Goat’s Milk Intake Prevents Obesity, Hepatic Steatosis and Insulin Resistance in Mice Fed A High-Fat Diet by Reducing Inflammatory Markers and Increasing Energy Expenditure and Mitochondrial Content in Skeletal Muscle

 ,
,  , ,
, , 
Abstract
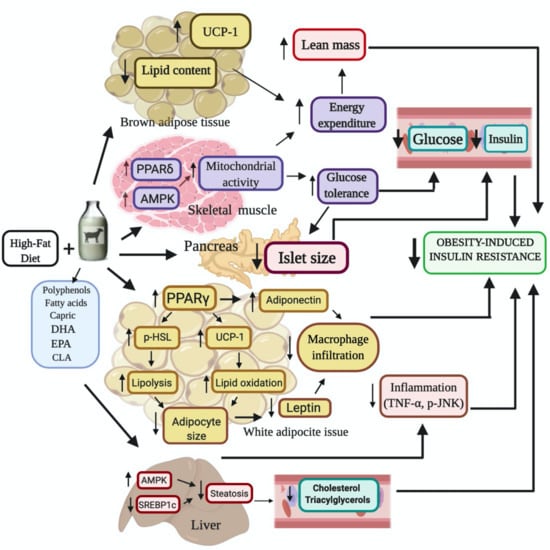
:
1. Introduction
2. Results
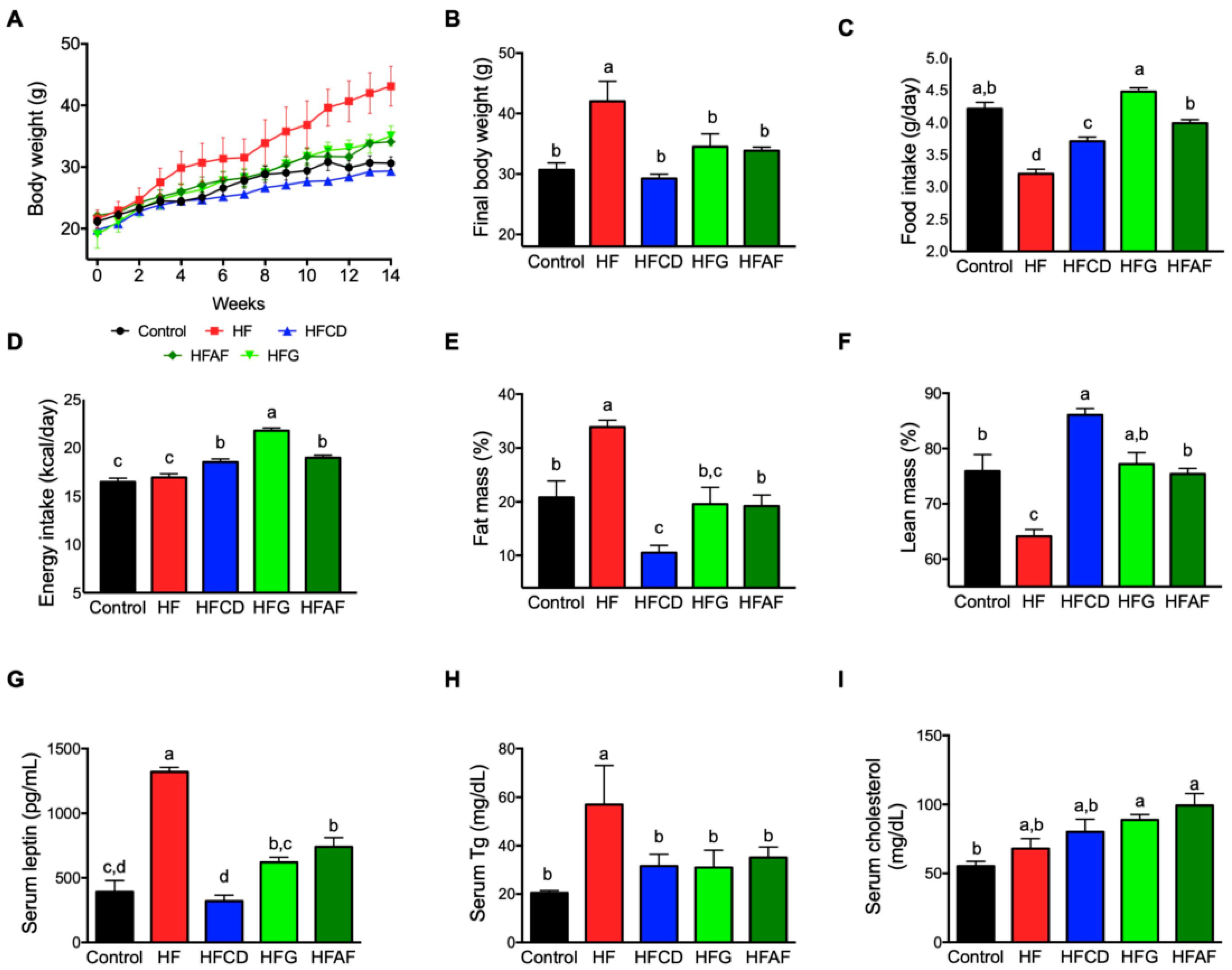
