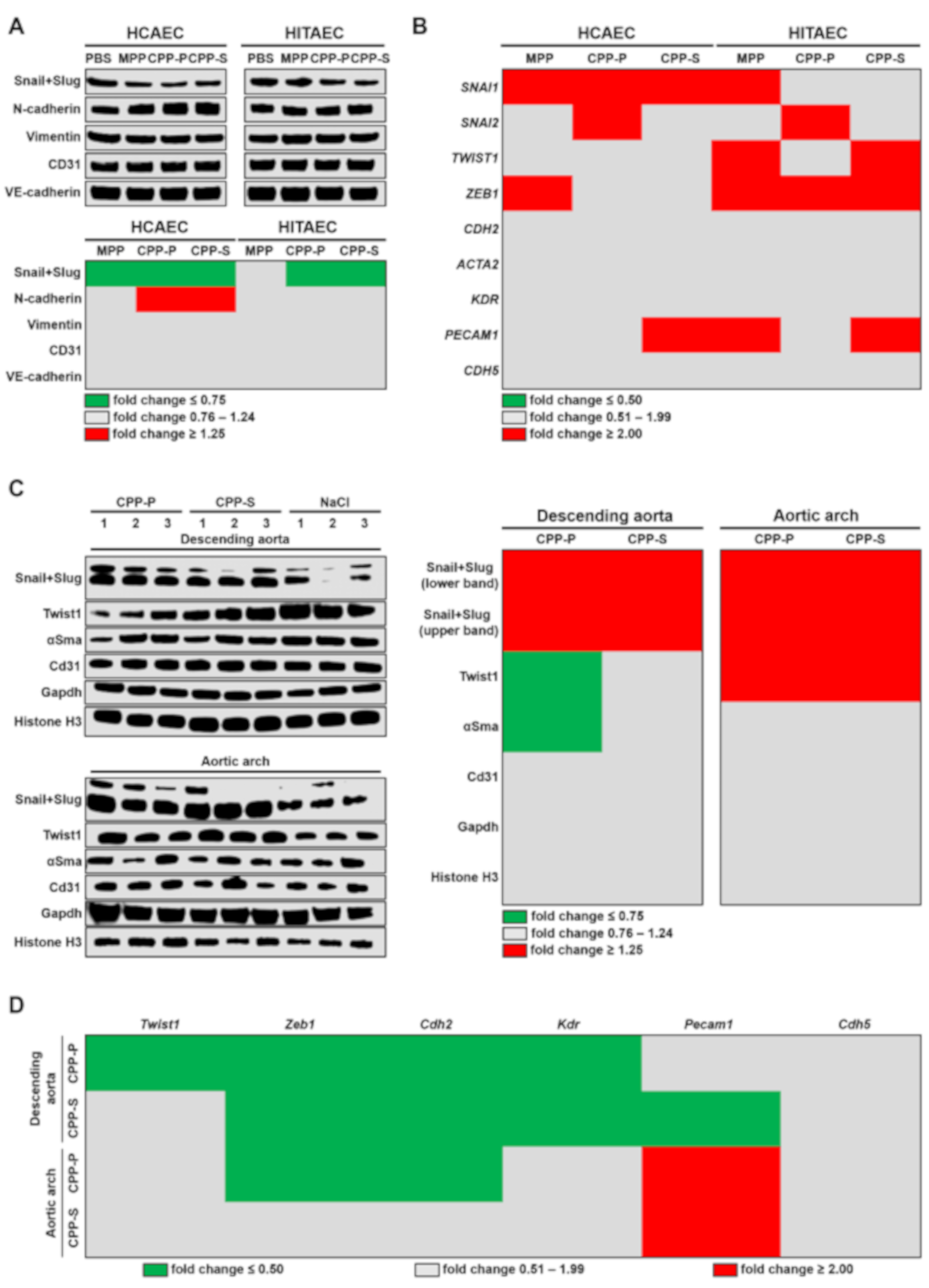
2.3. Circulation of CPPs May Contribute to the Development of EndoMT
2.4. Internalisation of CPPs Potentially Disturbs Endothelial Mechanotransduction
3. Discussion
4. Materials and Methods
4.1. Artificial Synthesis of CPPs and MPPs
4.2. Cell Culture and Flow Culture Model
4.3. Animal Model
4.4. RT-qPCR
4.5. Western Blotting
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CPPs | Calciprotein particles |
| MAC | Medial arterial calcification |
| ESRD | End-stage renal disease |
| ECs | Endothelial cells |
| CKD | Chronic kidney disease |
| EndoMT | Endothelial-to-mesenchymal transition |
| HCAEC | Human coronary artery endothelial cells |
| HITAEC | Human internal thoracic artery endothelial cells |
| PBMCs | Peripheral blood mononuclear cells |
| VCAM | Vascular cell adhesion molecule |
| ICAM | Intercellular cell adhesion molecule |
| KLF | Krüppel-like factor |
| YAP | Yes-associated protein |
| CPP-P | Primary calciprotein particles |
| CPP-S | Secondary calciprotein particles |
| MPPs | Magnesiprotein particles |
| FITC | Fluorescein isothiocyanate |
| PBS | Phosphate-buffered saline |
| EtBr | Ethidium bromide |
| SELE | E-selectin |
| RIPA | Radioimmunoprecipitation assay |
| RNA | Ribonucleic acid |
| CD | Cluster of differentiation |
| VE-cadherin | Vascular endothelial cadherin |
| GAPDH | Glyceraldehyde 3-phosphate dehydrogenase |
| Hist H3 | Histone H3 |
| RT-qPCR | Reverse transcription quantitative polymerase chain reaction |
| VSMCs | Vascular smooth muscle cells |
| N-cadherin | Neural cadherin |
| SNAI | Snail family transcriptional repressor |
| TWIST | Twist family basic helix-loop-helix transcription factor |
| ZEB | Zinc finger E-box binding homeobox |
| CDH | Cadherin |
| ACTA | Actin alpha |
| KDR | Kinase insert domain receptor |
| PECAM | Platelet endothelial cell adhesion molecule |
| αSMA | Alpha smooth muscle actin |
| NRF | Nuclear factor erythroid 2–related factor |
| pYAP | Phosphorylated YAP |
| TAZ | Transcriptional co-activator with PDZ binding motif |
| pTAZ | Phosphorylated TAZ |
| NFE2L2 | Nuclear factor erythroid 2 like 2 |
| WWTR | WW domain containing transcription regulator |
| MSR | Macrophage scavenger receptor |
| ApoE | Apolipoprotein E |
| LDLR | Low-density lipoprotein receptor |
| OD | Optical density |
| cDNA | Complementary DNA |
| ACTB | Beta actin |
| B2M | Beta-2-microglobulin |
| TBP | TATA-binding protein |
| PVDF | Polyvinylidene difluoride |
| FDR | False discovery rate |
References
