PTEN Is Required for The Anti-Epileptic Effects of AMPA Receptor Antagonists in Chronic Epileptic Rats

Abstract
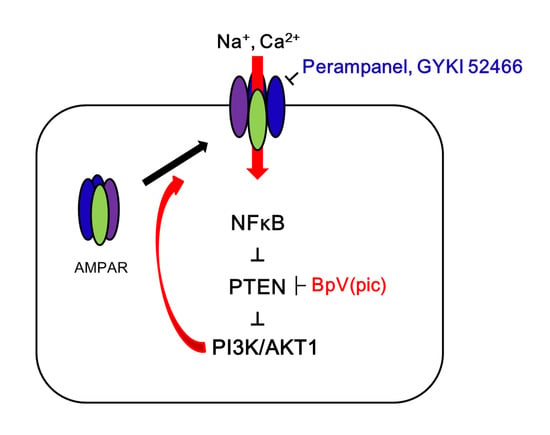
1. Introduction
2. Results
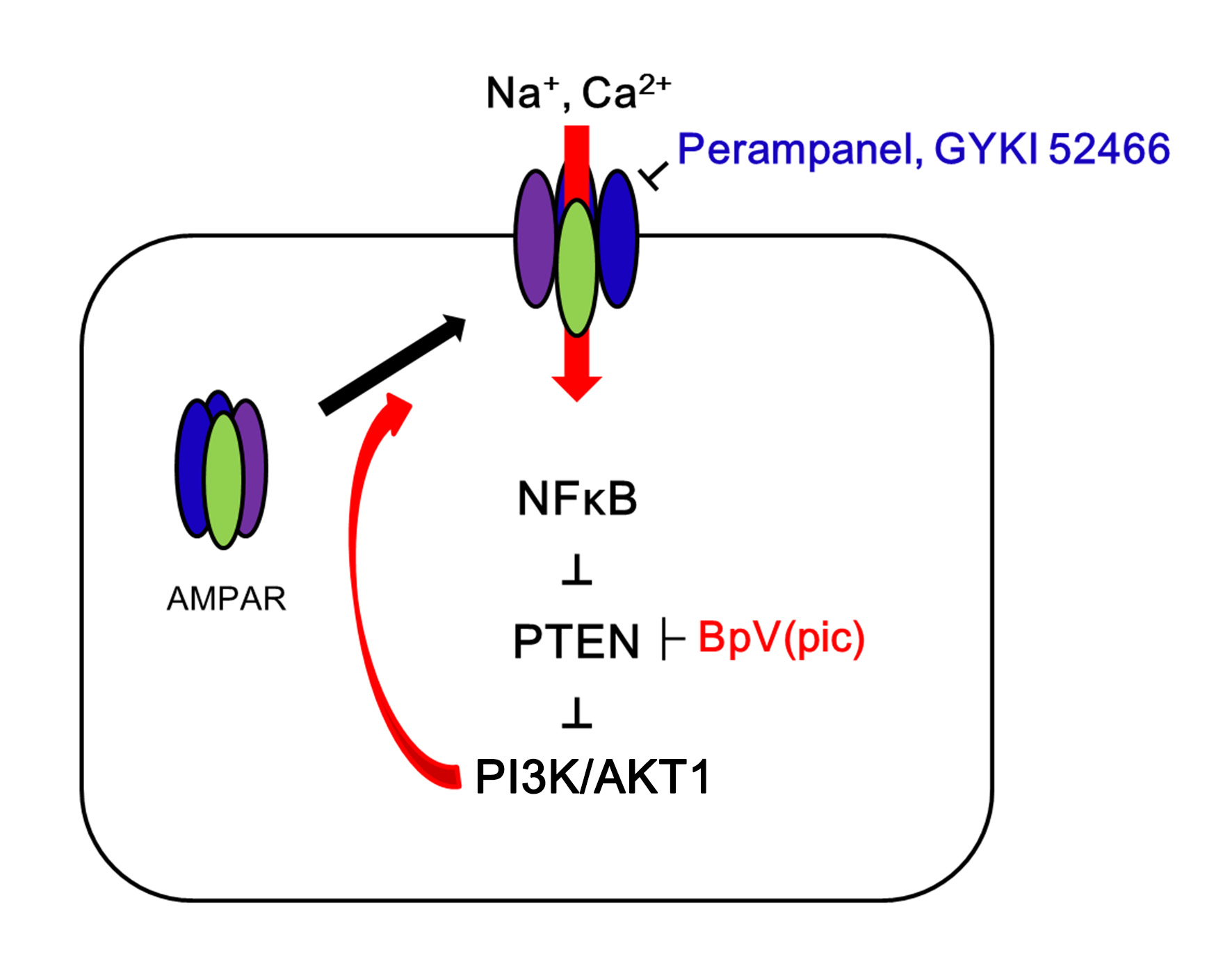
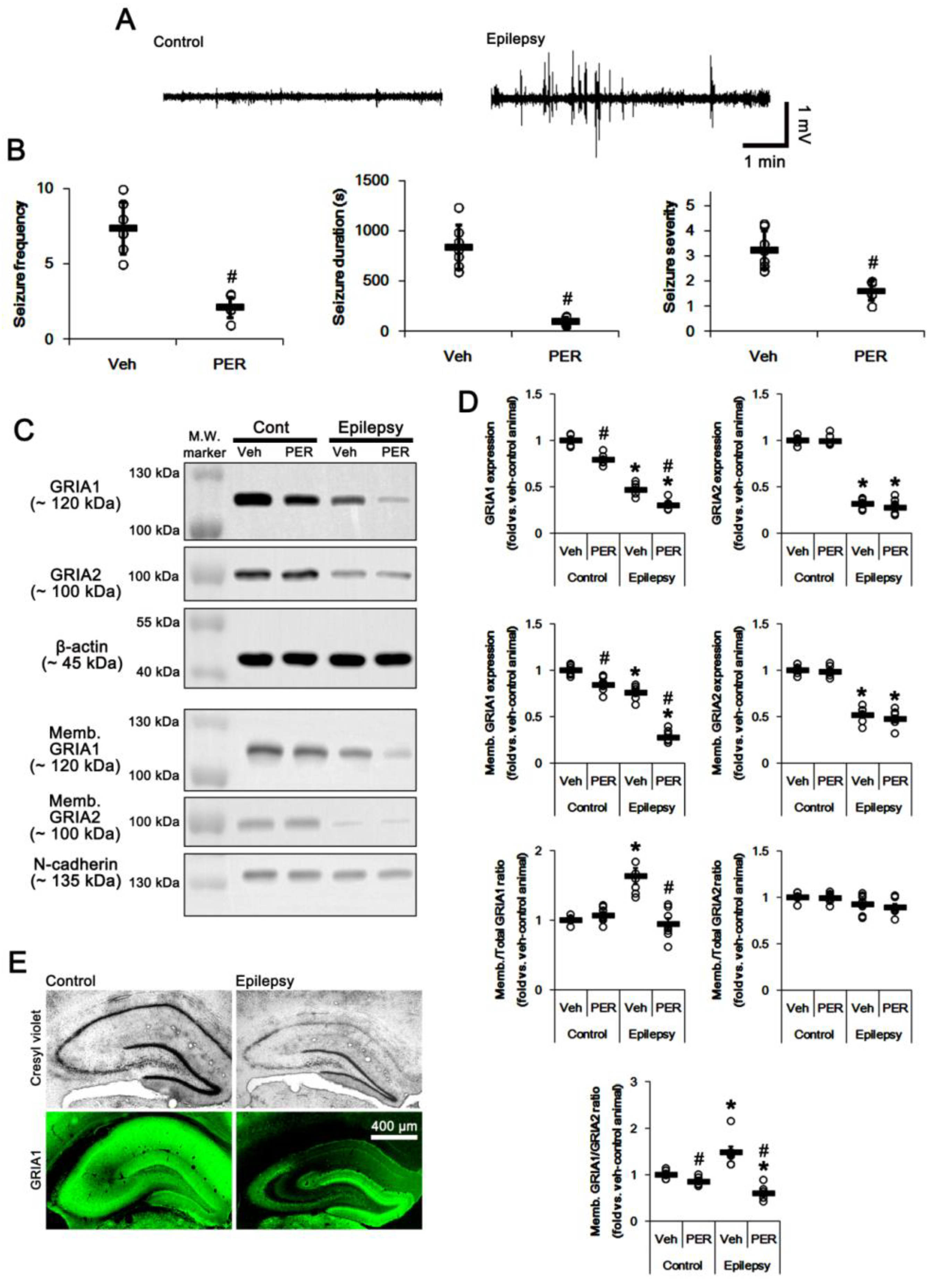
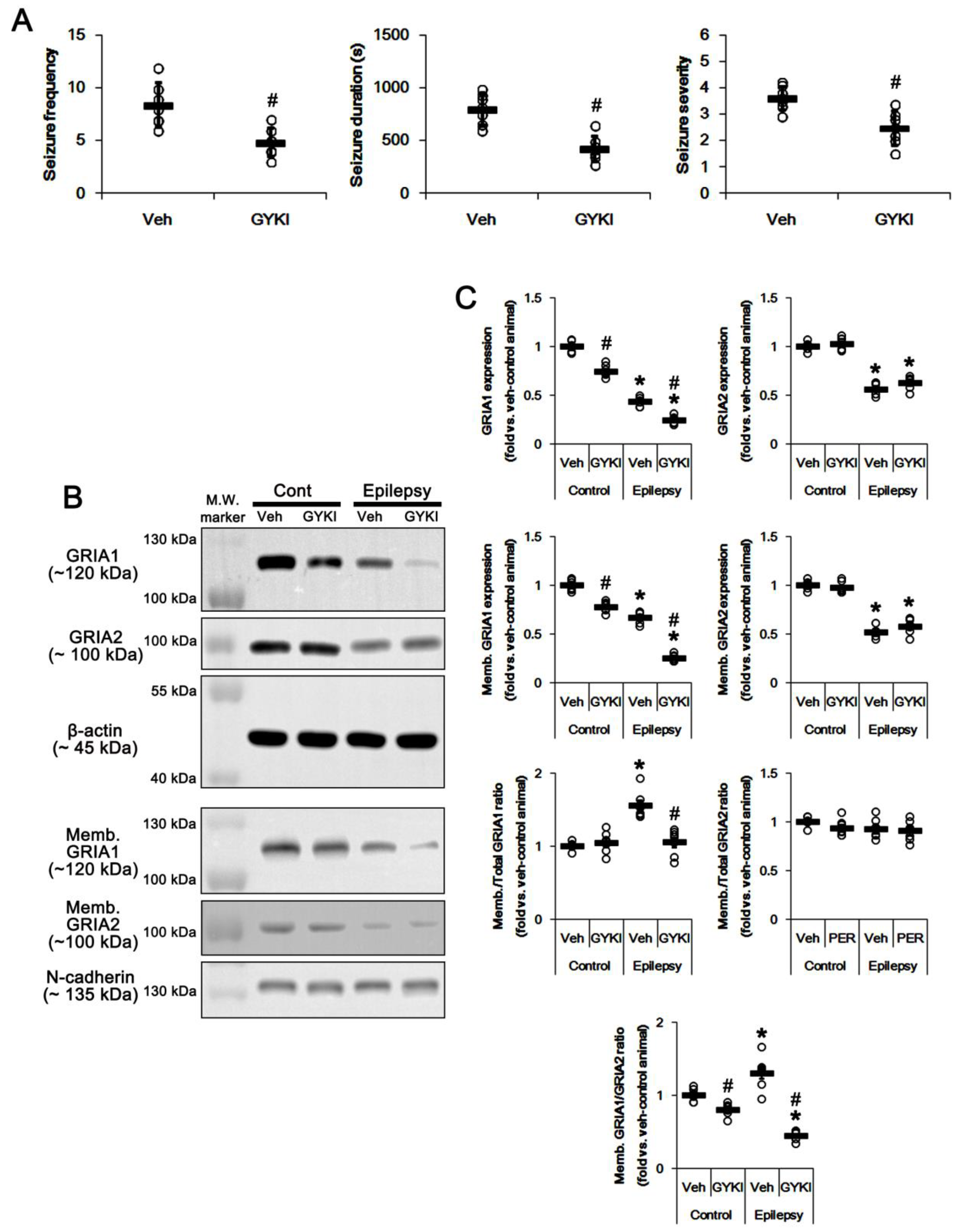
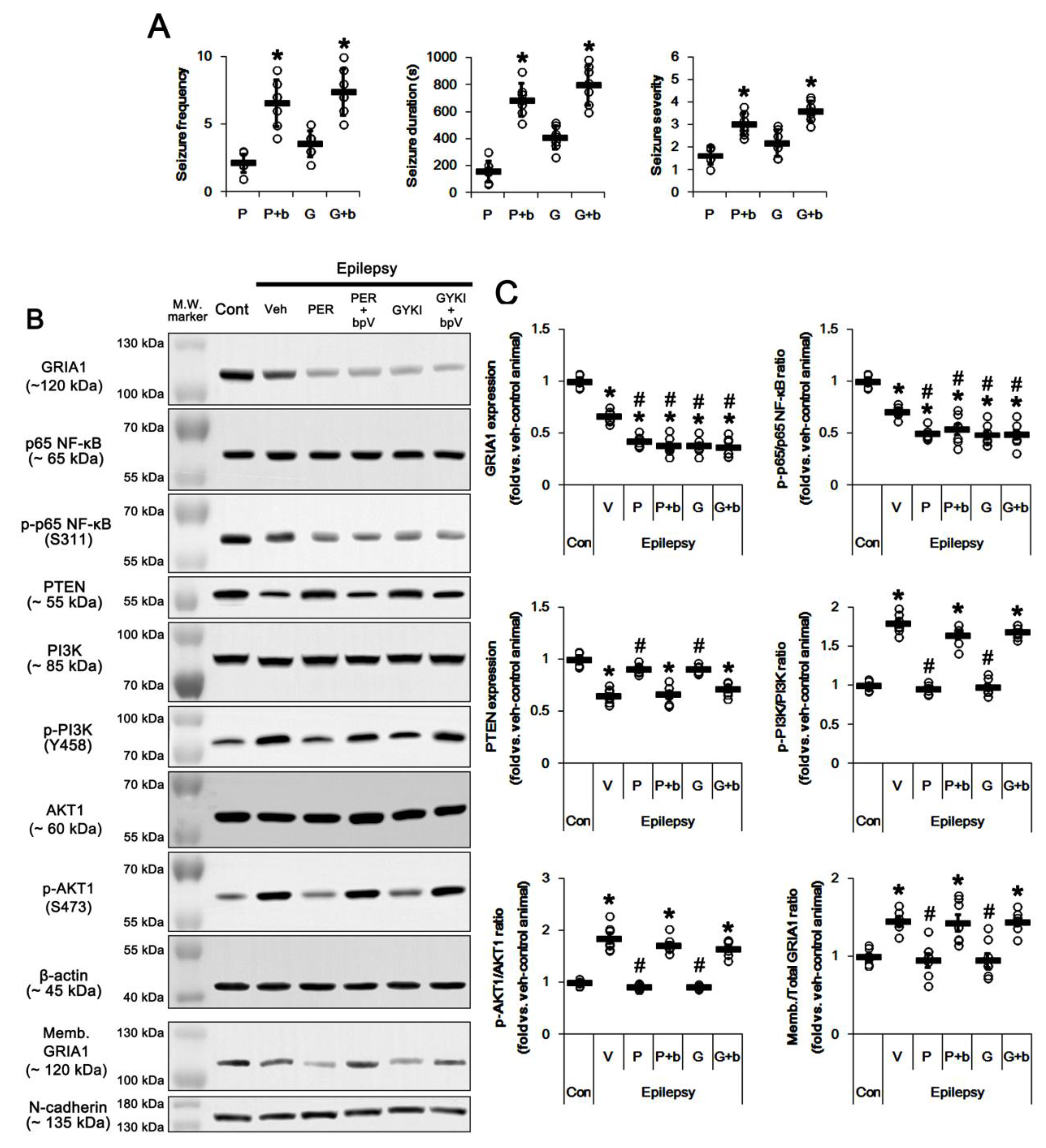
2.1. AMPAR Inhibition Reduces Surface GRIA1 Expression in the Epileptic Hippocampus
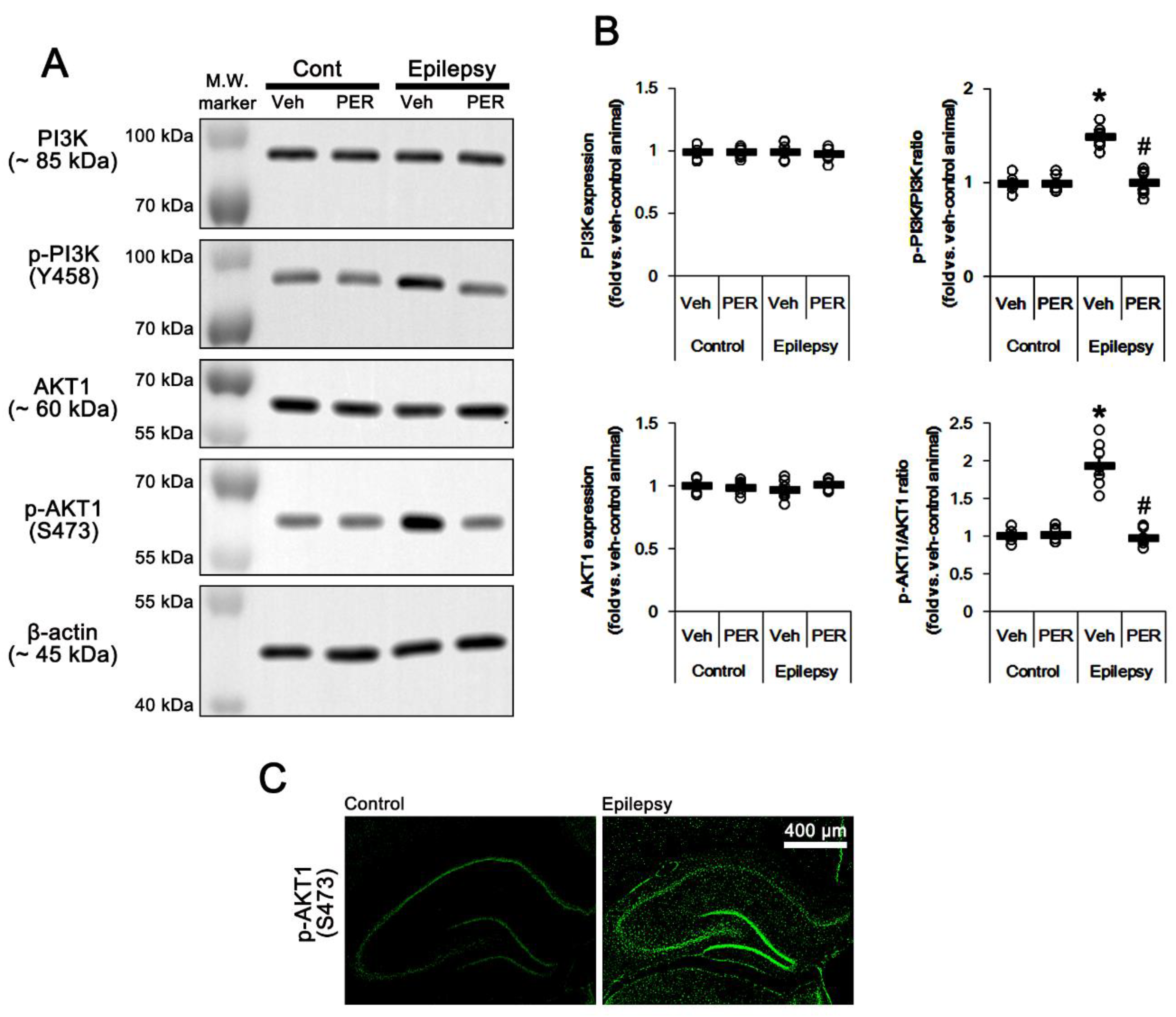
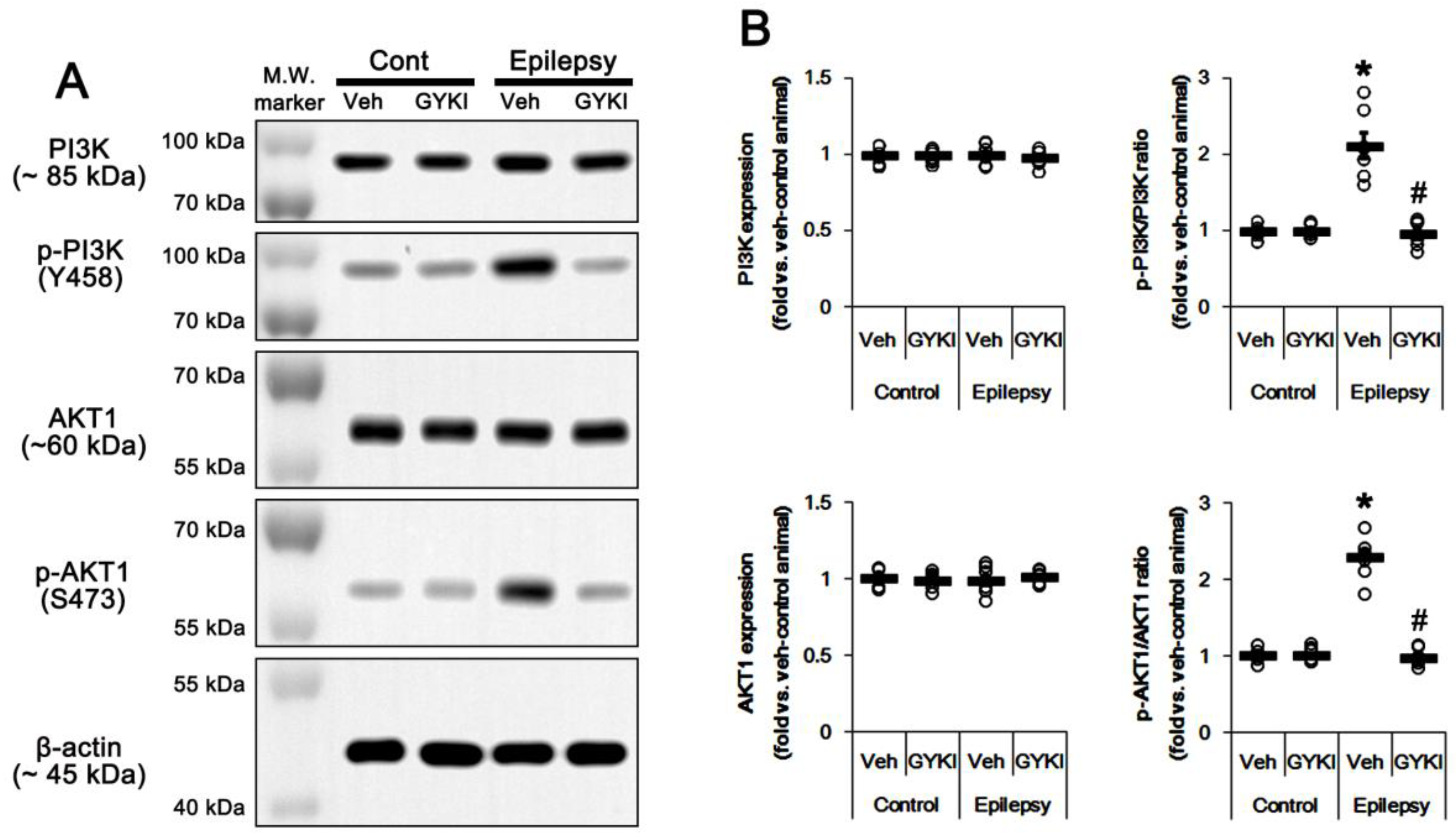
2.2. Blockade of AMPAR Abolishes the Upregulation of PI3K/AKT1 Activities in The Epileptic Hippocampus
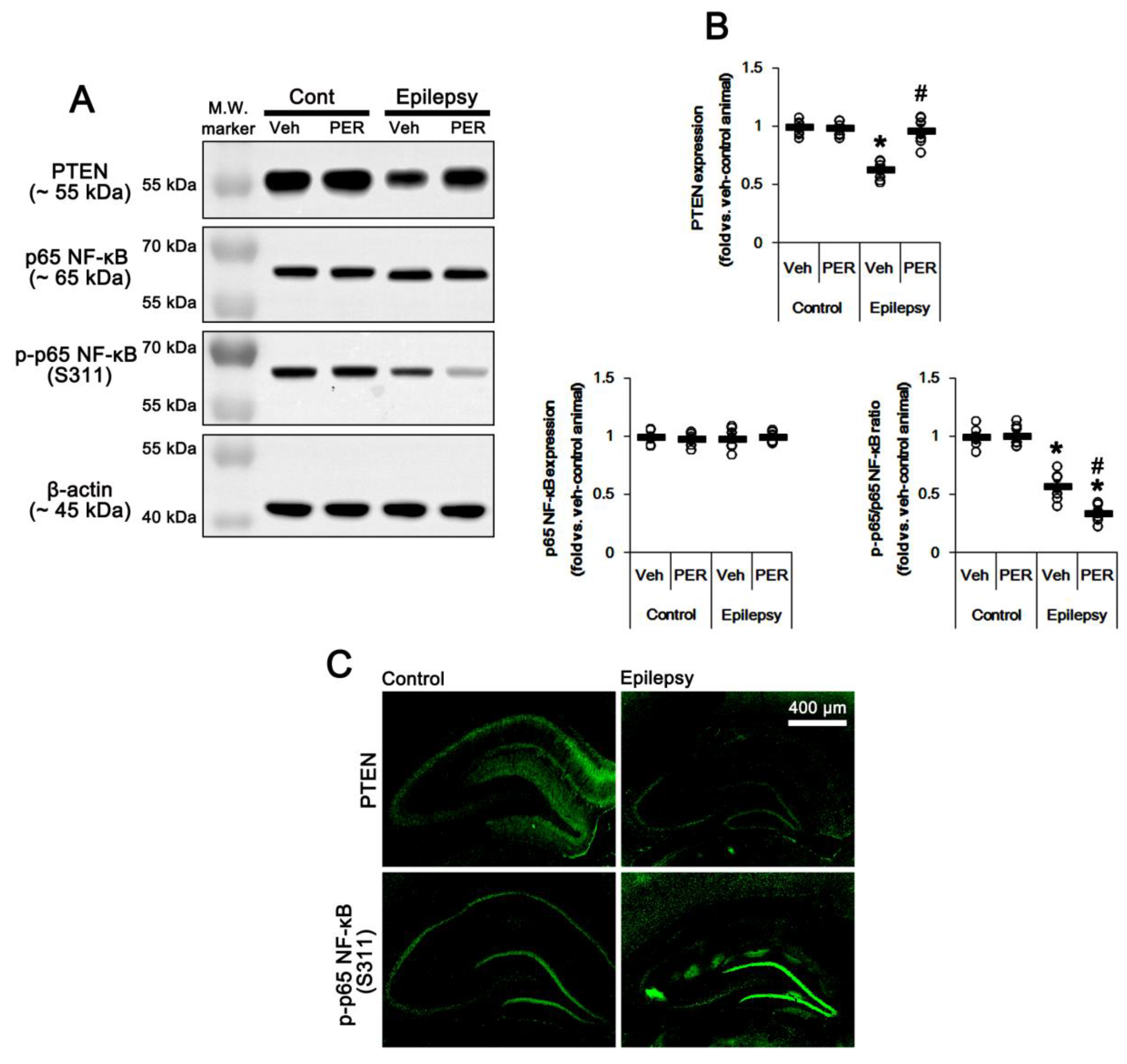
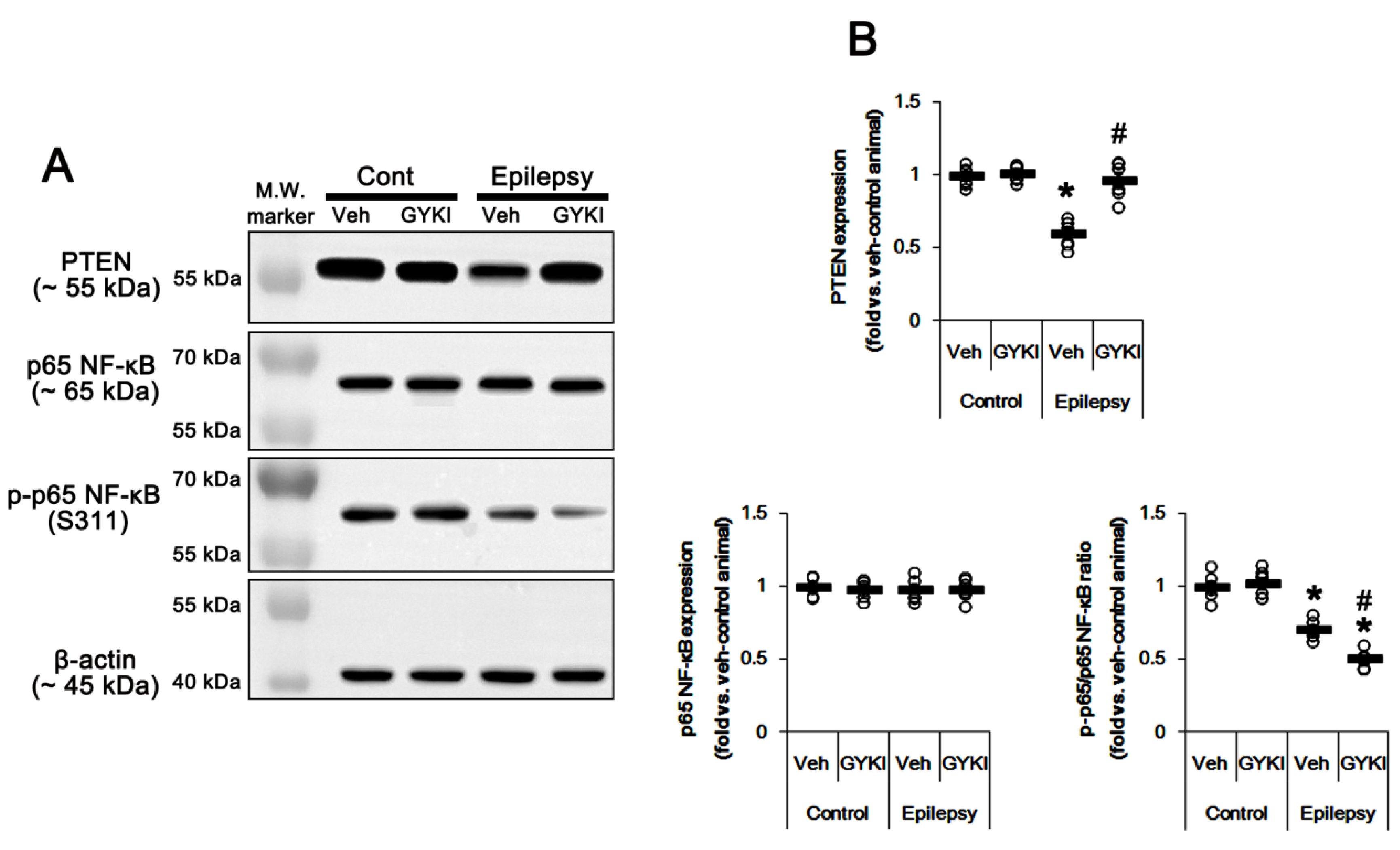
2.3. Blockade of AMPAR Attenuates NF-κB-Mediated PTEN Downregulation in the Epileptic Hippocampus
2.4. PTEN Inhibitor Abrogates the Anti-Epileptic Effects of Perampanel and GYKI 52,466 in Epileptic Rats
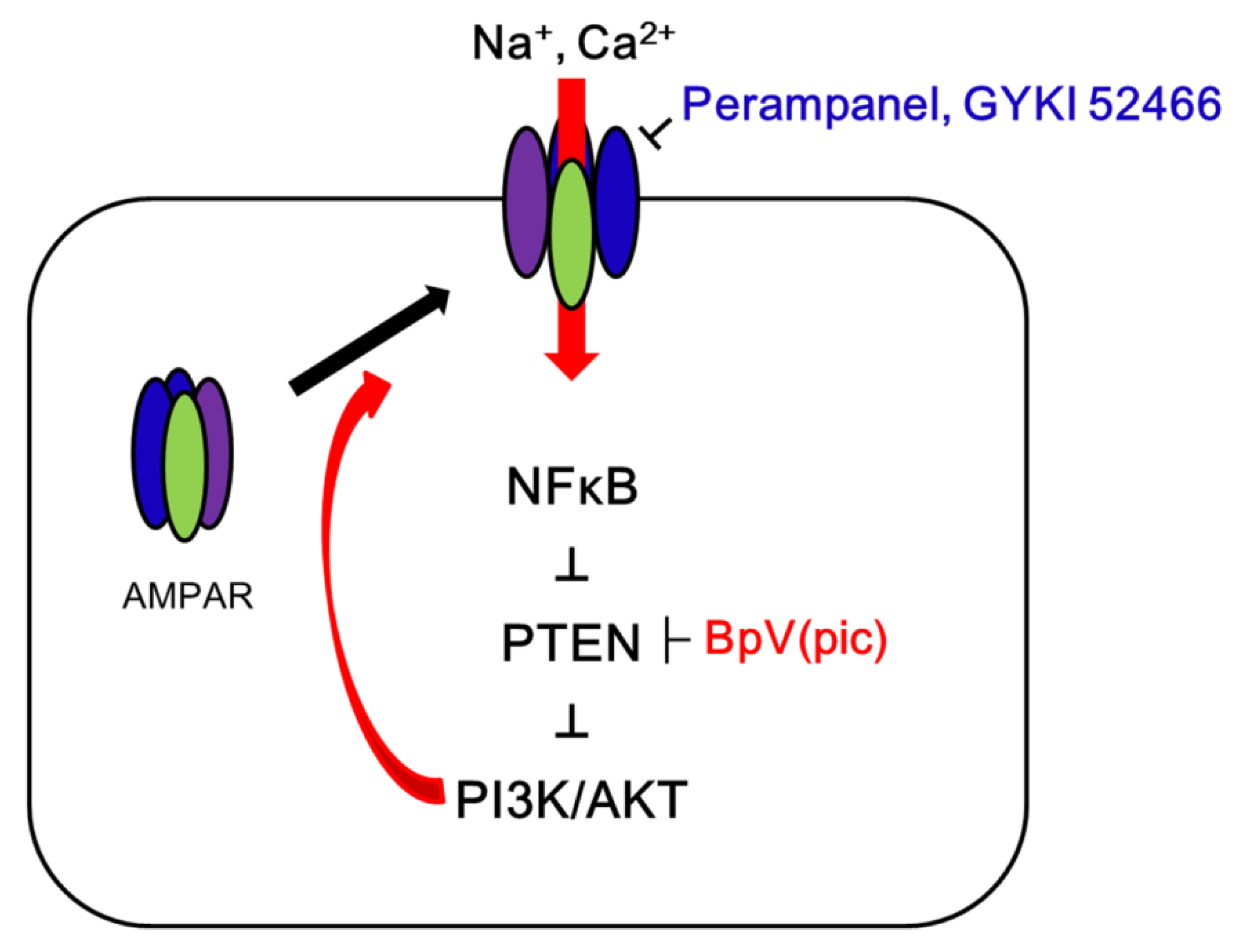
3. Discussion
4. Materials and Methods
4.1. Experimental Animals and Chemicals
4.2. Generation of Epileptic Rats
4.3. Electrode Implantation
4.4. Drug Trials and Quantification of Seizure Activity
4.5. Membrane Fraction and Western Blots
4.6. Tissue Processing and Immunohistochemistry
4.7. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AMPAR | α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor |
| BpV(pic) | Dipotassium bisperoxovanadium(pic) dihydrate |
| NF-κB | Nuclear factor-kappaB |
| PI3K | Phosphoinositide 3-kinase |
| PTEN | Phosphatase and tensin homolog deleted on chromosome 10 |
References
- Mathern, G.W.; Babb, T.L.; Pretorius, J.K.; Melendez, M.; Lévesque, M.F. The pathophysiologic relationships between lesion pathology, intracranial ictal EEG onsets, and hippocampal neuron losses in temporal lobe epilepsy. Epilepsy Res. 1995, 21, 133–147. [Google Scholar] [CrossRef]
- Wittner, L.; Maglóczky, Z.; Borhegyi, Z.; Halász, P.; Tóth, S.; Eross, L.; Szabó, Z.; Freund, T.F. Preservation of perisomatic inhibitory input of granule cells in the epileptic human dentate gyrus. Neuroscience 2001, 108, 587–600. [Google Scholar] [CrossRef]
- Banke, T.G.; Bowie, D.; Lee, H.; Huganir, R.L.; Schousboe, A.; Traynelis, S.F. Control of GluR1 AMPA receptor function by cAMP-dependent protein kinase. J. Neurosci. 2000, 20, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Rogawski, M.A. Revisiting AMPA receptors as an antiepileptic drug target. Epilepsy Curr. 2011, 11, 56–63. [Google Scholar] [CrossRef]
