Recurrent Glomerulonephritis after Renal Transplantation: The Clinical Problem

 ,
,  ,
, 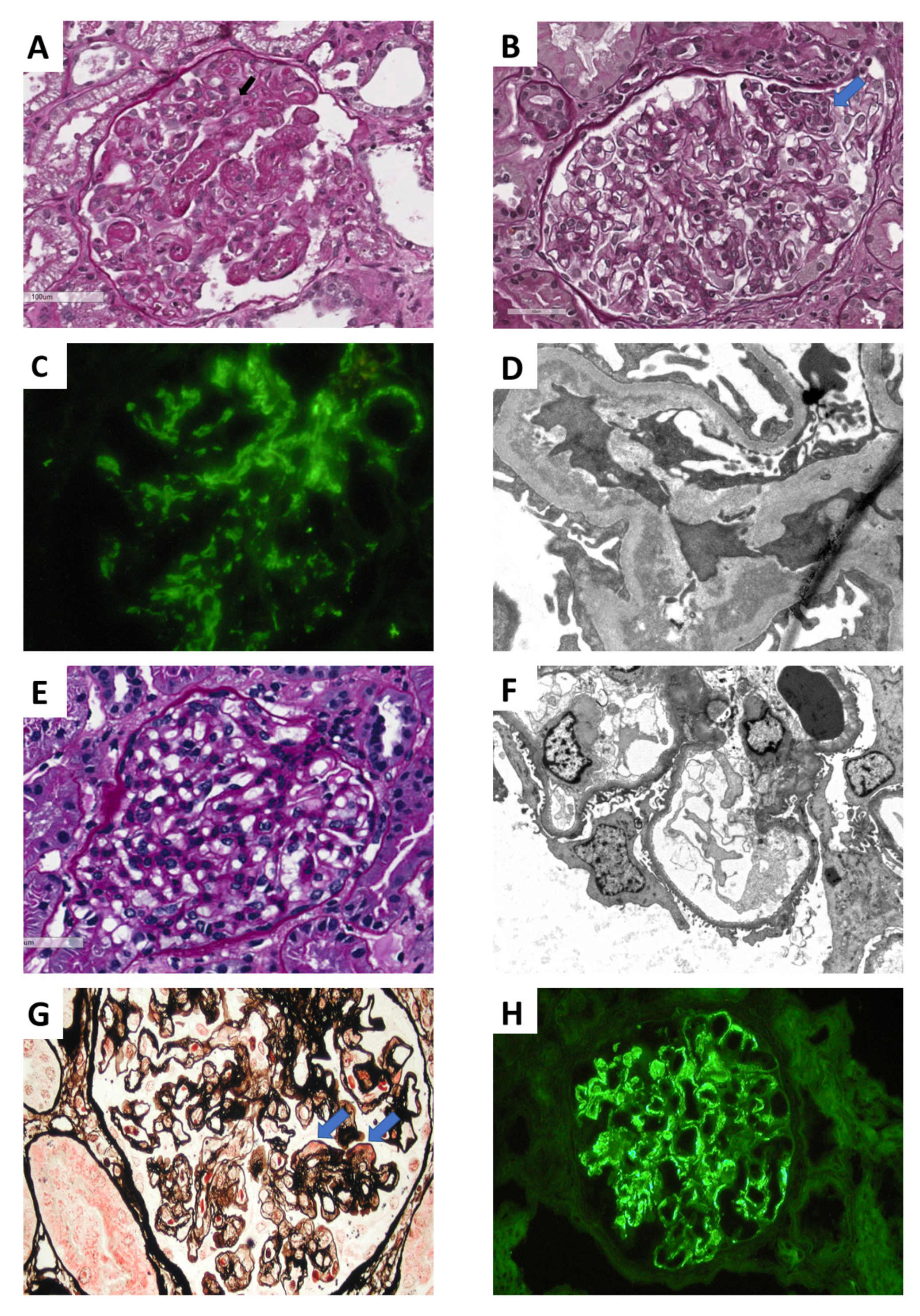{kind=link}
Abstract
1. Introduction
2. Epidemiology and Risk Factors
3. Clinical Features and Differential Diagnosis
4. Primary GN IgA Nephropathy (IgAN)
5. Focal and Segmental Glomerulosclerosis (FSGS)
5.1. Pathogenesis
5.2. Clinical Presentation of FSGS Recurrence
5.3. Risk Factors and Biomarkers of FSGS Recurrence
5.4. Treatment of Recurrent FSGS
6. Membrano-Proliferative Glomerulonephritis (MPGN)
7. Hemolytic Uremic Syndrome (HUS)
8. Membranous Nephropathy (MN)
9. Secondary GN
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ATG | Anti-thymocyte globulin |
| CALLA | Common Acute Lymphoblastic Leukemia Antigen |
| CNTFR | Ciliary neutrotrophic factor receptor |
| CR | Chronic rejection |
| DDD | Dense Deposit Disease |
| DSA | Donor-specific antibodies |
| ESKD | End-stage kidney disease |
| FSGS | Focal and segmental glomerulosclerosis |
| GBM | Glomerular basement membrane |
| Gd-IgA1 | Galactose-deficient IgA1 |
| GLEPP-1 | Glomerular Epithelial Cell Protein-1 |
| HLA | Human leukocyte antigen |
| HUS-TTP | Hemolytic uremic syndrome/thrombotic thrombocytopenic purpura |
| IgAN | IgA nephropathy |
| JAK/STAT | Janus kinase/signal transducer and activator of transcription |
| MAPK | Mitogen-activated protein kinase |
| MN | Membranous Nephropathy |
| MPGN | Membrano-proliferative GN |
| NEP | Neutral endopeptidase |
| PDGF | Platelet derived growth factor |
| PLA2R | Phospholipase A2 receptor |
| PTRO | Protein tyrosine phosphatase receptor type O |
| SLE | Systemic lupus erythematosus |
| suPAR | Soluble Urokinase-type Plasminogen Activator Receptor |
| TGF-β | Transforming growth factor |
| TNFSF13 | Tumor necrosis factor superfamily 13 |
| WT | Wilms’ tumor protein |
References
- Golgert, W.A.; Appel, G.B.; Hariharan, S. Recurrent glomerulonephritis after renal transplantation: An unsolved problem. Clin. J. Am. Soc. Nephrol. 2008, 3, 800–807. [Google Scholar] [CrossRef]
- Chadban, S. Glomerulonephritis recurrence in the renal graft. J. Am. Soc. Nephrol. 2001, 12, 394–402. [Google Scholar] [PubMed]
- Allen, P.J.; Chadban, S.J.; Craig, J.C.; Lim, W.H.; Allen, R.D.M.; Clayton, P.A.; Teixeira-Pinto, A.; Wong, G. Recurrent glomerulonephritis after kidney transplantation: Risk factors and allograft outcomes. Kidney Int. 2017, 92, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Briganti, E.M.; Russ, G.R.; McNeil, J.J.; Atkins, R.C.; Chadban, S.J. Risk of renal allograft loss from recurrent glomerulonephritis. N. Engl. J. Med. 2002, 347, 103–109. [Google Scholar] [CrossRef]
- Choy, B.Y.; Chan, T.M.; Lai, K.N. Recurrent glomerulonephritis after kidney transplantation. Am. J. Transplant. 2006, 6, 2535–2542. [Google Scholar] [CrossRef] [PubMed]
- Cosio, F.G.; Cattran, D.C. Recent advances in our understanding of recurrent primary glomerulonephritis after kidney transplantation. Kidney Int. 2017, 91, 304–314. [Google Scholar] [CrossRef] [PubMed]
- O’Shaughnessy, M.M.; Liu, S.; Montez-Rath, M.E.; Lenihan, C.R.; Lafayette, R.A.; Winkelmayer, W.C. Kidney Transplantation Outcomes across GN Subtypes in the United States. J. Am. Soc. Nephrol. 2017, 28, 632–644. [Google Scholar] [CrossRef]
- An, J.N.; Lee, J.P.; Oh, Y.J.; Oh, Y.K.; Ha, J.-W.; Chae, D.-W.; Kim, Y.S.; Lim, C.S. Incidence of post-transplant glomerulonephritis and its impact on graft outcome. Kidney Res. Clin. Pract. 2012, 31, 219–226. [Google Scholar] [CrossRef]
- Chailimpamontree, W.; Dmitrienko, S.; Li, G.; Balshaw, R.; Magil, A.; Shapiro, R.J.; Landsberg, D.; Gill, J.; Keown, P.A. Probability, predictors, and prognosis of posttransplantation glomerulonephritis. J. Am. Soc. Nephrol. 2009, 20, 843–851. [Google Scholar] [CrossRef]
- Cameron, J.S. Glomerulonephritis in renal transplants. Transplantation 1982, 34, 237–245. [Google Scholar] [CrossRef]
- Gesualdo, L.; Cormio, L.; Stallone, G.; Infante, B.; Di Palma, A.M.; Delli Carri, P.; Cignarelli, M.; Lamacchia, O.; Iannaccone, S.; Di Paolo, S.; et al. Percutaneous ultrasound-guided renal biopsy in supine antero-lateral position: A new approach for obese and non-obese patients. Nephrol. Dial. Transplant. 2008, 23, 971–976. [Google Scholar] [CrossRef] [PubMed]
- Hariharan, S. Recurrent and de novo diseases after renal transplantation. Semin. Dial. 2000, 13, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Fairhead, T.; Knoll, G. Recurrent glomerular disease after kidney transplantation. Curr. Opin. Nephrol. Hypertens. 2010, 19, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Hariharan, S.; Adams, M.B.; Brennan, D.C.; Davis, C.L.; First, M.R.; Johnson, C.P.; Ouseph, R.; Peddi, V.R.; Pelz, C.J.; Roza, A.M.; et al. Recurrent and de novo glomerular disease after renal transplantation: A report from Renal Allograft Disease Registry (RADR). Transplantation 1999, 68, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Hariharan, S.; Peddi, V.R.; Savin, V.J.; Johnson, C.P.; First, M.R.; Roza, A.M.; Adams, M.B. Recurrent and de novo renal diseases after renal transplantation: A report from the renal allograft disease registry. Am. J. Kidney Dis. 1998, 31, 928–931. [Google Scholar] [CrossRef] [PubMed]
- Andresdottir, M.B.; Hoitsma, A.J.; Assmann, K.J.; Koene, R.A.; Wetzels, J.F. The impact of recurrent glomerulonephritis on graft survival in recipients of human histocompatibility leucocyte antigen-identical living related donor grafts. Transplantation 1999, 68, 623–627. [Google Scholar] [CrossRef]
- Moroni, G.; Longhi, S.; Quaglini, S.; Rognoni, C.; Simonini, P.; Binda, V.; Montagnino, G.; Messa, P. The impact of recurrence of primary glomerulonephritis on renal allograft outcome. Clin. Transplant. 2014, 28, 368–376. [Google Scholar] [CrossRef]
- Jiang, S.H.; Kennard, A.L.; Walters, G.D. Recurrent glomerulonephritis following renal transplantation and impact on graft survival. BMC Nephrol. 2018, 19, 344. [Google Scholar] [CrossRef]