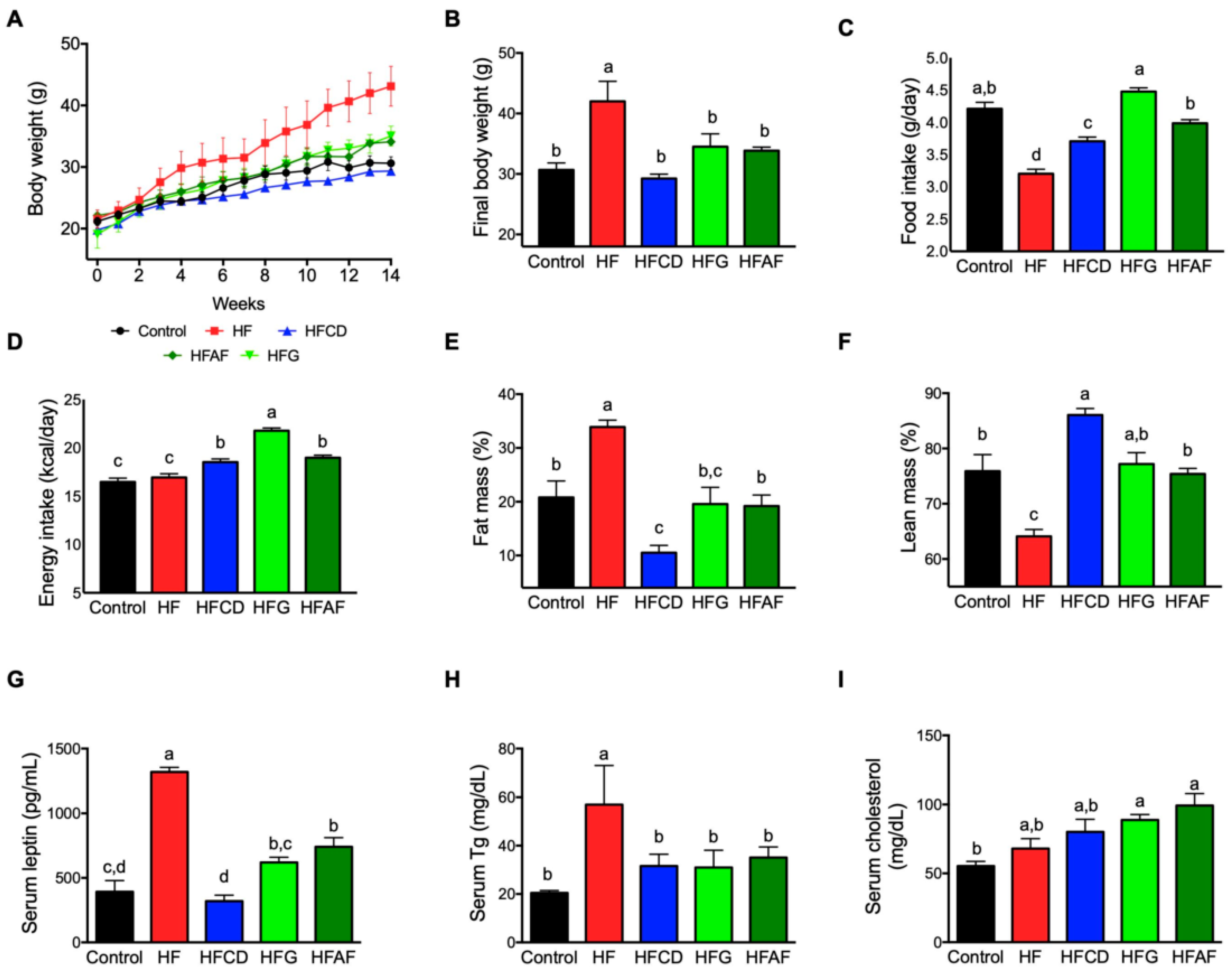
2.1. Goat’s Milk Intake Attenuates Body Weight Gain, Increases Food Intake, and Modifies Body Composition and Serum Parameters in Mice Fed A High-Fat Diet