- Lee, K.; Goodman, L.; Fourie, C.; Schenk, S.; Leitch, B.; Montgomery, J.M. AMPA Receptors as Therapeutic Targets for Neurological Disorders. Adv. Protein Chem. Struct. Biol. 2016, 103, 203–261. [Google Scholar] [CrossRef]
- Tang, F.R.; Chia, S.C.; Zhang, S.; Chen, P.M.; Gao, H.; Liu, C.P.; Khanna, S.; Lee, W.L. Glutamate receptor 1-immunopositive neurons in the gliotic CA1 area of the mouse hippocampus after pilocarpine-induced status epilepticus. Eur. J. Neurosci. 2005, 21, 2361–2374. [Google Scholar] [CrossRef]
- Lopes, M.W.; Lopes, S.C.; Costa, A.P.; Gonçalves, F.M.; Rieger, D.K.; Peres, T.V.; Eyng, H.; Prediger, R.D.; Diaz, A.P.; Nunes, J.C.; et al. Region-specific alterations of AMPA receptor phosphorylation and signaling pathways in the pilocarpine model of epilepsy. Neurochem. Int. 2015, 87, 22–33. [Google Scholar] [CrossRef]
- Ge, Y.X.; Tian, X.Z.; Lin, Y.Y.; Liu, X.Y. Chronic treatment with levetiracetam reverses deficits in hippocampal LTP in vivo in experimental temporal lobe epilepsy rats. Neurosci. Lett. 2016, 628, 194–200. [Google Scholar] [CrossRef]
- Kim, J.E.; Choi, H.C.; Song, H.K.; Kang, T.C. Perampanel affects up-stream regulatory signaling pathways of GluA1 phosphorylation in normal and epileptic rats. Front. Cell Neurosci. 2019, 13, 80. [Google Scholar] [CrossRef]
- Lin, C.H.; Yeh, S.H.; Lin, C.H.; Lu, K.T.; Leu, T.H.; Chang, W.C.; Gean, P.W. A role for the PI-3 kinase signaling pathway in fear conditioning and synaptic plasticity in the amygdala. Neuron 2001, 31, 841–851. [Google Scholar] [CrossRef]
- Arendt, K.L.; Royo, M.; Fernández-Monreal, M.; Knafo, S.; Petrok, C.N.; Martens, J.R.; Esteban, J.A. PIP3 controls synaptic function by maintaining AMPA receptor clustering at the postsynaptic membrane. Nat. Neurosci. 2010, 13, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.B.; Chen, Y.; Liu, X.; Tang, X.; Lee, C.W.; Mei, L.; Ye, K. PIKE-mediated PI3-kinase activity is required for AMPA receptor surface expression. EMBO J. 2001, 30, 4274–4286. [Google Scholar] [CrossRef] [PubMed]