- Heiss, A.; DuChesne, A.; Denecke, B.; Grötzinger, J.; Yamamoto, K.; Renné, T.; Jahnen-Dechent, W. Structural basis of calcification inhibition by alpha 2-HS glycoprotein/fetuin-A. Formation of colloidal calciprotein particles. J. Biol. Chem. 2003, 278, 13333–13341. [Google Scholar] [CrossRef]
- El-Abbadi, M.M.; Pai, A.S.; Leaf, E.M.; Yang, H.Y.; Bartley, B.A.; Quan, K.K.; Ingalls, C.M.; Liao, H.W.; Giachelli, C.M. Phosphate feeding induces arterial medial calcification in uremic mice: Role of serum phosphorus, fibroblast growth factor-23, and osteopontin. Kidney Int. 2009, 75, 1297–1307. [Google Scholar] [CrossRef] [PubMed]
- London, G.M.; Guérin, A.P.; Marchais, S.J.; Métivier, F.; Pannier, B.; Adda, H. Arterial media calcification in end-stage renal disease: Impact on all-cause and cardiovascular mortality. Nephrol. Dial. Transplant. 2003, 18, 1731–1740. [Google Scholar] [CrossRef] [PubMed]
- Masumoto, A.; Sonou, T.; Ohya, M.; Yashiro, M.; Nakashima, Y.; Okuda, K.; Iwashita, Y.; Mima, T.; Negi, S.; Shigematsu, T. Calcium Overload Accelerates Phosphate-Induced Vascular Calcification Via Pit-1, but not the Calcium-Sensing Receptor. J. Atheroscler. Thromb. 2017, 24, 716–724. [Google Scholar] [CrossRef] [PubMed]
- Sonou, T.; Ohya, M.; Yashiro, M.; Masumoto, A.; Nakashima, Y.; Ito, T.; Mima, T.; Negi, S.; Kimura-Suda, H.; Shigematsu, T. Magnesium prevents phosphate-induced vascular calcification via TRPM7 and Pit-1 in an aortic tissue culture model. Hypertens. Res. 2017, 40, 562–567. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Fujiyoshi, A.; Willcox, B.; Choo, J.; Vishnu, A.; Hisamatsu, T.; Ahuja, V.; Takashima, N.; Barinas-Mitchell, E.; Kadota, A.; et al. ERA JUMP Study Group. Increased Aortic Calcification Is Associated With Arterial Stiffness Progression in Multiethnic Middle-Aged Men. Hypertension 2017, 69, 102–108. [Google Scholar] [CrossRef]
- Sutliff, R.L.; Walp, E.R.; El-Ali, A.M.; Elkhatib, S.; Lomashvili, K.A.; O’Neill, W.C. Effect of medial calcification on vascular function in uremia. Am. J. Physiol. Renal. Physiol. 2011, 301, F78–F83. [Google Scholar] [CrossRef]
- Forsythe, R.O.; Dweck, M.R.; McBride, O.M.B.; Vesey, A.T.; Semple, S.I.; Shah, A.S.V.; Adamson, P.D.; Wallace, W.A.; Kaczynski, J.; Ho, W.; et al. (18)F-Sodium Fluoride Uptake in Abdominal Aortic Aneurysms: The SoFIA(3) Study. J. Am. Coll. Cardiol. 2018, 71, 513–523. [Google Scholar] [CrossRef]
- Buijs, R.V.; Willems, T.P.; Tio, R.A.; Boersma, H.H.; Tielliu, I.F.; Slart, R.H.; Zeebregts, C.J. Calcification as a risk factor for rupture of abdominal aortic aneurysm. Eur. J. Vasc. Endovasc. Surg. 2013, 46, 542–548. [Google Scholar] [CrossRef]