- Ohmacht, C.; Kliem, V.; Burg, M.; Nashan, B.; Schlitt, H.J.; Brunkhorst, R.; Koch, K.M.; Floege, J. Recurrent immunoglobulin A nephropathy after renal transplantation: A significant contributor to graft loss. Transplantation 1997, 64, 1493–1496. [Google Scholar] [CrossRef]
- Odorico, J.S.; Knechtle, S.J.; Rayhill, S.C.; Pirsch, J.D.; D’Alessandro, A.M.; Belzer, F.O.; Sollinger, H.W. The influence of native nephrectomy on the incidence of recurrent disease following renal transplantation for primary glomerulonephritis. Transplantation 1996, 61, 228–234. [Google Scholar] [CrossRef]
- Sakai, K.; Oguchi, H.; Muramatsu, M.; Shishido, S. Protocol graft biopsy in kidney transplantation. Nephrology (Carlton) 2018, 23 (Suppl. 2), 38–44. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.M.; Striegel, J.; Kim, Y.; Matas, A.J.; Najarian, J.S.; Mauer, S.M. Recurrence of steroid-resistant nephrotic syndrome in kidney transplants is associated with increased acute renal failure and acute rejection. Kidney Int. 1994, 45, 1440–1445. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Andresdottir, M.B.; Assmann, K.J.; Koene, R.A.; Wetzels, J.F. Immunohistological and ultrastructural differences between recurrent type I membranoproliferative glomerulonephritis and chronic transplant glomerulopathy. Am. J. Kidney Dis. 1998, 32, 582–588. [Google Scholar] [CrossRef]
- Roufosse, C.; Simmonds, N.; Clahsen-van Groningen, M.; Haas, M.; Henriksen, K.J.; Horsfield, C.; Loupy, A.; Mengel, M.; Perkowska-Ptasińska, A.; Rabant, M.; et al. A 2018 Reference Guide to the Banff Classification of Renal Allograft Pathology. Transplantation 2018, 102, 1795–1814. [Google Scholar] [CrossRef] [PubMed]
- Solez, K.; Colvin, R.B.; Racusen, L.C.; Sis, B.; Halloran, P.F.; Birk, P.E.; Campbell, P.M.; Cascalho, M.; Collins, A.B.; Demetris, A.J.; et al. Banff’05 Meeting Report: Differential diagnosis of chronic allograft injury and elimination of chronic allograft nephropathy (‘CAN’). Am. J. Transplant. 2007, 7, 518–526. [Google Scholar] [CrossRef] [PubMed]
- Naesens, M.; Kuypers, D.R.J.; Sarwal, M. Calcineurin inhibitor nephrotoxicity. Clin. J. Am. Soc. Nephrol. 2009, 4, 481–508. [Google Scholar] [CrossRef]
- Mengel, M.; Sis, B.; Halloran, P.F. SWOT analysis of Banff: Strengths, weaknesses, opportunities and threats of the international Banff consensus process and classification system for renal allograft pathology. Am. J. Transplant. 2007, 7, 2221–2226. [Google Scholar] [CrossRef]
- Racusen, L.C.; Solez, K.; Colvin, R.B.; Bonsib, S.M.; Castro, M.C.; Cavallo, T.; Croker, B.P.; Demetris, A.J.; Drachenberg, C.B.; Fogo, A.B.; et al. The Banff 97 working classification of renal allograft pathology. Kidney Int. 1999, 55, 713–723. [Google Scholar] [CrossRef]
- Filippone, E.J.; McCue, P.A.; Farber, J.L. Transplant glomerulopathy. Mod. Pathol. 2018, 31, 235–252. [Google Scholar] [CrossRef]
- Herrera, G.A.; Isaac, J.; Turbat-Herrera, E.A. Role of electron microscopy in transplant renal pathology. Ultrastruct. Pathol. 1997, 21, 481–498. [Google Scholar] [CrossRef]
- Jo, H.A.; Han, S.S.; Lee, S.; Kim, J.Y.; Yang, S.H.; Lee, H.; Yang, J.S.; Lee, J.P.; Joo, K.W.; Lim, C.S.; et al. The association of tumor necrosis factor superfamily 13 with recurrence of immunoglobulin A nephropathy in living related kidney transplantation. BMC Nephrol. 2019, 20, 33. [Google Scholar] [CrossRef] [PubMed]
- O’Shaughnessy, M.M.; Montez-Rath, M.E.; Lafayette, R.A.; Winkelmayer, W.C. Patient characteristics and outcomes by GN subtype in ESRD. Clin. J. Am. Soc. Nephrol. 2015, 10, 1170–1178. [Google Scholar] [CrossRef] [PubMed]
- Ponticelli, C.; Glassock, R.J. Posttransplant recurrence of primary glomerulonephritis. Clin. J. Am. Soc. Nephrol. 2010, 5, 2363–2372. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, F.; Gelpi, R.; Koskinen, P.; Manonelles, A.; Räisänen-Sokolowski, A.; Carrera, M.; Honkanen, E.; Grinyó, J.M.; Cruzado, J.M. IgA nephropathy recurs early in the graft when assessed by protocol biopsy. Nephrol. Dial. Transplant. 2012, 27, 2553–2558. [Google Scholar] [CrossRef] [PubMed]
- Lionaki, S.; Panagiotellis, K.; Melexopoulou, C.; Boletis, J.N. The clinical course of IgA nephropathy after kidney transplantation and its management. Transplant. Rev. (Orlando) 2017, 31, 106–114. [Google Scholar] [CrossRef]
- Moroni, G.; Belingheri, M.; Frontini, G.; Tamborini, F.; Messa, P. Immunoglobulin A Nephropathy. Recurrence After Renal Transplantation. Front. Immunol. 2019, 10, 1332. [Google Scholar] [CrossRef]
- Gharavi, A.G.; Moldoveanu, Z.; Wyatt, R.J.; Barker, C.V.; Woodford, S.Y.; Lifton, R.P.; Mestecky, J.; Novak, J.; Julian, B.A. Aberrant IgA1 glycosylation is inherited in familial and sporadic IgA nephropathy. J. Am. Soc. Nephrol. 2008, 19, 1008–1014. [Google Scholar] [CrossRef]
- Feehally, J.; Farrall, M.; Boland, A.; Gale, D.P.; Gut, I.; Heath, S.; Kumar, A.; Peden, J.F.; Maxwell, P.H.; Morris, D.L.; et al. HLA has strongest association with IgA nephropathy in genome-wide analysis. J. Am. Soc. Nephrol. 2010, 21, 1791–1797. [Google Scholar] [CrossRef]
- Moldoveanu, Z.; Wyatt, R.J.; Lee, J.Y.; Tomana, M.; Julian, B.A.; Mestecky, J.; Huang, W.-Q.; Anreddy, S.R.; Hall, S.; Hastings, M.C.; et al. Patients with IgA nephropathy have increased serum galactose-deficient IgA1 levels. Kidney Int. 2007, 71, 1148–1154. [Google Scholar] [CrossRef]
- Suzuki, H.; Kiryluk, K.; Novak, J.; Moldoveanu, Z.; Herr, A.B.; Renfrow, M.B.; Wyatt, R.J.; Scolari, F.; Mestecky, J.; Gharavi, A.G.; et al. The pathophysiology of IgA nephropathy. J. Am. Soc. Nephrol. 2011, 22, 1795–1803. [Google Scholar] [CrossRef]
- Jhee, J.H.; Kang, H.-Y.; Wu, M.; Nam, B.Y.; Chang, T.-I.; Jung, S.-Y.; Park, S.; Kim, H.; Yun, H.-R.; Kee, Y.K.; et al. Circulating CD89-IgA complex does not predict deterioration of kidney function in Korean patients with IgA nephropathy. Clin. Chem. Lab. Med. 2017, 56, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Moresco, R.N.; Speeckaert, M.M.; Delanghe, J.R. Diagnosis and monitoring of IgA nephropathy: The role of biomarkers as an alternative to renal biopsy. Autoimmun. Rev. 2015, 14, 847–853. [Google Scholar] [CrossRef] [PubMed]