- Backman, S.A.; Stambolic, V.; Suzuki, A.; Haight, J.; Elia, A.; Pretorius, J.; Tsao, M.S.; Shannon, P.; Bolon, B.; Ivy, G.O.; et al. Deletion of Pten in mouse brain causes seizures, ataxia and defects in soma size resembling Lhermitte-Duclos disease. Nat. Genet. 2001, 29, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Kwon, C.H.; Zhu, X.; Zhang, J.; Knoop, L.L.; Tharp, R.; Smeyne, R.J.; Eberhart, C.G.; Burger, P.C.; Baker, S.J. Pten regulates neuronal soma size: A mouse model of Lhermitte-Duclos disease. Nat. Genet. 2001, 29, 404–411. [Google Scholar] [CrossRef]
- Butler, M.G.; Dasouki, M.J.; Zhou, X.P.; Talebizadeh, Z.; Brown, M.; Takahashi, T.N.; Miles, J.H.; Wang, C.H.; Stratton, R.; Pilarski, R.; et al. Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J. Med. Genet. 2005, 42, 318–321. [Google Scholar] [CrossRef]
- Kwon, C.H.; Luikart, B.W.; Powell, C.M.; Zhou, J.; Matheny, S.A.; Zhang, W.; Li, Y.; Baker, S.J.; Parada, L.F. Pten regulates neuronal arborization and social interaction in mice. Neuron 2006, 50, 377–388. [Google Scholar] [CrossRef]
- Moult, P.R.; Cross, A.; Santos, S.D.; Carvalho, A.L.; Lindsay, Y.; Connolly, C.N.; Irving, A.J.; Leslie, N.R.; Harvey, J. Leptin regulates AMPA receptor trafficking via PTEN inhibition. J. Neurosci. 2010, 30, 4088–4101. [Google Scholar] [CrossRef]
- Egbenya, D.L.; Hussain, S.; Lai, Y.C.; Xia, J.; Anderson, A.E.; Davanger, S. Changes in synaptic AMPA receptor concentration and composition in chronic temporal lobe epilepsy. Mol. Cell. Neurosci. 2018, 92, 93–103. [Google Scholar] [CrossRef]
- Yu, Z.; Cheng, G.; Wen, X.; Wu, G.D.; Lee, W.T.; Pleasure, D. Tumor necrosis factor alpha increases neuronal vulnerability to excitotoxic necrosis by inducing expression of the AMPA-glutamate receptor subunit GluR1 via an acid sphingomyelinase- and NF-kappaB-dependent mechanism. Neurobiol. Dis. 2002, 11, 199–213. [Google Scholar] [CrossRef]
- Cheng, X.L.; Ding, F.; Li, H.; Tan, X.Q.; Liu, X.; Cao, J.M.; Gao, X. Activation of AMPA receptor promotes TNF-α release via the ROS-cSrc-NFκB signaling cascade in RAW264.7 macrophages. Biochem. Biophys. Res. Commun. 2015, 461, 275–280. [Google Scholar] [CrossRef]
- Duran, A.; Diaz-Meco, M.T.; Moscat, J. Essential role of RelA Ser311 phosphorylation by zetaPKC in NF-kappaB transcriptional activation. EMBO J. 2003, 22, 3910–3918. [Google Scholar] [CrossRef] [PubMed]