- Chowdhury, M.M.; Zieliński, L.P.; Sun, J.J.; Lambracos, S.; Boyle, J.R.; Harrison, S.C.; Rudd, J.H.F.; Coughlin, P.A. Editor’s Choice—Calcification of Thoracic and Abdominal Aneurysms is Associated with Mortality and Morbidity. Eur. J. Vasc. Endovasc. Surg. 2018, 55, 101–108. [Google Scholar] [CrossRef]
- Kutikhin, A.G.; Velikanova, E.A.; Mukhamadiyarov, R.A.; Glushkova, T.V.; Borisov, V.V.; Matveeva, V.G.; Antonova, L.V.; Filip’ev, D.E.; Golovkin, A.S.; Shishkova, D.K.; et al. Apoptosis-mediated endothelial toxicity but not direct calcification or functional changes in anti-calcification proteins defines pathogenic effects of calcium phosphate bions. Sci. Rep. 2016, 6, 27255. [Google Scholar] [CrossRef] [PubMed]
- Shishkova, D.; Velikanova, E.; Sinitsky, M.; Tsepokina, A.; Gruzdeva, O.; Bogdanov, L.; Kutikhin, A. Calcium Phosphate Bions Cause Intimal Hyperplasia in Intact Aortas of Normolipidemic Rats through Endothelial Injury. Int. J. Mol. Sci. 2019, 20, 5728. [Google Scholar] [CrossRef] [PubMed]
- Gimbrone, M.A., Jr.; García-Cardeña, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef] [PubMed]
- Pruijm, M.; Lu, Y.; Megdiche, F.; Piskunowicz, M.; Milani, B.; Stuber, M.; Bachtler, M.; Vogt, B.; Burnier, M.; Pasch, A. Serum calcification propensity is associated with renal tissue oxygenation and resistive index in patients with arterial hypertension or chronic kidney disease. J. Hypertens. 2017, 35, 2044–2052. [Google Scholar] [CrossRef]
- Nakazato, J.; Hoshide, S.; Wake, M.; Miura, Y.; Kuro-O, M.; Kario, K. Association of calciprotein particles measured by a new method with coronary artery plaque in patients with coronary artery disease: A cross-sectional study. J. Cardiol. 2019, 74, 428–435. [Google Scholar] [CrossRef]
- Bundy, J.D.; Cai, X.; Scialla, J.J.; Dobre, M.A.; Chen, J.; Hsu, C.Y.; Leonard, M.B.; Go, A.S.; Rao, P.S.; Lash, J.P.; et al. CRIC Study Investigators. Serum Calcification Propensity and Coronary Artery Calcification among Patients with CKD: The CRIC (Chronic Renal Insufficiency Cohort) Study. Am. J. Kidney Dis. 2019, 73, 806–814. [Google Scholar] [CrossRef]
- Eelderink, C.; Te Velde-Keyzer, C.A.; Frenay, A.S.; Vermeulen, E.A.; Bachtler, M.; Aghagolzadeh, P.; van Dijk, P.R.; Gansevoort, R.T.; Vervloet, M.G.; Hillebrands, J.L.; et al. NIGRAM2+ consortium. Serum Calcification Propensity and the Risk of Cardiovascular and All-Cause Mortality in the General Population: The PREVEND Study. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 1942–1951. [Google Scholar] [CrossRef]
- Pasch, A.; Block, G.A.; Bachtler, M.; Smith, E.R.; Jahnen-Dechent, W.; Arampatzis, S.; Chertow, G.M.; Parfrey, P.; Ma, X.; Floege, J. Blood Calcification Propensity, Cardiovascular Events, and Survival in Patients Receiving Hemodialysis in the EVOLVE Trial. Clin. J. Am. Soc. Nephrol. 2017, 12, 315–322. [Google Scholar] [CrossRef]