- Delanghe, S.E.; Speeckaert, M.M.; Segers, H.; Desmet, K.; Vande Walle, J.; Van Laecke, S.; Vanholder, R.; Delanghe, J.R. Soluble transferrin receptor in urine, a new biomarker for IgA nephropathy and Henoch-Schönlein purpura nephritis. Clin. Biochem. 2013, 46, 591–597. [Google Scholar] [CrossRef] [PubMed]
- Tamouza, H.; Chemouny, J.M.; Raskova Kafkova, L.; Berthelot, L.; Flamant, M.; Demion, M.; Mesnard, L.; Paubelle, E.; Walker, F.; Julian, B.A.; et al. The IgA1 immune complex-mediated activation of the MAPK/ERK kinase pathway in mesangial cells is associated with glomerular damage in IgA nephropathy. Kidney Int. 2012, 82, 1284–1296. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.-X.; Fukuda, N.; Endo, M.; Tahira, Y.; Yao, E.-H.; Matsuda, H.; Ueno, T.; Matsumoto, K. Complement 3 is involved in changing the phenotype of human glomerular mesangial cells. J. Cell. Physiol. 2007, 213, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Kiryluk, K.; Novak, J.; Gharavi, A.G. Pathogenesis of immunoglobulin A nephropathy: Recent insight from genetic studies. Annu. Rev. Med. 2013, 64, 339–356. [Google Scholar] [CrossRef]
- Han, S.S.; Yang, S.H.; Choi, M.; Kim, H.-R.; Kim, K.; Lee, S.; Moon, K.C.; Kim, J.Y.; Lee, H.; Lee, J.P.; et al. The Role of TNF Superfamily Member 13 in the Progression of IgA Nephropathy. J. Am. Soc. Nephrol. 2016, 27, 3430–3439. [Google Scholar] [CrossRef]
- He, B.; Xu, W.; Santini, P.A.; Polydorides, A.D.; Chiu, A.; Estrella, J.; Shan, M.; Chadburn, A.; Villanacci, V.; Plebani, A.; et al. Intestinal bacteria trigger T cell-independent immunoglobulin A(2) class switching by inducing epithelial-cell secretion of the cytokine APRIL. Immunity 2007, 26, 812–826. [Google Scholar] [CrossRef]
- Wyld, M.L.; Chadban, S.J. Recurrent IgA Nephropathy After Kidney Transplantation. Transplantation 2016, 100, 1827–1832. [Google Scholar] [CrossRef]
- Sofue, T.; Suzuki, H.; Ueda, N.; Kushida, Y.; Minamino, T. Post-transplant immunoglobulin A deposition and nephropathy in allografts. Nephrology (Carlton) 2018, 23 (Suppl. 2), 4–9. [Google Scholar] [CrossRef]
- Floege, J.; Gröne, H.J. Recurrent IgA nephropathy in the renal allograft: Not a benign condition. Nephrol. Dial. Transplant. 2013, 28, 1070–1073. [Google Scholar] [CrossRef]
- Berthelot, L.; Robert, T.; Vuiblet, V.; Tabary, T.; Braconnier, A.; Dramé, M.; Toupance, O.; Rieu, P.; Monteiro, R.C.; Touré, F. Recurrent IgA nephropathy is predicted by altered glycosylated IgA, autoantibodies and soluble CD89 complexes. Kidney Int. 2015, 88, 815–822. [Google Scholar] [CrossRef] [PubMed]
- Zagkotsis, G.; Vourlakou, C.; Paraskevopoulos, A.; Apostolou, T. Recurrence of crescentic IgA nephropathy after renal transplantation. CEN Case Rep. 2018, 7, 268–273. [Google Scholar] [CrossRef]
- Garnier, A.-S.; Duveau, A.; Demiselle, J.; Croué, A.; Subra, J.-F.; Sayegh, J.; Augusto, J.-F. Early post-transplant serum IgA level is associated with IgA nephropathy recurrence after kidney transplantation. PLoS ONE 2018, 13, e0196101. [Google Scholar] [CrossRef] [PubMed]
- Briggs, J.D.; Jones, E. Recurrence of glomerulonephritis following renal transplantation. Scientific Advisory Board of the ERA-EDTA Registry. European Renal Association-European Dialysis and Transplant Association. Nephrol. Dial. Transplant. 1999, 14, 564–565. [Google Scholar] [CrossRef] [PubMed]
- Avasare, R.S.; Rosenstiel, P.E.; Zaky, Z.S.; Tsapepas, D.S.; Appel, G.B.; Markowitz, G.S.; Bomback, A.S.; Canetta, P.A. Predicting Post-Transplant Recurrence of IgA Nephropathy: The Importance of Crescents. Am. J. Nephrol. 2017, 45, 99–106. [Google Scholar] [CrossRef]
- Li, L.-S.; Liu, Z.-H. Epidemiologic data of renal diseases from a single unit in China: Analysis based on 13,519 renal biopsies. Kidney Int. 2004, 66, 920–923. [Google Scholar] [CrossRef]
- Lim, E.C.; Chia, D.; Gjertson, D.W.; Koka, P.; Terasaki, P.I. In vitro studies to explain high renal allograft survival in IgA nephropathy patients. Transplantation 1993, 55, 996–999. [Google Scholar] [CrossRef]
- Moroni, G.; Longhi, S.; Quaglini, S.; Gallelli, B.; Banfi, G.; Montagnino, G.; Messa, P. The long-term outcome of renal transplantation of IgA nephropathy and the impact of recurrence on graft survival. Nephrol. Dial. Transplant. 2013, 28, 1305–1314. [Google Scholar] [CrossRef]
- Odum, J.; Peh, C.A.; Clarkson, A.R.; Bannister, K.M.; Seymour, A.E.; Gillis, D.; Thomas, A.C.; Mathew, T.H.; Woodroffe, A.J. Recurrent mesangial IgA nephritis following renal transplantation. Nephrol. Dial. Transplant. 1994, 9, 309–312. [Google Scholar]
- Jeong, H.J.; Kim, Y.S.; Kwon, K.H.; Kim, S., II; Kim, M.S.; Choi, K.H.; Lee, H.Y.; Han, D.S.; Park, K. Glomerular crescents are responsible for chronic graft dysfunction in post-transplant IgA nephropathy. Pathol. Int. 2004, 54, 837–842. [Google Scholar] [CrossRef] [PubMed]
- Coppo, R.; Troyanov, S.; Bellur, S.; Cattran, D.; Cook, H.T.; Feehally, J.; Roberts, I.S.D.; Morando, L.; Camilla, R.; Tesar, V.; et al. Validation of the Oxford classification of IgA nephropathy in cohorts with different presentations and treatments. Kidney Int. 2014, 86, 828–836. [Google Scholar] [CrossRef] [PubMed]
- Trimarchi, H.; Barratt, J.; Cattran, D.C.; Cook, H.T.; Coppo, R.; Haas, M.; Liu, Z.-H.; Roberts, I.S.D.; Yuzawa, Y.; Zhang, H.; et al. Oxford Classification of IgA nephropathy 2016: An update from the IgA Nephropathy Classification Working Group. Kidney Int. 2017, 91, 1014–1021. [Google Scholar] [CrossRef] [PubMed]
- Lim, B.J.; Joo, D.J.; Kim, M.S.; Kim, Y.S.; Kim, S., II; Kim, Y.; Huh, K.H.; Koh, M.J.; Jeong, H.J. Usefulness of Oxford classification in assessing immunoglobulin A nephropathy after transplantation. Transplantation 2013, 95, 1491–1497. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Go, H.; Baek, C.H.; Kim, Y.H.; Kim, Y.C.; Yang, S.H.; Lee, J.P.; Min, S.-I.; Ha, J.; Song, E.Y.; et al. Clinical importance of the updated Oxford classification in allograft IgA nephropathy. Am. J. Transplant. 2019, 19, 2855–2864. [Google Scholar] [CrossRef]