- Ryu, H.J.; Kim, J.E.; Yeo, S.I.; Kim, M.J.; Jo, S.M.; Kang, T.C. ReLA/P65-serine 536 nuclear factor-kappa B phosphorylation is related to vulnerability to status epilepticus in the rat hippocampus. Neuroscience 2011, 187, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Kim, D.S.; Ryu, H.J.; Kim, W.I.; Kim, M.J.; Kim, D.W.; Choi, S.Y.; Kang, T.C. The effect of P2 × 7 receptor activation on nuclear factor-κB phosphorylation induced by status epilepticus in the rat hippocampus. Hippocampus 2013, 23, 500–514. [Google Scholar] [CrossRef] [PubMed]
- Vasudevan, K.M.; Gurumurthy, S.; Rangnekar, V.M. Suppression of PTEN expression by NF-kappa B prevents apoptosis. Mol. Cell Biol. 2004, 24, 1007–1021. [Google Scholar] [CrossRef]
- Lewerenz, J.; Baxter, P.; Kassubek, R.; Albrecht, P.; Van Liefferinge, J.; Westhoff, M.A.; Halatsch, M.E.; Karpel-Massler, G.; Meakin, P.J.; Hayes, J.D.; et al. Phosphoinositide 3-kinases upregulate system xc(-) via eukaryotic initiation factor 2α and activating transcription factor 4-A pathway active in glioblastomas and epilepsy. Antioxid. Redox. Signal. 2014, 20, 2907–2922. [Google Scholar] [CrossRef]
- Xiao, Z.; Peng, J.; Yang, L.; Kong, H.; Yin, F. Interleukin-1β plays a role in the pathogenesis of mesial temporal lobe epilepsy through the PI3K/Akt/mTOR signaling pathway in hippocampal neurons. J. Neuroimmunol. 2015, 282, 110–117. [Google Scholar] [CrossRef]
- Carter, A.N.; Born, H.A.; Levine, A.T.; Dao, A.T.; Zhao, A.J.; Lee, W.L.; Anderson, A.E. Wortmannin Attenuates Seizure-Induced Hyperactive PI3K/Akt/mTOR Signaling, Impaired Memory, and Spine Dysmorphology in Rats. eNeuro 2017, 4, ENEURO.0354–16.2017. [Google Scholar] [CrossRef]
- Mazumder, A.G.; Patial, V.; Singh, D. Mycophenolate mofetil contributes to downregulation of the hippocampal interleukin type 2 and 1β mediated PI3K/AKT/mTOR pathway hyperactivation and attenuates neurobehavioral comorbidities in a rat model of temporal lobe epilepsy. Brain Behav. Immun. 2019, 75, 84–93. [Google Scholar] [CrossRef]
- Marsh, D.J.; Kum, J.B.; Lunetta, K.L.; Bennett, M.J.; Gorlin, R.J.; Ahmed, S.F.; Bodurtha, J.; Crowe, C.; Curtis, M.A.; Dasouki, M.; et al. PTEN mutation spectrum and genotype-phenotype correlations in Bannayan-Riley-Ruvalcaba syndrome suggest a single entity with Cowden syndrome. Hum. Mol. Genet. 1999, 8, 1461–1472. [Google Scholar] [CrossRef]