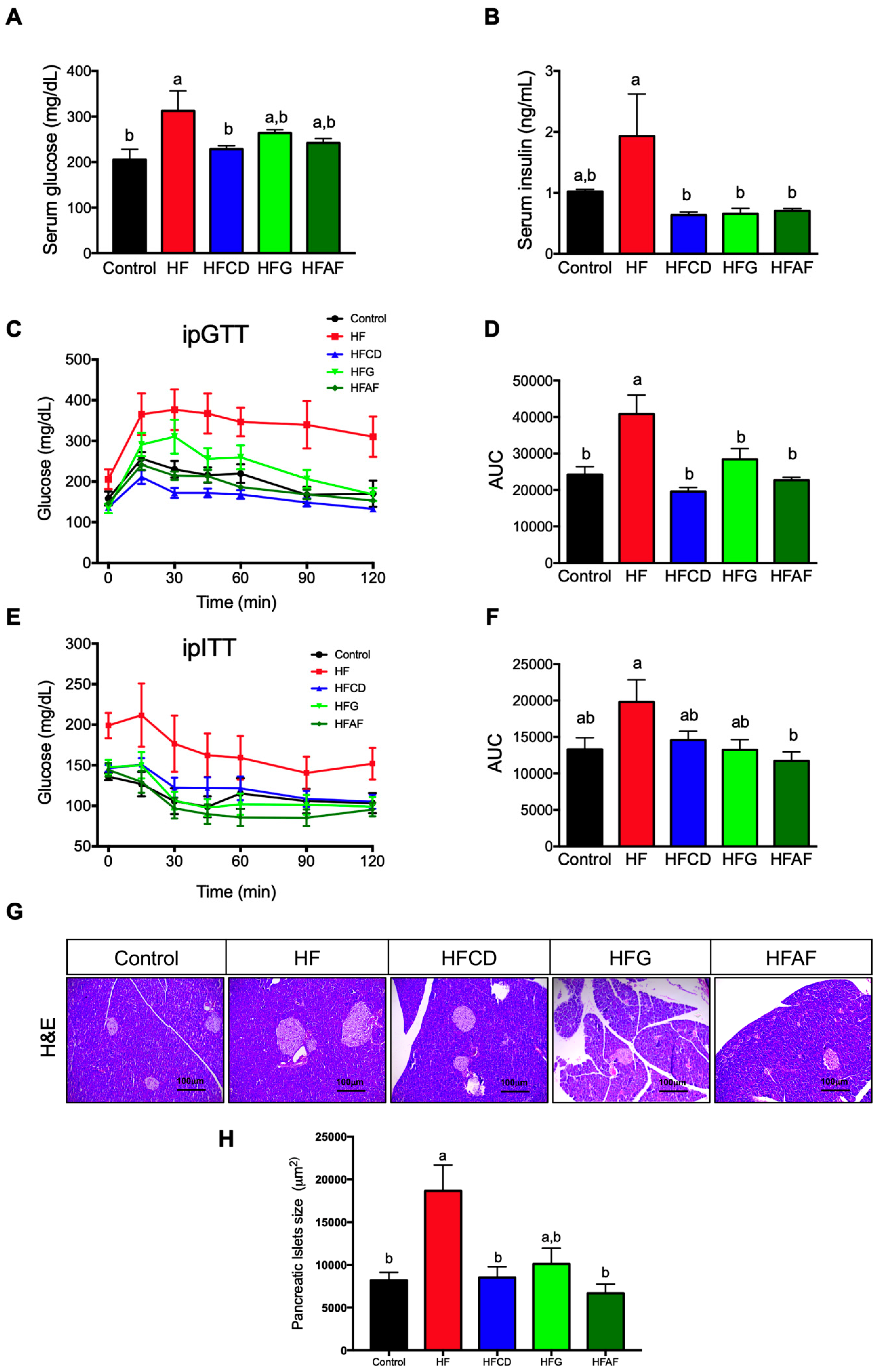
2.2. Goat’s Milk Consumption Prevents Insulin Resistance and Pancreatic Islets Hypertrophy in Mice Fed A High-Fat Diet