- Barbour, S.; Beaulieu, M.; Gill, J.; Espino-Hernandez, G.; Reich, H.N.; Levin, A. The need for improved uptake of the KDIGO glomerulonephritis guidelines into clinical practice in Canada: A survey of nephrologists. Clin. Kidney J. 2014, 7, 538–545. [Google Scholar] [CrossRef]
- Oka, K.; Imai, E.; Moriyama, T.; Akagi, Y.; Ando, A.; Hori, M.; Okuyama, A.; Toki, K.; Kyo, M.; Kokado, Y.; et al. A clinicopathological study of IgA nephropathy in renal transplant recipients: Beneficial effect of angiotensin-converting enzyme inhibitor. Nephrol. Dial. Transplant. 2000, 15, 689–695. [Google Scholar] [CrossRef]
- Berthoux, F.; El Deeb, S.; Mariat, C.; Diconne, E.; Laurent, B.; Thibaudin, L. Antithymocyte globulin (ATG) induction therapy and disease recurrence in renal transplant recipients with primary IgA nephropathy. Transplantation 2008, 85, 1505–1507. [Google Scholar] [CrossRef]
- Kessler, M.; Hiesse, C.; Hestin, D.; Mayeux, D.; Boubenider, K.; Charpentier, B. Recurrence of immunoglobulin A nephropathy after renal transplantation in the cyclosporine era. Am. J. Kidney Dis. 1996, 28, 99–104. [Google Scholar] [CrossRef]
- Mulay, A.V.; van Walraven, C.; Knoll, G.A. Impact of immunosuppressive medication on the risk of renal allograft failure due to recurrent glomerulonephritis. Am. J. Transplant. 2009, 9, 804–811. [Google Scholar] [CrossRef]
- Clayton, P.; McDonald, S.; Chadban, S. Steroids and recurrent IgA nephropathy after kidney transplantation. Am. J. Transplant. 2011, 11, 1645–1649. [Google Scholar] [CrossRef] [PubMed]
- Mundel, P.; Reiser, J. Proteinuria: An enzymatic disease of the podocyte? Kidney Int. 2010, 77, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.-F.; Wang, S.-X.; Zhang, Y.-K.; Zhao, M.-H.; Zou, W.-Z. Ultrastructural features and expression of cytoskeleton proteins of podocyte from patients with minimal change disease and focal segmental glomerulosclerosis. Ren. Fail. 2008, 30, 477–483. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ponticelli, C. Recurrence of focal segmental glomerular sclerosis (FSGS) after renal transplantation. Nephrol. Dial. Transplant. 2010, 25, 25–31. [Google Scholar] [CrossRef]
- Uffing, A.; Pérez-Sáez, M.J.; La Manna, G.; Comai, G.; Fischman, C.; Farouk, S.; Manfro, R.C.; Bauer, A.C.; Lichtenfels, B.; Mansur, J.B.; et al. A large, international study on post-transplant glomerular diseases: The TANGO project. BMC Nephrol. 2018. [Google Scholar] [CrossRef]
- Kienzl-Wagner, K.; Waldegger, S.; Schneeberger, S. Disease Recurrence—The Sword of Damocles in Kidney Transplantation for Primary Focal Segmental Glomerulosclerosis. Front. Immunol. 2019, 10, 1669. [Google Scholar] [CrossRef]
- Savin, V.J.; Sharma, M.; Zhou, J.; Genochi, D.; Sharma, R.; Srivastava, T.; Ilahe, A.; Budhiraja, P.; Gupta, A.; McCarthy, E.T. Multiple Targets for Novel Therapy of FSGS Associated with Circulating Permeability Factor. Biomed. Res. Int. 2017, 2017, 6232616. [Google Scholar] [CrossRef]
- Marshall, C.B.; Shankland, S.J. Cell cycle regulatory proteins in podocyte health and disease. Nephron. Exp. Nephrol. 2007, 106, e51–e59. [Google Scholar] [CrossRef]
- Bariéty, J.; Bruneval, P.; Hill, G.S.; Mandet, C.; Jacquot, C.; Meyrier, A. Transdifferentiation of epithelial glomerular cells. J. Am. Soc. Nephrol. 2003, 14 (Suppl. 1), S42–S47. [Google Scholar] [CrossRef][Green Version]
- Charba, D.S.; Wiggins, R.C.; Goyal, M.; Wharram, B.L.; Wiggins, J.E.; McCarthy, E.T.; Sharma, R.; Sharma, M.; Savin, V.J. Antibodies to protein tyrosine phosphatase receptor type O (PTPro) increase glomerular albumin permeability (P(alb)). Am. J. Physiol. Renal Physiol. 2009, 297, F138–F144. [Google Scholar] [CrossRef][Green Version]
- Smeets, B.; Dijkman, H.B.P.M.; Wetzels, J.F.M.; Steenbergen, E.J. Lessons from studies on focal segmental glomerulosclerosis: An important role for parietal epithelial cells? J. Pathol. 2006, 210, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Matsusaka, T.; Nakayama, M.; Asano, T.; Watanabe, T.; Ichikawa, I.; Nagata, M. Genetic podocyte lineage reveals progressive podocytopenia with parietal cell hyperplasia in a murine model of cellular/collapsing focal segmental glomerulosclerosis. Am. J. Pathol. 2009, 174, 1675–1682. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, A.; Higo, S.; Fujita, E.; Mii, A.; Kaneko, T. Focal segmental glomerulosclerosis after renal transplantation. Clin. Transplant. 2011, 25 (Suppl. 2), 6–14. [Google Scholar] [CrossRef] [PubMed]
- Machuca, E.; Benoit, G.; Antignac, C. Genetics of nephrotic syndrome: Connecting molecular genetics to podocyte physiology. Hum. Mol. Genet. 2009, 18, R185–R194. [Google Scholar] [CrossRef]
- D’Agati, V.D.; Fogo, A.B.; Bruijn, J.A.; Jennette, J.C. Pathologic classification of focal segmental glomerulosclerosis: A working proposal. Am. J. Kidney Dis. 2004, 43, 368–382. [Google Scholar] [CrossRef]
- D’Agati, V.D.; Kaskel, F.J.; Falk, R.J. Focal segmental glomerulosclerosis. N. Engl. J. Med. 2011, 365, 2398–2411. [Google Scholar] [CrossRef]
- Gigante, M.; D’Altilia, M.; Montemurno, E.; Diella, S.; Bruno, F.; Netti, G.S.; Ranieri, E.; Stallone, G.; Infante, B.; Grandaliano, G.; et al. Branchio-Oto-Renal Syndrome (BOR) associated with focal glomerulosclerosis in a patient with a novel EYA1 splice site mutation. BMC Nephrol. 2013. [Google Scholar] [CrossRef]
- Rudnicki, M. FSGS Recurrence in Adults after Renal Transplantation. Biomed. Res. Int. 2016, 2016, 3295618. [Google Scholar] [CrossRef]
- Maas, R.J.H.; Deegens, J.K.J.; van den Brand, J.A.J.G.; Cornelissen, E.A.M.; Wetzels, J.F.M. A retrospective study of focal segmental glomerulosclerosis: Clinical criteria can identify patients at high risk for recurrent disease after first renal transplantation. BMC Nephrol. 2013, 14, 47. [Google Scholar] [CrossRef]
- Cibrik, D.M.; Kaplan, B.; Campbell, D.A.; Meier-Kriesche, H.-U. Renal allograft survival in transplant recipients with focal segmental glomerulosclerosis. Am. J. Transplant. 2003, 3, 64–67. [Google Scholar] [CrossRef][Green Version]
- Vinai, M.; Waber, P.; Seikaly, M.G. Recurrence of focal segmental glomerulosclerosis in renal allograft: An in-depth review. Pediatr. Transplant. 2010, 14, 314–325. [Google Scholar] [CrossRef]