- Dahle, D.O.; Åsberg, A.; Hartmann, A.; Holdaas, H.; Bachtler, M.; Jenssen, T.G.; Dionisi, M.; Pasch, A. Serum Calcification Propensity Is a Strong and Independent Determinant of Cardiac and All-Cause Mortality in Kidney Transplant Recipients. Am. J. Transplant. 2016, 16, 204–212. [Google Scholar] [CrossRef]
- Gordon, E.; Schimmel, L.; Frye, M. The Importance of Mechanical Forces for in vitro Endothelial Cell Biology. Front. Physiol. 2020, 11, 684. [Google Scholar] [CrossRef]
- Cho, J.G.; Lee, A.; Chang, W.; Lee, M.S.; Kim, J. Endothelial to Mesenchymal Transition Represents a Key Link in the Interaction between Inflammation and Endothelial Dysfunction. Front. Immunol. 2018, 9, 294. [Google Scholar] [CrossRef] [PubMed]
- Baratchi, S.; Khoshmanesh, K.; Woodman, O.L.; Potocnik, S.; Peter, K.; McIntyre, P. Molecular Sensors of Blood Flow in Endothelial Cells. Trends Mol. Med. 2017, 23, 850–868. [Google Scholar] [CrossRef] [PubMed]
- Jensen, H.A.; Mehta, J.L. Endothelial cell dysfunction as a novel therapeutic target in atherosclerosis. Expert Rev. Cardiovasc. Ther. 2016, 14, 1021–1033. [Google Scholar] [CrossRef] [PubMed]
- Cahill, P.A.; Redmond, E.M. Vascular endothelium—Gatekeeper of vessel health. Atherosclerosis 2016, 248, 97–109. [Google Scholar] [CrossRef]
- Chen, P.Y.; Schwartz, M.A.; Simons, M. Endothelial-to-Mesenchymal Transition, Vascular Inflammation, and Atherosclerosis. Front. Cardiovasc. Med. 2020, 7, 53. [Google Scholar] [CrossRef]
- Li, Y.; Lui, K.O.; Zhou, B. Reassessing endothelial-to-mesenchymal transition in cardiovascular diseases. Nat. Rev. Cardiol. 2018, 15, 445–456. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. Effects of shear stress on endothelial cells: Go with the flow. Acta Physiol. 2017, 219, 382–408. [Google Scholar] [CrossRef]
- Baeyens, N.; Bandyopadhyay, C.; Coon, B.G.; Yun, S.; Schwartz, M.A. Endothelial fluid shear stress sensing in vascular health and disease. J. Clin. Investig. 2016, 126, 821–828. [Google Scholar] [CrossRef]
- Le, N.T.; Sandhu, U.G.; Quintana-Quezada, R.A.; Hoang, N.M.; Fujiwara, K.; Abe, J.I. Flow signaling and atherosclerosis. Cell Mol. Life Sci. 2017, 74, 1835–1858. [Google Scholar] [CrossRef]
- Bundy, J.D.; Cai, X.; Mehta, R.C.; Scialla, J.J.; de Boer, I.H.; Hsu, C.Y.; Go, A.S.; Dobre, M.A.; Chen, J.; Rao, P.S.; et al. CRIC Study Investigators. Serum Calcification Propensity and Clinical Events in CKD. Clin. J. Am. Soc. Nephrol. 2019, 14, 1562–1571. [Google Scholar] [CrossRef]