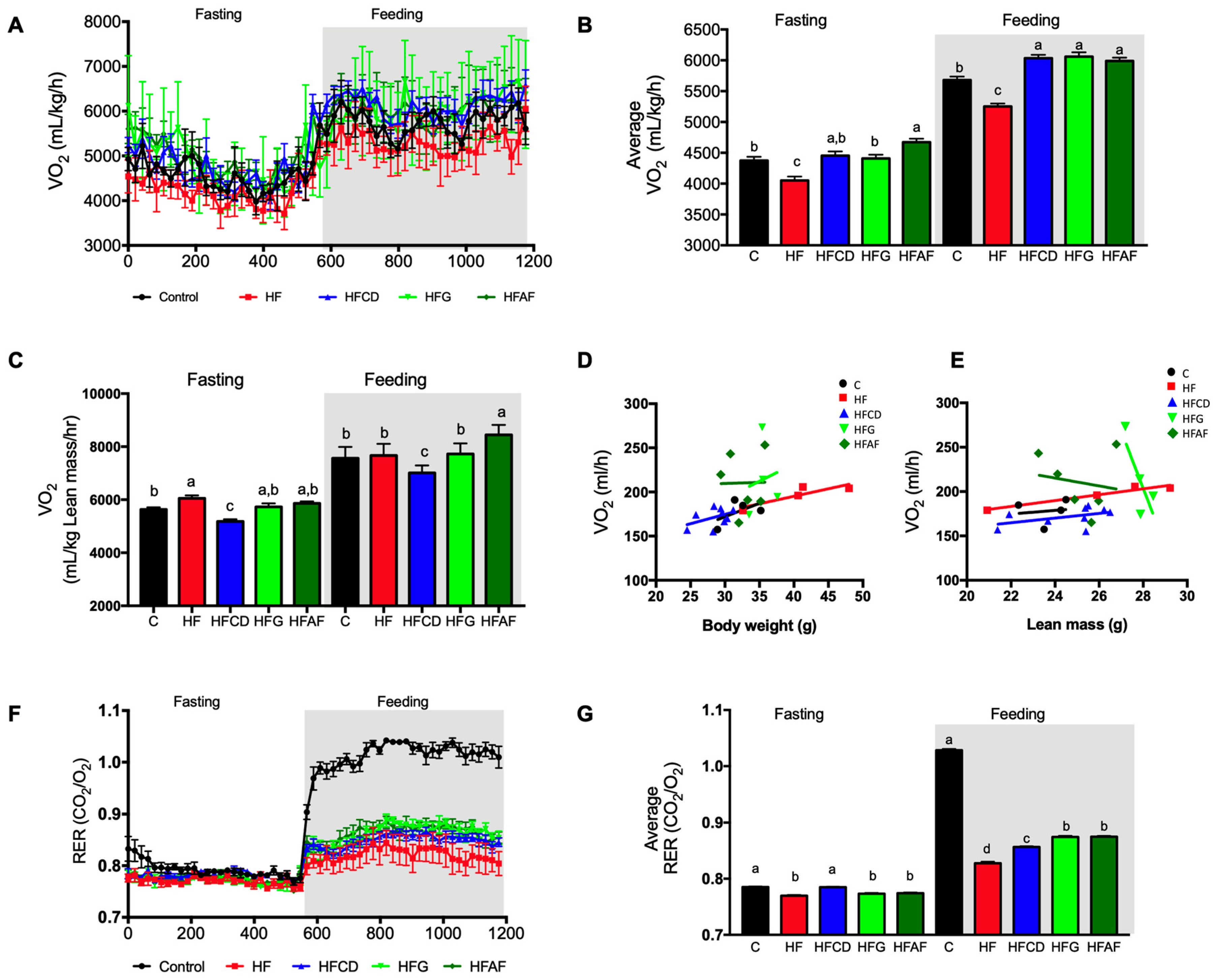
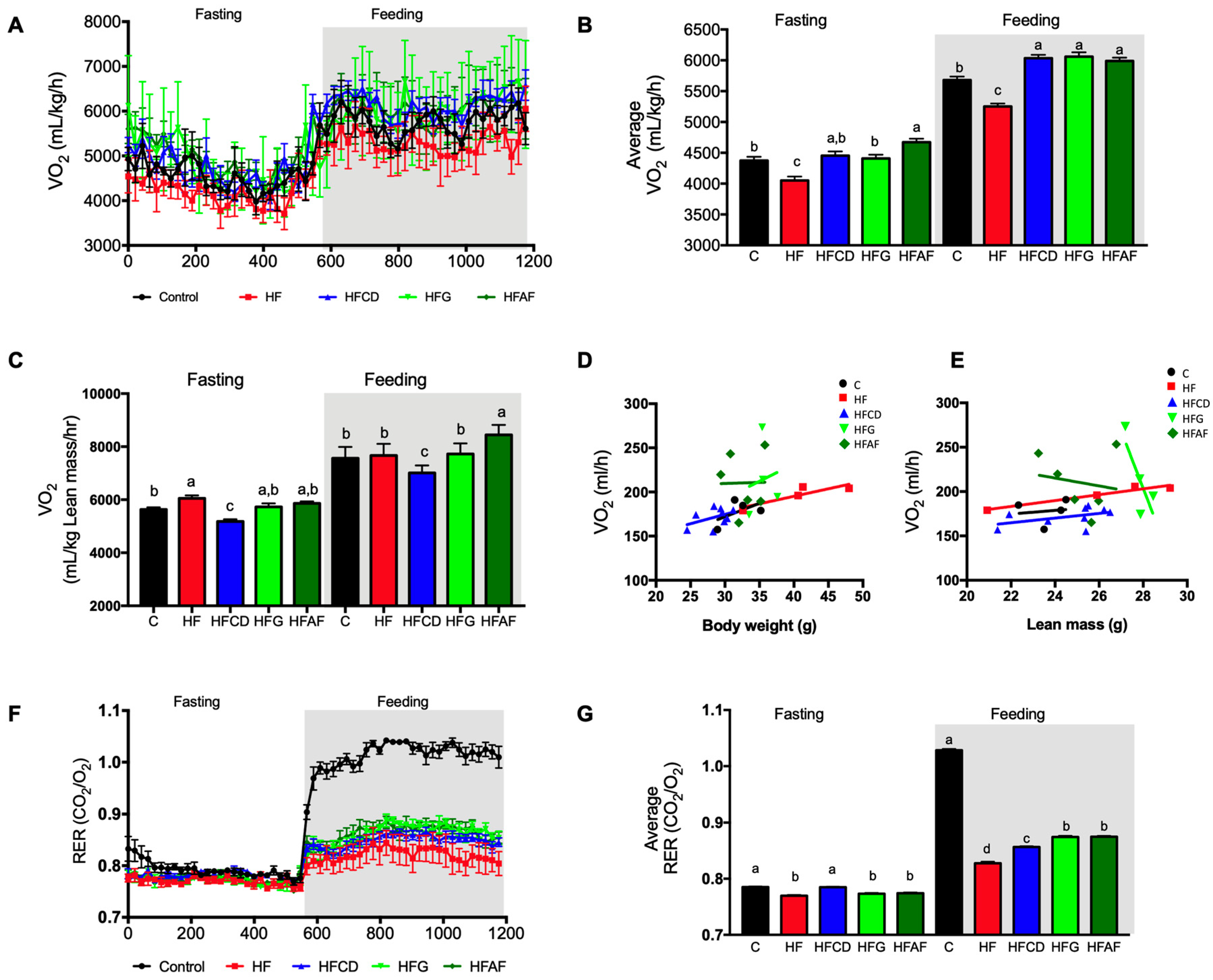
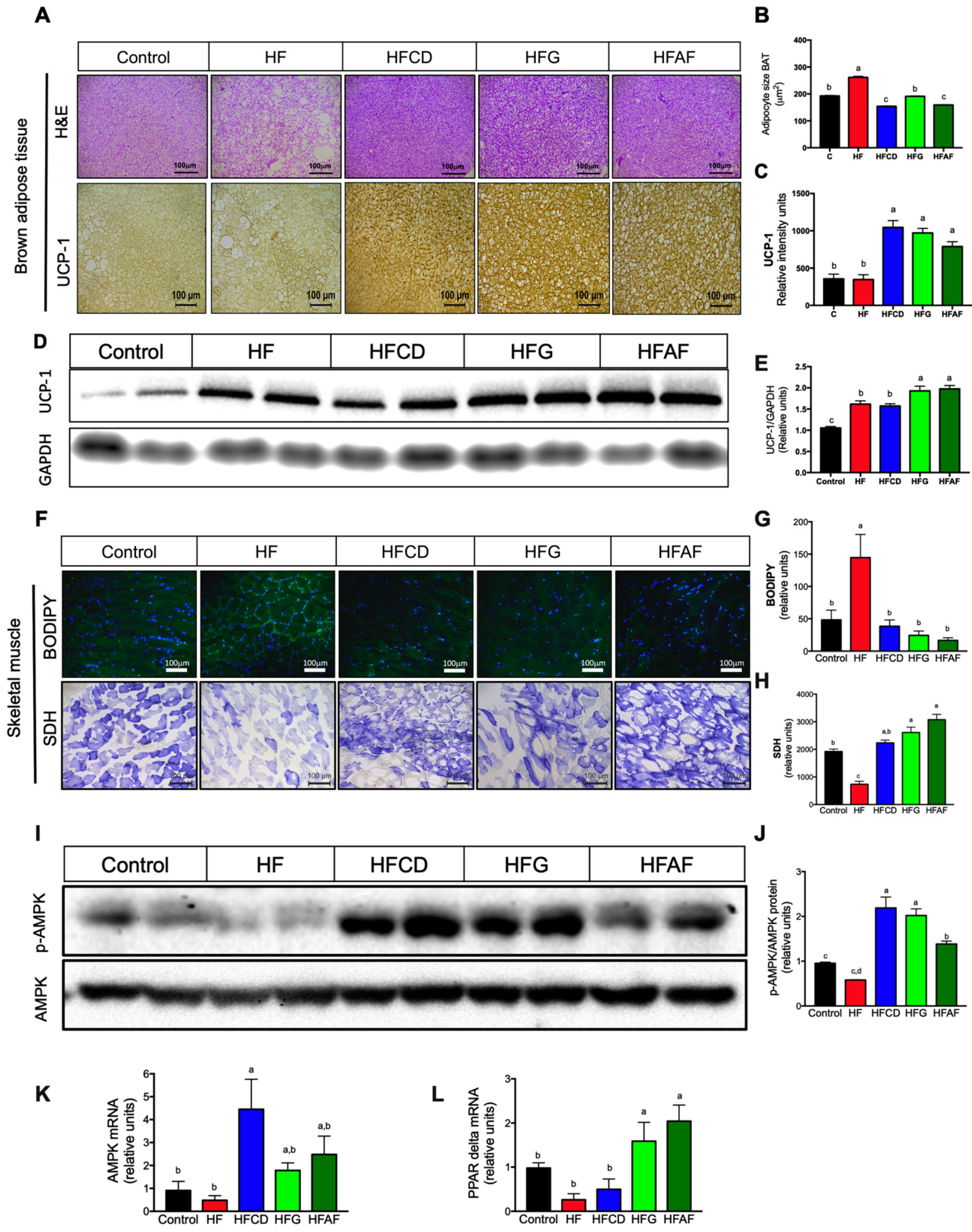
2.3. Goat’s Milk Consumption Increases Whole-Body Energy Expenditure