- LaSarge, C.L.; Pun, R.Y.; Muntifering, M.B.; Danzer, S.C. Disrupted hippocampal network physiology following PTEN deletion from newborn dentate granule cells. Neurobiol. Dis. 2016, 96, 105–114. [Google Scholar] [CrossRef]
- Santos, V.R.; Pun, R.Y.K.; Arafa, S.R.; LaSarge, C.L.; Rowley, S.; Khademi, S.; Bouley, T.; Holland, K.D.; Garcia-Cairasco, N.; Danzer, S.C. PTEN deletion increases hippocampal granule cell excitability in male and female mice. Neurobiol. Dis. 2017, 108, 339–351. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Zhang, C.; Zhang, G. Nuclear factor kappaB activation is mediated by NMDA and non-NMDA receptor and L-type voltage-gated Ca(2+) channel following severe global ischemia in rat hippocampus. Brain Res. 2002, 933, 23–30. [Google Scholar] [CrossRef]
- De Erausquin, G.A.; Hyrc, K.; Dorsey, D.A.; Mamah, D.; Dokucu, M.; Mascó, D.H.; Walton, T.; Dikranian, K.; Soriano, M.; García Verdugo, J.M.; et al. Nuclear translocation of nuclear transcription factor-kappa B by alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors leads to transcription of p53 and cell death in dopaminergic neurons. Mol. Pharmacol. 2003, 63, 784–790. [Google Scholar] [CrossRef] [PubMed]
- Meffert, M.K.; Chang, J.M.; Wiltgen, B.J.; Fanselow, M.S.; Baltimore, D. NF-kappa B functions in synaptic signaling and behavior. Nat. Neurosci. 2003, 6, 1072–1078. [Google Scholar] [CrossRef] [PubMed]
- Cruise, L.; Ho, L.K.; Veitch, K.; Fuller, G.; Morris, B.J. Kainate receptors activate NF-kappaB via MAP kinase in striatal neurones. Neuroreport 2000, 11, 395–398. [Google Scholar] [CrossRef] [PubMed]
- O’Mahony, A.; Raber, J.; Montano, M.; Foehr, E.; Han, V.; Lu, S.M.; Kwon, H.; LeFevour, A.; Chakraborty-Sett, S.; Greene, W.C. NF-kappaB/Rel regulates inhibitory and excitatory neuronal function and synaptic plasticity. Mol. Cell. Biol. 2006, 26, 7283–7298. [Google Scholar] [CrossRef]
- Kim, J.E.; Kang, T.C. TRPC3- and ET(B) receptor-mediated PI3K/AKT activation induces vasogenic edema formation following status epilepticus. Brain Res. 2017, 1672, 58–64. [Google Scholar] [CrossRef]