- Lorenz, G.; Steubl, D.; Kemmner, S.; Pasch, A.; Koch-Sembdner, W.; Pham, D.; Haller, B.; Bachmann, Q.; Mayer, C.C.; Wassertheurer, S.; et al. Worsening calcification propensity precedes all-cause and cardiovascular mortality in haemodialyzed patients. Sci. Rep. 2017, 7, 13368. [Google Scholar] [CrossRef] [PubMed]
- Keyzer, C.A.; de Borst, M.H.; van den Berg, E.; Jahnen-Dechent, W.; Arampatzis, S.; Farese, S.; Bergmann, I.P.; Floege, J.; Navis, G.; Bakker, S.J.; et al. Calcification Propensity and Survival among Renal Transplant Recipients. J. Am. Soc. Nephrol. 2016, 27, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Bostom, A.; Pasch, A.; Madsen, T.; Roberts, M.B.; Franceschini, N.; Steubl, D.; Garimella, P.S.; Ix, J.H.; Tuttle, K.R.; Ivanova, A.; et al. Serum Calcification Propensity and Fetuin-A: Biomarkers of Cardiovascular Disease in Kidney Transplant Recipients. Am. J. Nephrol. 2018, 48, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Kutikhin, A.G.; Sinitsky, M.Y.; Yuzhalin, A.E.; Velikanova, E.A. Shear stress: An essential driver of endothelial progenitor cells. J. Mol. Cell Cardiol. 2018, 118, 46–69. [Google Scholar] [CrossRef]
- Cea Soriano, L.; Johansson, S.; Stefansson, B.; Rodríguez, L.A. Cardiovascular events and all-cause mortality in a cohort of 57,946 patients with type 2 diabetes: Associations with renal function and cardiovascular risk factors. Cardiovasc. Diabetol. 2015, 14, 38. [Google Scholar] [CrossRef]
- Ohishi, M.; Tatara, Y.; Ito, N.; Takeya, Y.; Onishi, M.; Maekawa, Y.; Kato, N.; Kamide, K.; Rakugi, H. The combination of chronic kidney disease and increased arterial stiffness is a predictor for stroke and cardiovascular disease in hypertensive patients. Hypertens. Res. 2011, 34, 1209–1215. [Google Scholar] [CrossRef]
- Dumaine, R.L.; Montalescot, G.; Steg, P.G.; Ohman, E.M.; Eagle, K.; Bhatt, D.L.; REACH Registry Investigators. Renal function, atherothrombosis extent, and outcomes in high-risk patients. Am. Heart J. 2009, 158, 141–148.e1. [Google Scholar] [CrossRef]
- Bentzon, J.F.; Otsuka, F.; Virmani, R.; Falk, E. Mechanisms of plaque formation and rupture. Circ. Res. 2014, 114, 1852–1866. [Google Scholar] [CrossRef]
- Otsuka, F.; Yahagi, K.; Sakakura, K.; Virmani, R. Why is the mammary artery so special and what protects it from atherosclerosis? Ann. Cardiothorac. Surg. 2013, 2, 519–526. [Google Scholar] [CrossRef]
- Perrotta, I.; Sciangula, A.; Concistrè, G.; Mazzulla, S.; Aquila, S.; Agnino, A. Internal mammary artery atherosclerosis: An ultrastructural study of two cases. Ultrastruct. Pathol. 2014, 38, 199–203. [Google Scholar] [CrossRef]