and Attenuates Metabolic Inflexibility by an Increase in UCP-1 in BAT and Oxidative Fibers in Muscle of Mice Fed A High-Fat Diet
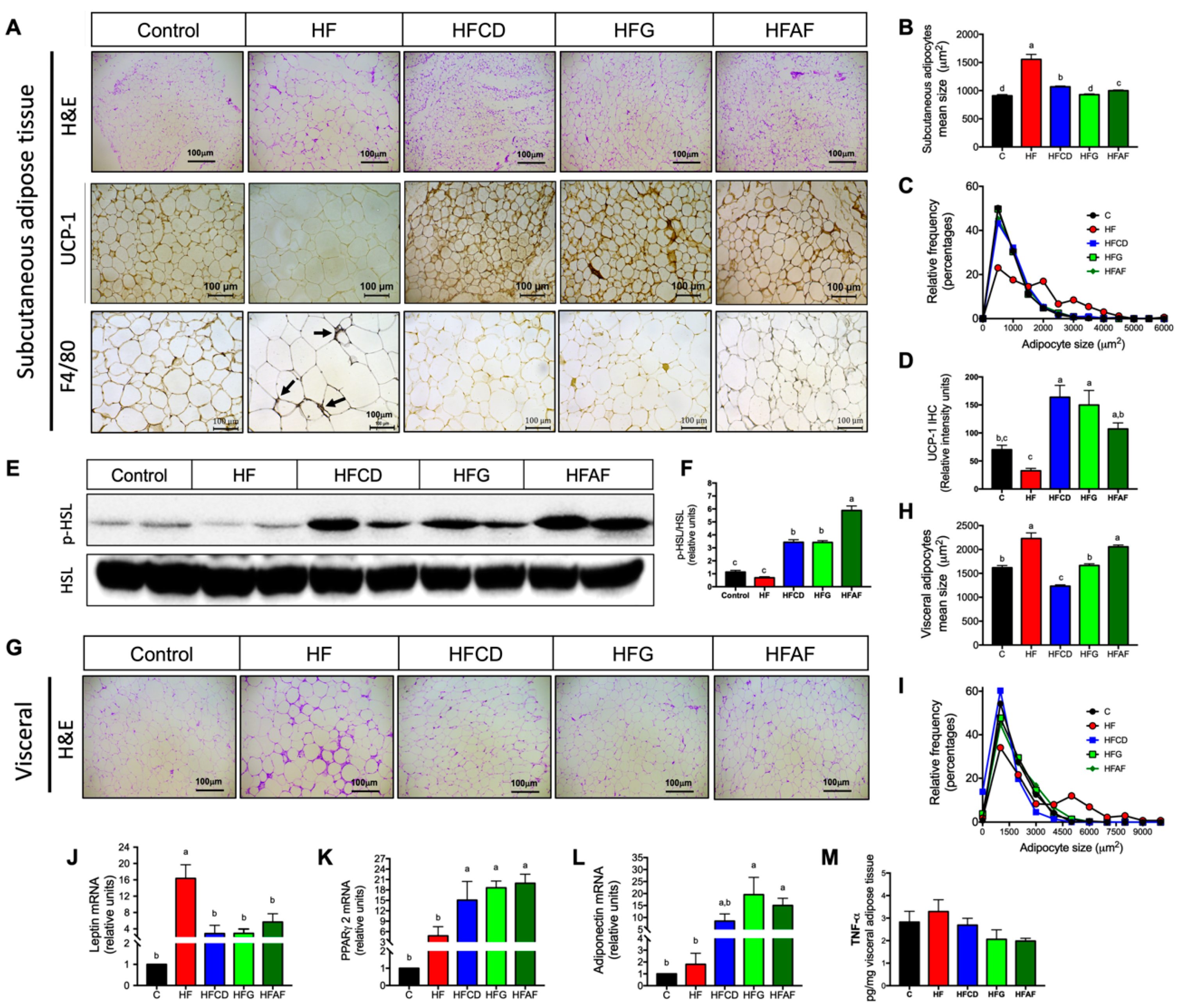