- Kim, M.J.; Park, H.; Choi, S.H.; Kong, M.J.; Kim, J.E.; Kang, T.C. CDDO-Me attenuates vasogenic edema and astroglial death by regulating NF-κB p65 phosphorylations and Nrf2 expression following status epilepticus. Int. J. Mol. Sci. 2019, 20, 4862. [Google Scholar] [CrossRef]
- Koul, D.; Takada, Y.; Shen, R.; Aggarwal, B.B.; Yung, W.K. PTEN enhances TNF-induced apoptosis through modulation of nuclear factor-kappaB signaling pathway in human glioma cells. Biochem. Biophys. Res. Commun. 2006, 350, 463–471. [Google Scholar] [CrossRef]
- Du, J.; Gray, N.A.; Falke, C.A.; Chen, W.; Yuan, P.; Szabo, S.T.; Einat, H.; Manji, H.K. Modulation of synaptic plasticity by antimanic agents: The role of AMPA glutamate receptor subunit 1 synaptic expression. J. Neurosci. 2004, 24, 6578–6589. [Google Scholar] [CrossRef][Green Version]
- Pellegrini-Giampietro, D.E.; Gorter, J.A.; Bennett, M.V.; Zukin, R.S. The GluR2 (GluR-B) hypothesis: Ca(2+)-permeable AMPA receptors in neurological disorders. Trends Neurosci. 1997, 20, 464–470. [Google Scholar] [CrossRef]
- Salpietro, V.; Dixon, C.L.; Guo, H.; Bello, O.D.; Vandrovcova, J.; Efthymiou, S.; Maroofian, R.; Heimer, G.; Burglen, L.; Valence, S.; et al. AMPA receptor GluA2 subunit defects are a cause of neurodevelopmental disorders. Nat. Commun. 2019, 10, 3094. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Sun, X.; Li, J.; Jia, R.; Yuan, F.; Wei, D.; Jiang, W. Melatonin Alleviates the Epilepsy-Associated Impairments in Hippocampal LTP and Spatial Learning Through Rescue of Surface GluR2 Expression at Hippocampal CA1 Synapses. Neurochem. Res. 2017, 42, 1438–1448. [Google Scholar] [CrossRef] [PubMed]
- Lorgen, J.Ø.; Egbenya, D.L.; Hammer, J.; Davanger, S. PICK1 facilitates lasting reduction in GluA2 concentration in the hippocampus during chronic epilepsy. Epilepsy Res. 2017, 137, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Rajasekaran, K.; Todorovic, M.; Kapur, J. Calcium-permeable AMPA receptors are expressed in a rodent model of status epilepticus. Ann. Neurol. 2012, 72, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Tian, M.; Liu, L.; Wong, T.P.; Gong, B.; Wu, D.; Cho, T.; Lin, S.; Kast, J.; Lu, J.; et al. p97 regulates GluA1 homomeric AMPA receptor formation and plasma membrane expression. Nat. Commun. 2019, 10, 4089. [Google Scholar] [CrossRef] [PubMed]