- Heiss, A.; Eckert, T.; Aretz, A.; Richtering, W.; van Dorp, W.; Schäfer, C.; Jahnen-Dechent, W. Hierarchical role of fetuin-A and acidic serum proteins in the formation and stabilization of calcium phosphate particles. J. Biol. Chem. 2008, 283, 14815–14825. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.R.; Hewitson, T.D.; Hanssen, E.; Holt, S.G. Biochemical transformation of calciprotein particles in uraemia. Bone 2018, 110, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Koppert, S.; Büscher, A.; Babler, A.; Ghallab, A.; Buhl, E.M.; Latz, E.; Hengstler, J.G.; Smith, E.R.; Jahnen-Dechent, W. Cellular Clearance and Biological Activity of Calciprotein Particles Depend on Their Maturation State and Crystallinity. Front. Immunol. 2018, 9, 1991. [Google Scholar] [CrossRef] [PubMed]
- van Setten, P.A.; van Hinsbergh, V.W.; van der Velden, T.J.; van de Kar, N.C.; Vermeer, M.; Mahan, J.D.; Assmann, K.J.; van den Heuvel, L.P.; Monnens, L.A. Effects of TNF alpha on verocytotoxin cytotoxicity in purified human glomerular microvascular endothelial cells. Kidney Int. 1997, 51, 1245–1256. [Google Scholar] [CrossRef]
- Eslami, P.; Seo, J.H.; Lardo, A.C.; Chen, M.Y.; Mittal, R. Flow Dynamics in the Aortic Arch and Its Effect on the Arterial Input Function in Cardiac Computed Tomography. J. Biomech. Eng. 2019, 141, 1045011–1045018. [Google Scholar] [CrossRef]
- Endo, S.; Goldsmith, H.L.; Karino, T. Flow patterns and preferred sites of atherosclerotic lesions in the human aorta—II. Abdominal aorta. Biorheology 2014, 51, 257–274. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer Sequence | Reverse Primer Sequence |
|---|---|---|
| Human primers | ||
| VCAM1 | 5′-CGTCTTGGTCAGCCCTTCCT-3′ | 5′-ACATTCATATACTCCCGCATCCTTC-3′ |
| ICAM1 | 5′-TTGGGCATAGAGACCCCGTT-3′ | 5′-GCACATTGCTCAGTTCATACACC-3′ |
| SELE | 5′-GCACAGCCTTGTCCAACC-3′ | 5′-ACCTCACCAAACCCTTCG-3′ |
| SNAI1 | 5′-CAGACCCACTCAGATGTCAAGAA-3′ | 5′-GGGCAGGTATGGAGAGGAAGA-3′ |
| SNAI2 | 5′-ACTCCGAAGCCAAATGACAA-3′ | 5′-CTCTCTCTGTGGGTGTGTGT-3′ |
| TWIST1 | 5′-GTCCGCAGTCTTACGAGGAG-3′ | 5′-GCTTGAGGGTCTGAATCTTGCT-3′ |
| ZEB1 | 5′-GATGATGAATGCGAGTCAGATGC-3′ | 5′-ACAGCAGTGTCTTGTTGTTGT-3′ |
| CDH2 | 5′-GCTTCTGGTGAAATCGCATTA-3′ | 5′-AGTCTCTCTTCTGCCTTTGTAG-3′ |
| ACTA2 | 5′-GTGTTGCCCCTGAAGAGCAT-3′ | 5′-GCTGGGACATTGAAAGTCTCA-3′ |
| KDR | 5′-TGCCTACCTCACCTGTTTC-3′ | 5′-GGCTCTTTCGCTTACTGTTC-3′ |
| PECAM1 | 5′-TGGCGCATGCCTGTAGTA-3′ | 5′-TCCGTTTCCTGGGTTCAA-3′ |
| CDH5 | 5′-AAGCGTGAGTCGCAAGAATG-3′ | 5′-TCTCCAGGTTTTCGCCAGTG-3′ |
| KLF2 | 5′-CAGCACTGGTCTGGTTGCTTG-3′ | 5′-ACCCACTGCACACGATGCTT-3′ |
| KLF4 | 5′-GAAAAGGACCGCCACCCACA-3′ | 5′-AGCGGGCGAATTTCCATCCA-3′ |
| NFE2L2 | 5′-GCACATCCAGTCAGAAACCAGT-3′ | 5′-ACTGAAACGTAGCCGAAGAAAC-3′ |