2.4. Goat’s Milk Intake Prevent White Adipose Tissue Hypertrophy and Modulates Adipokine mRNA in Adipocytes of Mice Fed A High-Fat Diet
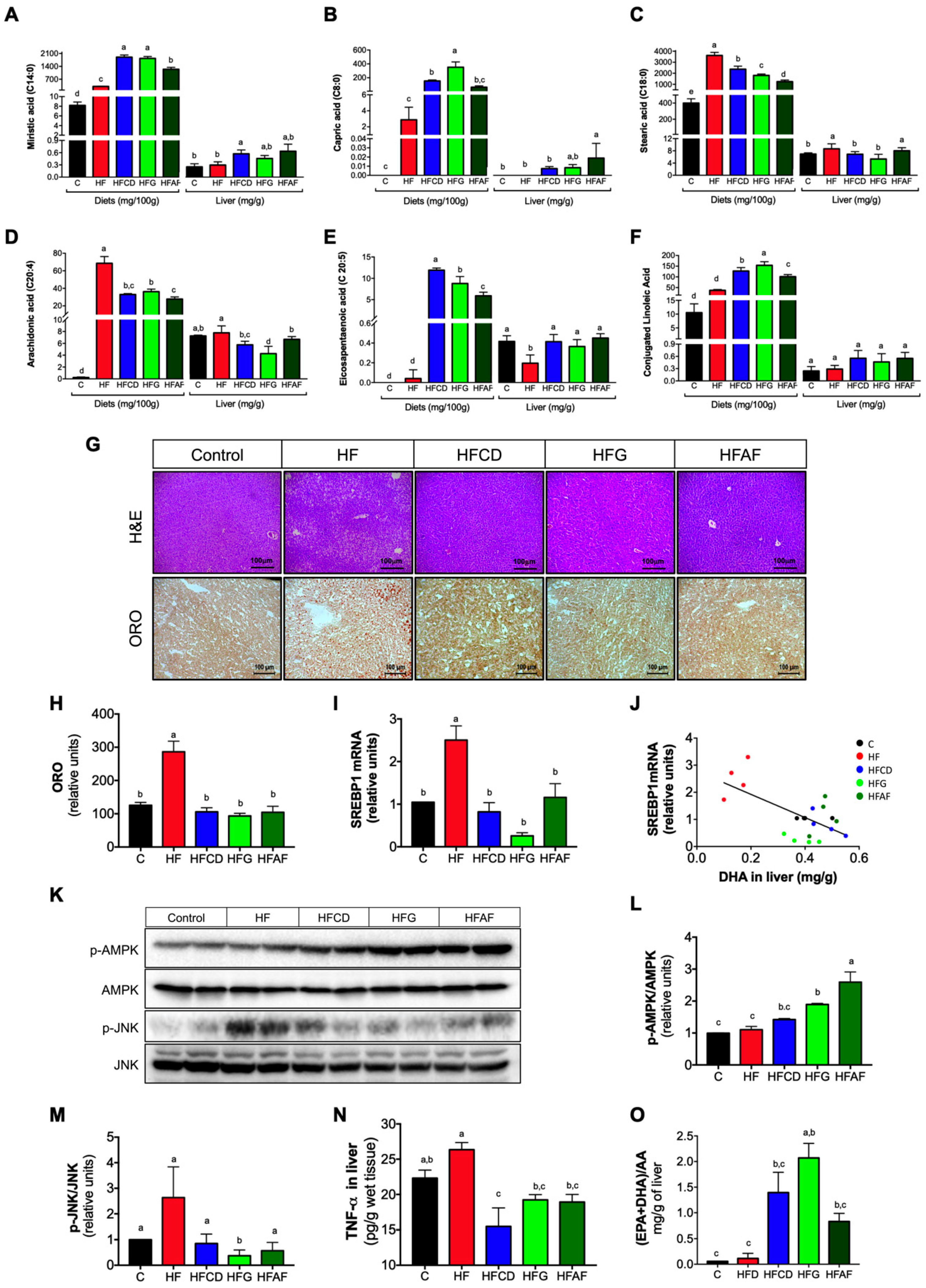