- Cochat, P.; Fargue, S.; Mestrallet, G.; Jungraithmayr, T.; Koch-Nogueira, P.; Ranchin, B.; Zimmerhackl, L.B. Disease recurrence in paediatric renal transplantation. Pediatr. Nephrol. 2009, 24, 2097–2108. [Google Scholar] [CrossRef] [PubMed]
- Tada, M.; Jimi, S.; Hisano, S.; Sasatomi, Y.; Oshima, K.; Matsuoka, H.; Takebayashi, S. Histopathological evidence of poor prognosis in patients with vesicoureteral reflux. Pediatr. Nephrol. 2001, 16, 482–487. [Google Scholar] [CrossRef] [PubMed]
- Schachter, M.E.; Monahan, M.; Radhakrishnan, J.; Crew, J.; Pollak, M.; Ratner, L.; Valeri, A.M.; Stokes, M.B.; Appel, G.B. Recurrent focal segmental glomerulosclerosis in the renal allograft: Single center experience in the era of modern immunosuppression. Clin. Nephrol. 2010, 74, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Baum, M.A. Outcomes after renal transplantation for FSGS in children. Pediatric Transplant. 2004, 8, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Cochat, P.; Schell, M.; Ranchin, B.; Boueva, A.; Saïd, M.H. Management of recurrent nephrotic syndrome after kidney transplantation in children. Clin. Nephrol. 1996, 46, 17–20. [Google Scholar]
- Stokes, M.B.; De Palma, J. Post-transplantation nephrotic syndrome. Kidney Int. 2006, 69, 1088–1091. [Google Scholar] [CrossRef]
- Shalhoub, R.J. Pathogenesis of lipoid nephrosis: A disorder of T-cell function. Lancet 1974, 2, 556–560. [Google Scholar] [CrossRef]
- Savin, V.J.; Sharma, R.; Sharma, M.; McCarthy, E.T.; Swan, S.K.; Ellis, E.; Lovell, H.; Warady, B.; Gunwar, S.; Chonko, A.M.; et al. Circulating factor associated with increased glomerular permeability to albumin in recurrent focal segmental glomerulosclerosis. N. Engl. J. Med. 1996, 334, 878–883. [Google Scholar] [CrossRef]
- Dantal, J.; Godfrin, Y.; Koll, R.; Perretto, S.; Naulet, J.; Bouhours, J.F.; Soulillou, J.P. Antihuman immunoglobulin affinity immunoadsorption strongly decreases proteinuria in patients with relapsing nephrotic syndrome. J. Am. Soc. Nephrol. 1998, 9, 1709–1715. [Google Scholar]
- McCarthy, E.T.; Sharma, M.; Savin, V.J. Circulating permeability factors in idiopathic nephrotic syndrome and focal segmental glomerulosclerosis. Clin. J. Am. Soc. Nephrol. 2010, 5, 2115–2121. [Google Scholar] [CrossRef] [PubMed]
- Crosson, J.T. Focal segmental glomerulosclerosis and renal transplantation. Transplant. Proc. 2007, 39, 737–743. [Google Scholar] [CrossRef] [PubMed]
- Newstead, C.G. Recurrent disease in renal transplants. Nephrol. Dial. Transplant. 2003, 18 (Suppl. 6), vi68–vi74. [Google Scholar] [CrossRef] [PubMed]
- Hickson, L.J.; Gera, M.; Amer, H.; Iqbal, C.W.; Moore, T.B.; Milliner, D.S.; Cosio, F.G.; Larson, T.S.; Stegall, M.D.; Ishitani, M.B.; et al. Kidney transplantation for primary focal segmental glomerulosclerosis: Outcomes and response to therapy for recurrence. Transplantation 2009, 87, 1232–1239. [Google Scholar] [CrossRef] [PubMed]
- Dall’Amico, R.; Ghiggeri, G.; Carraro, M.; Artero, M.; Ghio, L.; Zamorani, E.; Zennaro, C.; Basile, G.; Montini, G.; Rivabella, L.; et al. Prediction and treatment of recurrent focal segmental glomerulosclerosis after renal transplantation in children. Am. J. Kidney Dis. 1999, 34, 1048–1055. [Google Scholar] [CrossRef]
- Couser, W. Recurrent glomerulonephritis in the renal allograft: An update of selected areas. Exp. Clin. Transplant. 2005, 3, 283–288. [Google Scholar]
- Choi, K.H.; Kim, S.I.; Yoon, S.Y.; Kim, J.H.; Kang, S.W.; Ha, S.K.; Lee, H.Y.; Han, D.S.; Kim, Y.S.; Park, K.; et al. Long-term outcome of kidney transplantation in adult recipients with focal segmental glomerulosclerosis. Yonsei Med. J. 2001, 42, 209–214. [Google Scholar] [CrossRef]
- Fujisawa, M.; Iijima, K.; Ishimura, T.; Higuchi, A.; Isotani, S.; Yoshiya, K.; Arakawa, S.; Hamami, G.; Matsumoto, O.; Yoshikawa, N.; et al. Long-term outcome of focal segmental glomerulosclerosis after Japanese pediatric renal transplantation. Pediatr. Nephrol. 2002, 17, 165–168. [Google Scholar] [CrossRef]
- Abbott, K.C.; Sawyers, E.S.; Oliver, J.D.; Ko, C.W.; Kirk, A.D.; Welch, P.G.; Peters, T.G.; Agodoa, L.Y. Graft loss due to recurrent focal segmental glomerulosclerosis in renal transplant recipients in the United States. Am. J. Kidney Dis. 2001, 37, 366–373. [Google Scholar] [CrossRef]
- Banfi, G.; Colturi, C.; Montagnino, G.; Ponticelli, C. The recurrence of focal segmental glomerulosclerosis in kidney transplant patients treated with cyclosporine. Transplantation 1990, 50, 594–596. [Google Scholar] [CrossRef]
- Swaminathan, S.; Lager, D.J.; Qian, X.; Stegall, M.D.; Larson, T.S.; Griffin, M.D. Collapsing and non-collapsing focal segmental glomerulosclerosis in kidney transplants. Nephrol. Dial. Transplant. 2006, 21, 2607–2614. [Google Scholar] [CrossRef] [PubMed]
- Nehus, E.J.; Goebel, J.W.; Succop, P.S.; Abraham, E.C. Focal segmental glomerulosclerosis in children: Multivariate analysis indicates that donor type does not alter recurrence risk. Transplantation 2013, 96, 550–554. [Google Scholar] [CrossRef]
- Raafat, R.; Travis, L.B.; Kalia, A.; Diven, S. Role of transplant induction therapy on recurrence rate of focal segmental glomerulosclerosis. Pediatr. Nephrol. 2000, 14, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Pascual, J.; Mezrich, J.D.; Djamali, A.; Leverson, G.; Chin, L.T.; Torrealba, J.; Bloom, D.; Voss, B.; Becker, B.N.; Knechtle, S.J.; et al. Alemtuzumab induction and recurrence of glomerular disease after kidney transplantation. Transplantation 2007, 83, 1429–1434. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; El Hindi, S.; Li, J.; Fornoni, A.; Goes, N.; Sageshima, J.; Maiguel, D.; Karumanchi, S.A.; Yap, H.-K.; Saleem, M.; et al. Circulating urokinase receptor as a cause of focal segmental glomerulosclerosis. Nat. Med. 2011, 17, 952–960. [Google Scholar] [CrossRef] [PubMed]
- Morath, C.; Wei, C.; Macher-Goeppinger, S.; Schwenger, V.; Zeier, M.; Reiser, J. Management of severe recurrent focal segmental glomerulosclerosis through circulating soluble urokinase receptor modification. Am. J. Ther. 2013, 20, 226–229. [Google Scholar] [CrossRef] [PubMed]