| YAP1 | 5′-AGAACTGCTTCGGCAGGCAA-3′ | 5′-CCACCATCCTGCTCCAGTGT-3′ |
| WWTR1 | 5′-CGTCAGTTCCACACCAGTGC-3′ | 5′-GGTTCTGCTGGCTCAGGGTA-3′ |
| ACTB | 5′-CATCGAGCACGGCATCGTCA-3′ | 5′-TAGCACAGCCTGGACAGCAAC-3′ |
| GAPDH | 5′-AGCCACATCGCTCAGACAC-3′ | 5′-GCCCAATACGACCAAATCC-3′ |
| B2M | 5′-TCCATCCGACATTGAAGTTG-3′ | 5′-CGGCAGGCATACTCATCTT-3′ |
| Rat primers | ||
| Vcam1 | 5′-GGAAATGCCACCCTCACCTTA-3′ | 5′-TCCAGGGGAGATGTCAACAGT-3′ |
| Icam1 | 5′-CGACATTGGGGAAGACAGCAG-3′ | 5′-TCCACTCGCTCTGGGAACG-3′ |
| Twist1 | 5′-ATGTCCGCGTCCCACTAGCA-3′ | 5′-CCCCACGCCCTGATTCTTGT-3′ |
| Zeb1 | 5′-CCAGTGAAGGTGATCCAGCCA-3′ | 5′-CTTTTTGGGTGGCGTGCAGT-3′ |
| Cdh2 | 5′-ACCCAGGAAAAGTGGCAGGT-3′ | 5′-GCTGTGCTTGGCGAGTTGTC-3′ |
| Kdr | 5′-TGGTCCTTGCCTCAGAAGAGC-3′ | 5′-GCTGGTCTGGTTGGAGCCTT-3′ |
| Pecam1 | 5′-CCGTCCAGGTGTGCGAAATG-3′ | 5′-GGGCGCAGTCCCATTTACTG-3′ |
| Cdh5 | 5′-ACAAGGACGTGGTGCCAGTA-3′ | 5′-GGGCATCCCATTGTCGGAGA-3′ |
| Klf2 | 5′-CACCAACTGCGGCAAGACCT-3′ | 5′-GTAGTGGCGGGTAAGCTCGTCA-3′ |
| Klf4 | 5′-GACTATGCAGGCTGTGGCAAA-3′ | 5′-CGGTAGTGCCTGGTCAGTTCA-3′ |
| Nfe2l2 | 5′-CAGTGGATCTGTCAGCTACTCC-3′ | 5′-CTCTCAACGTGGCTGGGAAT-3′ |
| Yap1 | 5′-TTTCGGCAGGCAATACGGAAT-3′ | 5′-TGCTCCAGTGAGGGCAACTG-3′ |
| Wwtr1 | 5′-GTCAACACGCCTGCCATGAA-3′ | 5′-TGCTCTGCTCCCGTGAATGA-3′ |
| Actb | 5′-ACAACCTTCTTGCAGCTCCTC-3′ | 5′-CCATACCCACCATCACACCCT-3′ |
| Tbp | 5′-TGCCAAGTGTGAGCCTCTCC-3′ | 5′-TGGGTTATCGTCACGCACCAT-3′ |
| B2m | 5′-GGTGACCGTGATCTTTCTGGTG-3′ | 5′-TGAGGAAGTTGGGCTTCCCATT-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shishkova, D.; Markova, V.; Sinitsky, M.; Tsepokina, A.; Velikanova, E.; Bogdanov, L.; Glushkova, T.; Kutikhin, A. Calciprotein Particles Cause Endothelial Dysfunction under Flow. Int. J. Mol. Sci. 2020, 21, 8802. https://doi.org/10.3390/ijms21228802
Shishkova D, Markova V, Sinitsky M, Tsepokina A, Velikanova E, Bogdanov L, Glushkova T, Kutikhin A. Calciprotein Particles Cause Endothelial Dysfunction under Flow. International Journal of Molecular Sciences. 2020; 21(22):8802. https://doi.org/10.3390/ijms21228802
Chicago/Turabian StyleShishkova, Daria, Victoria Markova, Maxim Sinitsky, Anna Tsepokina, Elena Velikanova, Leo Bogdanov, Tatiana Glushkova, and Anton Kutikhin. 2020. "Calciprotein Particles Cause Endothelial Dysfunction under Flow" International Journal of Molecular Sciences 21, no. 22: 8802. https://doi.org/10.3390/ijms21228802
APA StyleShishkova, D., Markova, V., Sinitsky, M., Tsepokina, A., Velikanova, E., Bogdanov, L., Glushkova, T., & Kutikhin, A. (2020). Calciprotein Particles Cause Endothelial Dysfunction under Flow. International Journal of Molecular Sciences, 21(22), 8802. https://doi.org/10.3390/ijms21228802