- Schweitzer, K.; Pralow, A.; Naumann, M. p97/VCP promotes Cullin-RING-ubiquitin-ligase/proteasome-dependent degradation of IκBα and the preceding liberation of RelA from ubiquitinated IκBα. J. Cell Mol. Med. 2016, 20, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Crouin, C.; Arnaud, M.; Gesbert, F.; Camonis, J.; Bertoglio, J. A yeast two-hybrid study of human p97/Gab2 interactions with its SH2 domain-containing binding partners. FEBS Lett. 2001, 495, 148–153. [Google Scholar] [CrossRef]
- Zhang, X.; Lavoie, G.; Méant, A.; Aubert, L.; Cargnello, M.; Haman, A.; Hoang, T.; Roux, P.P. Extracellular Signal-Regulated Kinases 1 and 2 Phosphorylate Gab2 To Promote a Negative-Feedback Loop That Attenuates Phosphoinositide 3-Kinase/Akt Signaling. Mol. Cell Biol. 2017, 37, e00357–00316. [Google Scholar] [CrossRef]
- Pan, X.L.; Ren, R.J.; Wang, G.; Tang, H.D.; Chen, S.D. The Gab2 in signal transduction and its potential role in the pathogenesis of Alzheimer’s disease. Neurosci. Bull. 2010, 26, 241–246. [Google Scholar] [CrossRef]
- Zhang, Y.; Guo, O.; Huo, Y.; Wang, G.; Man, H.Y. Amyloid-β Induces AMPA Receptor Ubiquitination and Degradation in Primary Neurons and Human Brains of Alzheimer’s Disease. J. Alzheimers Dis. 2018, 62, 1789–1801. [Google Scholar] [CrossRef] [PubMed]
- Zhan, A.; Xu, X.; Chen, L.; Wang, X.; Yanfeng, X.; Dan, W.; Zhan, Y.; Shi, Q. Decreased expression of Gab2 in patients with temporal lobe epilepsy and pilocarpine-induced rat model. Synapse 2014, 68, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Ko, A.R.; Kang, T.C. Blockade of endothelin B receptor improves the efficacy of levetiracetam in chronic epileptic rats. Seizure 2015, 31, 133–140. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kim, J.E.; Choi, H.C.; Song, H.K.; Kang, T.C. Blockade of AMPA receptor regulates mitochondrial dynamics by modulating ERK1/2 and PP1/PP2A-mediated DRP1-S616 phosphorylations in the normal rat hippocampus. Front. Cell Neurosci. 2019, 13, 179. [Google Scholar] [CrossRef]
- Sury, M.D.; Vorlet-Fawer, L.; Agarinis, C.; Yousefi, S.; Grandgirard, D.; Leib, S.L.; Christen, S. Restoration of Akt activity by the bisperoxovanadium compound bpV(pic) attenuates hippocampal apoptosis in experimental neonatal pneumococcal meningitis. Neurobiol. Dis. 2011, 41, 201–208. [Google Scholar] [CrossRef]