- Thunø, M.; Macho, B.; Eugen-Olsen, J. suPAR: The molecular crystal ball. Dis. Markers 2009, 27, 157–172. [Google Scholar] [CrossRef]
- Delville, M.; Sigdel, T.K.; Wei, C.; Li, J.; Hsieh, S.-C.; Fornoni, A.; Burke, G.W.; Bruneval, P.; Naesens, M.; Jackson, A.; et al. A circulating antibody panel for pretransplant prediction of FSGS recurrence after kidney transplantation. Sci. Transl. Med. 2014, 6, 256ra136. [Google Scholar] [CrossRef]
- Montagnino, G.; Colturi, C.; Banfi, G.; Aroldi, A.; Tarantino, A.; Ponticelli, C. Membranous nephropathy in cyclosporine-treated renal transplant recipients. Transplantation 1989, 47, 725–727. [Google Scholar] [CrossRef]
- Ingulli, E.; Tejani, A. Incidence, treatment, and outcome of recurrent focal segmental glomerulosclerosis posttransplantation in 42 allografts in children—A single-center experience. Transplantation 1991, 51, 401–405. [Google Scholar] [CrossRef]
- Le Berre, L.; Godfrin, Y.; Perretto, S.; Smit, H.; Buzelin, F.; Kerjaschki, D.; Usal, C.; Cuturi, C.; Soulillou, J.P.; Dantal, J. The Buffalo/Mna rat, an animal model of FSGS recurrence after renal transplantation. Transplant. Proc. 2001, 33, 3338–3340. [Google Scholar] [CrossRef]
- Faul, C.; Donnelly, M.; Merscher-Gomez, S.; Chang, Y.H.; Franz, S.; Delfgaauw, J.; Chang, J.-M.; Choi, H.Y.; Campbell, K.N.; Kim, K.; et al. The actin cytoskeleton of kidney podocytes is a direct target of the antiproteinuric effect of cyclosporine A. Nat. Med. 2008, 14, 931–938. [Google Scholar] [CrossRef]
- Sethi, S.; Fervenza, F.C. Membranoproliferative glomerulonephritis—A new look at an old entity. N. Engl. J. Med. 2012, 366, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.-C.; Fornoni, A.; Weins, A.; Hakroush, S.; Maiguel, D.; Sageshima, J.; Chen, L.; Ciancio, G.; Faridi, M.H.; Behr, D.; et al. Abatacept in B7-1-positive proteinuric kidney disease. N. Engl. J. Med. 2013, 369, 2416–2423. [Google Scholar] [CrossRef] [PubMed]
- Savin, V.J.; McCarthy, E.T.; Sharma, R.; Charba, D.; Sharma, M. Galactose binds to focal segmental glomerulosclerosis permeability factor and inhibits its activity. Transl. Res. 2008, 151, 288–292. [Google Scholar] [CrossRef]
- Leroy, S.; Guigonis, V.; Bruckner, D.; Emal-Aglae, V.; Deschênes, G.; Bensman, A.; Ulinski, T. Successful anti-TNFalpha treatment in a child with posttransplant recurrent focal segmental glomerulosclerosis. Am. J. Transplant. 2009, 9, 858–861. [Google Scholar] [CrossRef]
- Sethi, S.; Fervenza, F.C. Membranoproliferative glomerulonephritis: Pathogenetic heterogeneity and proposal for a new classification. Semin. Nephrol. 2011, 31, 341–348. [Google Scholar] [CrossRef]
- Pickering, M.C.; D’Agati, V.D.; Nester, C.M.; Smith, R.J.; Haas, M.; Appel, G.B.; Alpers, C.E.; Bajema, I.M.; Bedrosian, C.; Braun, M.; et al. C3 glomerulopathy: Consensus report. Kidney Int. 2013, 84, 1079–1089. [Google Scholar] [CrossRef]
- Sethi, S.; Fervenza, F.C.; Zhang, Y.; Nasr, S.H.; Leung, N.; Vrana, J.; Cramer, C.; Nester, C.M.; Smith, R.J.H. Proliferative glomerulonephritis secondary to dysfunction of the alternative pathway of complement. Clin. J. Am. Soc. Nephrol. 2011, 6, 1009–1017. [Google Scholar] [CrossRef]
- Licht, C.; Fremeaux-Bacchi, V. Hereditary and acquired complement dysregulation in membranoproliferative glomerulonephritis. Thromb. Haemost. 2009, 101, 271–278. [Google Scholar]
- Medjeral-Thomas, N.R.; O’Shaughnessy, M.M.; O’Regan, J.A.; Traynor, C.; Flanagan, M.; Wong, L.; Teoh, C.W.; Awan, A.; Waldron, M.; Cairns, T.; et al. C3 glomerulopathy: Clinicopathologic features and predictors of outcome. Clin. J. Am. Soc. Nephrol. 2014, 9, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Servais, A.; Noël, L.-H.; Roumenina, L.T.; Le Quintrec, M.; Ngo, S.; Dragon-Durey, M.-A.; Macher, M.-A.; Zuber, J.; Karras, A.; Provot, F.; et al. Acquired and genetic complement abnormalities play a critical role in dense deposit disease and other C3 glomerulopathies. Kidney Int. 2012, 82, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Barbour, S.; Gill, J.S. Advances in the understanding of complement mediated glomerular disease: Implications for recurrence in the transplant setting. Am. J. Transplant. 2015, 15, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Müller, D.; Rudolph, B.; Hartmann, A.; Kuwertz-Bröking, E.; Wu, K.; Kirschfink, M.; Skerka, C.; Zipfel, P.F. Combined C3b and factor B autoantibodies and MPGN type II. N. Engl. J. Med. 2011, 365, 2340–2342. [Google Scholar] [CrossRef]
- Masani, N.; Jhaveri, K.D.; Fishbane, S. Update on membranoproliferative GN. Clin. J. Am. Soc. Nephrol. 2014, 9, 600–608. [Google Scholar] [CrossRef]
- Zand, L.; Lorenz, E.C.; Cosio, F.G.; Fervenza, F.C.; Nasr, S.H.; Gandhi, M.J.; Smith, R.J.H.; Sethi, S. Clinical findings, pathology, and outcomes of C3GN after kidney transplantation. J. Am. Soc. Nephrol. 2014, 25, 1110–1117. [Google Scholar] [CrossRef]
- Lorenz, E.C.; Sethi, S.; Leung, N.; Dispenzieri, A.; Fervenza, F.C.; Cosio, F.G. Recurrent membranoproliferative glomerulonephritis after kidney transplantation. Kidney Int. 2010, 77, 721–728. [Google Scholar] [CrossRef]
- Moroni, G.; Casati, C.; Quaglini, S.; Gallelli, B.; Banfi, G.; Montagnino, G.; Messa, P. Membranoproliferative glomerulonephritis type I in renal transplantation patients: A single-center study of a cohort of 68 renal transplants followed up for 11 years. Transplantation 2011, 91, 1233–1239. [Google Scholar] [CrossRef]
- Zuber, J.; Fakhouri, F.; Roumenina, L.T.; Loirat, C.; Frémeaux-Bacchi, V. Use of eculizumab for atypical haemolytic uraemic syndrome and C3 glomerulopathies. Nat. Rev. Nephrol. 2012, 8, 643–657. [Google Scholar] [CrossRef]
- Giordano, M.; Castellano, G.; Messina, G.; Divella, C.; Bellantuono, R.; Puteo, F.; Colella, V.; Depalo, T.; Gesualdo, L. Preservation of renal function in atypical hemolytic uremic syndrome by eculizumab: A case report. Pediatrics 2012, 130. [Google Scholar] [CrossRef]
- Giordano, P.; Netti, G.S.; Santangelo, L.; Castellano, G.; Carbone, V.; Torres, D.D.; Martino, M.; Sesta, M.; Di Cuonzo, F.; Resta, M.C.; et al. A pediatric neurologic assessment score may drive the eculizumab-based treatment of Escherichia coli-related hemolytic uremic syndrome with neurological involvement. Pediatr. Nephrol. 2019, 34. [Google Scholar] [CrossRef] [PubMed]
- Zuber, J.; Le Quintrec, M.; Krid, S.; Bertoye, C.; Gueutin, V.; Lahoche, A.; Heyne, N.; Ardissino, G.; Chatelet, V.; Noël, L.-H.; et al. Eculizumab for atypical hemolytic uremic syndrome recurrence in renal transplantation. Am. J. Transplant. 2012, 12, 3337–3354. [Google Scholar] [CrossRef] [PubMed]
- McCaughan, J.A.; O’Rourke, D.M.; Courtney, A.E. Recurrent dense deposit disease after renal transplantation: An emerging role for complementary therapies. Am. J. Transplant. 2012, 12, 1046–1051. [Google Scholar] [CrossRef] [PubMed]
- Habbig, S.; Mihatsch, M.J.; Heinen, S.; Beck, B.; Emmel, M.; Skerka, C.; Kirschfink, M.; Hoppe, B.; Zipfel, P.F.; Licht, C. C3 deposition glomerulopathy due to a functional factor H defect. Kidney Int. 2009, 75, 1230–1234. [Google Scholar] [CrossRef]
- Kavanagh, D.; Richards, A.; Goodship, T.; Jalanko, H. Transplantation in atypical hemolytic uremic syndrome. Semin. Thromb. Hemost. 2010, 36, 653–659. [Google Scholar] [CrossRef]
- Netti, G.S.; Santangelo, L.; Paulucci, L.; Piscopo, G.; Torres, D.D.; Carbone, V.; Giordano, P.; Spadaccino, F.; Castellano, G.; Stallone, G.; et al. Low C3 Serum Levels Predict Severe Forms of STEC-HUS With Neurologic Involvement. Front. Med. 2020, 7, 357. [Google Scholar] [CrossRef]
- Jokiranta, T.S. HUS and atypical HUS. Blood 2017, 129, 2847–2856. [Google Scholar] [CrossRef]
- Salvadori, M.; Bertoni, E. Update on hemolytic uremic syndrome: Diagnostic and therapeutic recommendations. World J. Nephrol. 2013, 2, 56–76. [Google Scholar] [CrossRef]
- Okumi, M.; Tanabe, K. Prevention and treatment of atypical haemolytic uremic syndrome after kidney transplantation. Nephrology (Carlton) 2016, 21 (Suppl. 1), 9–13. [Google Scholar] [CrossRef][Green Version]
- Le Quintrec, M.; Zuber, J.; Moulin, B.; Kamar, N.; Jablonski, M.; Lionet, A.; Chatelet, V.; Mousson, C.; Mourad, G.; Bridoux, F.; et al. Complement genes strongly predict recurrence and graft outcome in adult renal transplant recipients with atypical hemolytic and uremic syndrome. Am. J. Transplant. 2013, 13, 663–675. [Google Scholar] [CrossRef]
- Frémeaux-Bacchi, V.; Miller, E.C.; Liszewski, M.K.; Strain, L.; Blouin, J.; Brown, A.L.; Moghal, N.; Kaplan, B.S.; Weiss, R.A.; Lhotta, K.; et al. Mutations in complement C3 predispose to development of atypical hemolytic uremic syndrome. Blood 2008, 112, 4948–4952. [Google Scholar] [CrossRef]
- Simone, S.; Rascio, F.; Castellano, G.; Divella, C.; Chieti, A.; Ditonno, P.; Battaglia, M.; Crovace, A.; Staffieri, F.; Oortwijn, B.; et al. Complement-dependent NADPH oxidase enzyme activation in renal ischemia/reperfusion injury. Free Radic. Biol. Med. 2014, 74. [Google Scholar] [CrossRef] [PubMed]
- Noris, M.; Remuzzi, G. Atypical hemolytic-uremic syndrome. N. Engl. J. Med. 2009, 361, 1676–1687. [Google Scholar] [CrossRef] [PubMed]
- Sellier-Leclerc, A.-L.; Fremeaux-Bacchi, V.; Dragon-Durey, M.-A.; Macher, M.-A.; Niaudet, P.; Guest, G.; Boudailliez, B.; Bouissou, F.; Deschenes, G.; Gie, S.; et al. Differential impact of complement mutations on clinical characteristics in atypical hemolytic uremic syndrome. J. Am. Soc. Nephrol. 2007, 18, 2392–2400. [Google Scholar] [CrossRef] [PubMed]
- Bresin, E.; Rurali, E.; Caprioli, J.; Sanchez-Corral, P.; Fremeaux-Bacchi, V.; Rodriguez de Cordoba, S.; Pinto, S.; Goodship, T.H.J.; Alberti, M.; Ribes, D.; et al. Combined complement gene mutations in atypical hemolytic uremic syndrome influence clinical phenotype. J. Am. Soc. Nephrol. 2013, 24, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, S.C.; Karpman, D.; Vaziri-Sani, F.; Kristoffersson, A.-C.; Salomon, R.; Provot, F.; Fremeaux-Bacchi, V.; Trouw, L.A.; Blom, A.M. A mutation in factor I that is associated with atypical hemolytic uremic syndrome does not affect the function of factor I in complement regulation. Mol. Immunol. 2007, 44, 1835–1844. [Google Scholar] [CrossRef]
- Loirat, C.; Fremeaux-Bacchi, V. Hemolytic uremic syndrome recurrence after renal transplantation. Pediatr. Transplant. 2008, 12, 619–629. [Google Scholar] [CrossRef]
- Richards, A.; Kavanagh, D.; Atkinson, J.P. Inherited complement regulatory protein deficiency predisposes to human disease in acute injury and chronic inflammatory statesthe examples of vascular damage in atypical hemolytic uremic syndrome and debris accumulation in age-related macular degenerati. Adv. Immunol. 2007, 96, 141–177. [Google Scholar] [CrossRef]
- Dragon-Durey, M.-A.; Loirat, C.; Cloarec, S.; Macher, M.-A.; Blouin, J.; Nivet, H.; Weiss, L.; Fridman, W.H.; Frémeaux-Bacchi, V. Anti-Factor H autoantibodies associated with atypical hemolytic uremic syndrome. J. Am. Soc. Nephrol. 2005, 16, 555–563. [Google Scholar] [CrossRef]
- Trachtman, H.; Sethna, C.; Epstein, R.; D’Souza, M.; Rubin, L.G.; Ginocchio, C.C. Atypical hemolytic uremic syndrome associated with H1N1 influenza A virus infection. Pediatr. Nephrol. 2011, 26, 145–146. [Google Scholar] [CrossRef]
- Noris, M.; Remuzzi, G. Thrombotic microangiopathy after kidney transplantation. Am. J. Transplant. 2010, 10, 1517–1523. [Google Scholar] [CrossRef] [PubMed]
- Saland, J.M.; Ruggenenti, P.; Remuzzi, G. Liver-kidney transplantation to cure atypical hemolytic uremic syndrome. J. Am. Soc. Nephrol. 2009, 20, 940–949. [Google Scholar] [CrossRef] [PubMed]
- Larrea, C.F.; Cofan, F.; Oppenheimer, F.; Campistol, J.M.; Escolar, G.; Lozano, M. Efficacy of eculizumab in the treatment of recurrent atypical hemolytic-uremic syndrome after renal transplantation. Transplantation 2010, 89, 903–904. [Google Scholar] [CrossRef] [PubMed]
- Matar, D.; Naqvi, F.; Racusen, L.C.; Carter-Monroe, N.; Montgomery, R.A.; Alachkar, N. Atypical hemolytic uremic syndrome recurrence after kidney transplantation. Transplantation 2014, 98, 1205–1212. [Google Scholar] [CrossRef]