- Grande, V.; Manassero, G.; Vercelli, A. Neuroprotective and anti-inflammatory roles of the phosphatase and tensin homolog deleted on chromosome ten (PTEN) inhibition in a mouse model of temporal lobe epilepsy. PLoS ONE 2014, 9, e114554. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antigen | Host | Manufacturer (Catalog Number) | Dilution Used |
|---|---|---|---|
| AKT1 | Rabbit | Cell signaling (#9272) | 1:1000 (WB) |
| 1:100 (IH) | |||
| GRIA1 | Mouse | Synaptic systems (182011) | 1:1000 (WB) |
| GRIA2 | Sigma (AB1768-I) | 1:1000 (WB) | |
| N-cadherin | Rabbit | Abcam (ab18203) | 1:4000 (WB) |
| NF-κB RelA p65 | Rabbit | Abcam (ab16502) | 1:1000 (WB) |
| 1:200 (IH) | |||
| p-AKT1-S473 | Rabbit | Cell signaling (#4060) | 1:1000 (WB) |
| 1:100 (IH) | |||
| p-NF-κB RelA p65-S311 | Rabbit | Abcam (ab194926) | 1:2000 (WB) |
| p-PI3K-Y458 | Rabbit | Cell signaling (#4228S) | 1:1000 (WB) |
| p-PTEN-S380/Y382/Y383 | Rabbit | Cell signaling (#9549) | 1:1000 (WB) |
| PI3K | Rabbit | Cell signaling (#4292S) | 1:1000 (WB) |
| 1:200 (IH) | |||
| PTEN | Rabbit | Abcam (ab170941) | 1:5000 (WB) |
| β-actin | Mouse | Sigma (#A5316) | 1:5000 (WB) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.-E.; Park, H.; Lee, J.-E.; Kim, T.-H.; Kang, T.-C. PTEN Is Required for The Anti-Epileptic Effects of AMPA Receptor Antagonists in Chronic Epileptic Rats. Int. J. Mol. Sci. 2020, 21, 5643. https://doi.org/10.3390/ijms21165643
Kim J-E, Park H, Lee J-E, Kim T-H, Kang T-C. PTEN Is Required for The Anti-Epileptic Effects of AMPA Receptor Antagonists in Chronic Epileptic Rats. International Journal of Molecular Sciences. 2020; 21(16):5643. https://doi.org/10.3390/ijms21165643
Chicago/Turabian StyleKim, Ji-Eun, Hana Park, Ji-Eun Lee, Tae-Hyun Kim, and Tae-Cheon Kang. 2020. "PTEN Is Required for The Anti-Epileptic Effects of AMPA Receptor Antagonists in Chronic Epileptic Rats" International Journal of Molecular Sciences 21, no. 16: 5643. https://doi.org/10.3390/ijms21165643
APA StyleKim, J.-E., Park, H., Lee, J.-E., Kim, T.-H., & Kang, T.-C. (2020). PTEN Is Required for The Anti-Epileptic Effects of AMPA Receptor Antagonists in Chronic Epileptic Rats. International Journal of Molecular Sciences, 21(16), 5643. https://doi.org/10.3390/ijms21165643