- Dabade, T.S.; Grande, J.P.; Norby, S.M.; Fervenza, F.C.; Cosio, F.G. Recurrent idiopathic membranous nephropathy after kidney transplantation: A surveillance biopsy study. Am. J. Transplant. 2008, 8, 1318–1322. [Google Scholar] [CrossRef]
- Kowalewska, J. Pathology of recurrent diseases in kidney allografts: Membranous nephropathy and focal segmental glomerulosclerosis. Curr. Opin. Organ Transplant. 2013, 18, 313–318. [Google Scholar] [CrossRef]
- Beck, L.H.J.; Bonegio, R.G.B.; Lambeau, G.; Beck, D.M.; Powell, D.W.; Cummins, T.D.; Klein, J.B.; Salant, D.J. M-type phospholipase A2 receptor as target antigen in idiopathic membranous nephropathy. N. Engl. J. Med. 2009, 361, 11–21. [Google Scholar] [CrossRef]
- Kattah, A.; Ayalon, R.; Beck, L.H.J.; Sethi, S.; Sandor, D.G.; Cosio, F.G.; Gandhi, M.J.; Lorenz, E.C.; Salant, D.J.; Fervenza, F.C. Anti-phospholipase A2 receptor antibodies in recurrent membranous nephropathy. Am. J. Transplant. 2015, 15, 1349–1359. [Google Scholar] [CrossRef]
- Cosyns, J.P.; Couchoud, C.; Pouteil-Noble, C.; Squifflet, J.P.; Pirson, Y. Recurrence of membranous nephropathy after renal transplantation: Probability, outcome and risk factors. Clin. Nephrol. 1998, 50, 144–153. [Google Scholar]
- Dahan, K.; Debiec, H.; Plaisier, E.; Cachanado, M.; Rousseau, A.; Wakselman, L.; Michel, P.-A.; Mihout, F.; Dussol, B.; Matignon, M.; et al. Rituximab for Severe Membranous Nephropathy: A 6-Month Trial with Extended Follow-Up. J. Am. Soc. Nephrol. 2017, 28, 348–358. [Google Scholar] [CrossRef]
- Hruskova, Z.; Tesar, V.; Geetha, D. Renal Transplantation in Antineutrophil Cytoplasmic Antibody-Associated Vasculitis: Current Perspectives. Kidney Blood Press. Res. 2020, 45, 157–165. [Google Scholar] [CrossRef]
- Gera, M.; Griffin, M.D.; Specks, U.; Leung, N.; Stegall, M.D.; Fervenza, F.C. Recurrence of ANCA-associated vasculitis following renal transplantation in the modern era of immunosupression. Kidney Int. 2007, 71, 1296–1301. [Google Scholar] [CrossRef]
- Nachman, P.H.; Segelmark, M.; Westman, K.; Hogan, S.L.; Satterly, K.K.; Jennette, J.C.; Falk, R. Recurrent ANCA-associated small vessel vasculitis after transplantation: A pooled analysis. Kidney Int. 1999, 56, 1544–1550. [Google Scholar] [CrossRef]
- Goral, S.; Ynares, C.; Shappell, S.B.; Snyder, S.; Feurer, I.D.; Kazancioglu, R.; Fogo, A.B.; Helderman, J.H. Recurrent lupus nephritis in renal transplant recipients revisited: It is not rare. Transplantation 2003, 75, 651–656. [Google Scholar] [CrossRef] [PubMed]
- Ponticelli, C.; Moroni, G.; Glassock, R.J. Recurrence of secondary glomerular disease after renal transplantation. Clin. J. Am. Soc. Nephrol. 2011, 6, 1214–1221. [Google Scholar] [CrossRef] [PubMed]
- Norby, G.E.; Strøm, E.H.; Midtvedt, K.; Hartmann, A.; Gilboe, I.-M.; Leivestad, T.; Stenstrøm, J.; Holdaas, H. Recurrent lupus nephritis after kidney transplantation: A surveillance biopsy study. Ann. Rheum. Dis. 2010, 69, 1484–1487. [Google Scholar] [CrossRef] [PubMed]
- Lionaki, S.; Skalioti, C.; Boletis, J.N. Kidney transplantation in patients with systemic lupus erythematosus. World J. Transplant. 2014, 4, 176–182. [Google Scholar] [CrossRef]
- Rahman, A.; Isenberg, D.A. Systemic lupus erythematosus. N. Engl. J. Med. 2008, 358, 929–939. [Google Scholar] [CrossRef]
- Rascio, F.; Pontrelli, P.; Accetturo, M.; Oranger, A.; Gigante, M.; Castellano, G.; Gigante, M.; Zito, A.; Zaza, G.; Lupo, A.; et al. A type i interferon signature characterizes chronic antibody-mediated rejection in kidney transplantation. J. Pathol. 2015, 237, 72–84. [Google Scholar] [CrossRef]
- Castellano, G.; Cafiero, C.; Divella, C.; Sallustio, F.; Gigante, M.; Pontrelli, P.; De Palma, G.; Rossini, M.; Grandaliano, G.; Gesualdo, L. Local synthesis of interferon-alpha in lupus nephritis is associated with type I interferons signature and LMP7 induction in renal tubular epithelial cells. Arthritis Res. Ther. 2015, 17. [Google Scholar] [CrossRef]
- Zarember, K.A.; Godowski, P.J. Tissue expression of human Toll-like receptors and differential regulation of Toll-like receptor mRNAs in leukocytes in response to microbes, their products, and cytokines. J. Immunol. 2002, 168, 554–561. [Google Scholar] [CrossRef] [PubMed]
- Cook, H.T.; Botto, M. Mechanisms of Disease: The complement system and the pathogenesis of systemic lupus erythematosus. Nat. Clin. Pract. Rheumatol. 2006, 2, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Ortega, L.M.; Schultz, D.R.; Lenz, O.; Pardo, V.; Contreras, G.N. Review: Lupus nephritis: Pathologic features, epidemiology and a guide to therapeutic decisions. Lupus 2010, 19, 557–574. [Google Scholar] [CrossRef] [PubMed]
- Thibaud, V.; Rioux-Leclercq, N.; Vigneau, C.; Morice, S. Recurrence of Goodpasture syndrome without circulating anti-glomerular basement membrane antibodies after kidney transplant, a case report. BMC Nephrol. 2019, 20, 6. [Google Scholar] [CrossRef]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Infante, B.; Rossini, M.; Leo, S.; Troise, D.; Netti, G.S.; Ranieri, E.; Gesualdo, L.; Castellano, G.; Stallone, G. Recurrent Glomerulonephritis after Renal Transplantation: The Clinical Problem. Int. J. Mol. Sci. 2020, 21, 5954. https://doi.org/10.3390/ijms21175954
Infante B, Rossini M, Leo S, Troise D, Netti GS, Ranieri E, Gesualdo L, Castellano G, Stallone G. Recurrent Glomerulonephritis after Renal Transplantation: The Clinical Problem. International Journal of Molecular Sciences. 2020; 21(17):5954. https://doi.org/10.3390/ijms21175954
Chicago/Turabian StyleInfante, Barbara, Michele Rossini, Serena Leo, Dario Troise, Giuseppe Stefano Netti, Elena Ranieri, Loreto Gesualdo, Giuseppe Castellano, and Giovanni Stallone. 2020. "Recurrent Glomerulonephritis after Renal Transplantation: The Clinical Problem" International Journal of Molecular Sciences 21, no. 17: 5954. https://doi.org/10.3390/ijms21175954
APA StyleInfante, B., Rossini, M., Leo, S., Troise, D., Netti, G. S., Ranieri, E., Gesualdo, L., Castellano, G., & Stallone, G. (2020). Recurrent Glomerulonephritis after Renal Transplantation: The Clinical Problem. International Journal of Molecular Sciences, 21(17), 5954. https://doi.org/10.3390/ijms21175954