2.5. The Anti-Inflammatory Fatty Acid Profile of Goat Milk’s Prevents Hepatic Lipid Deposition and Inflammatory Markers and Increases AMPK Phosphorylation
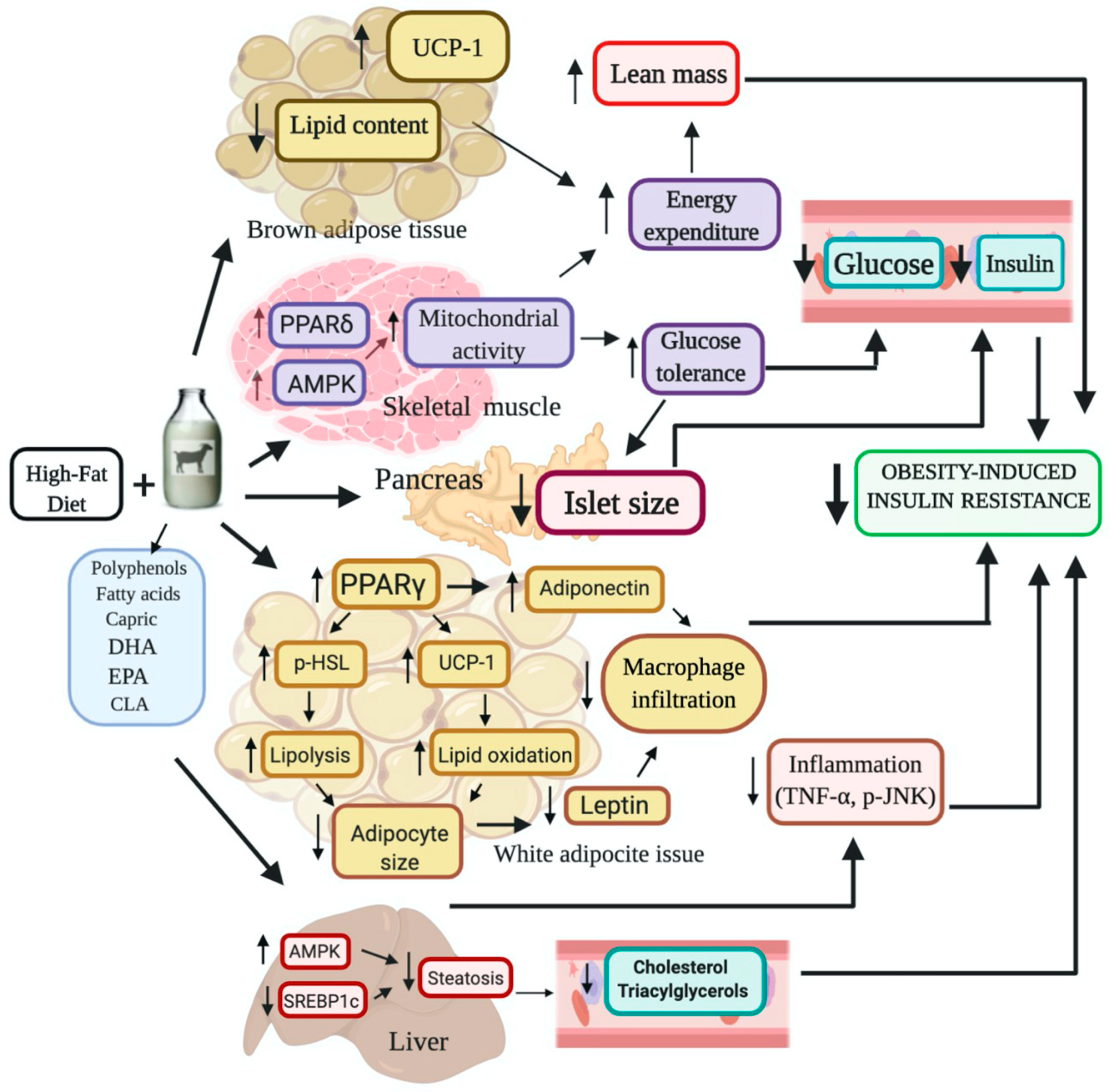
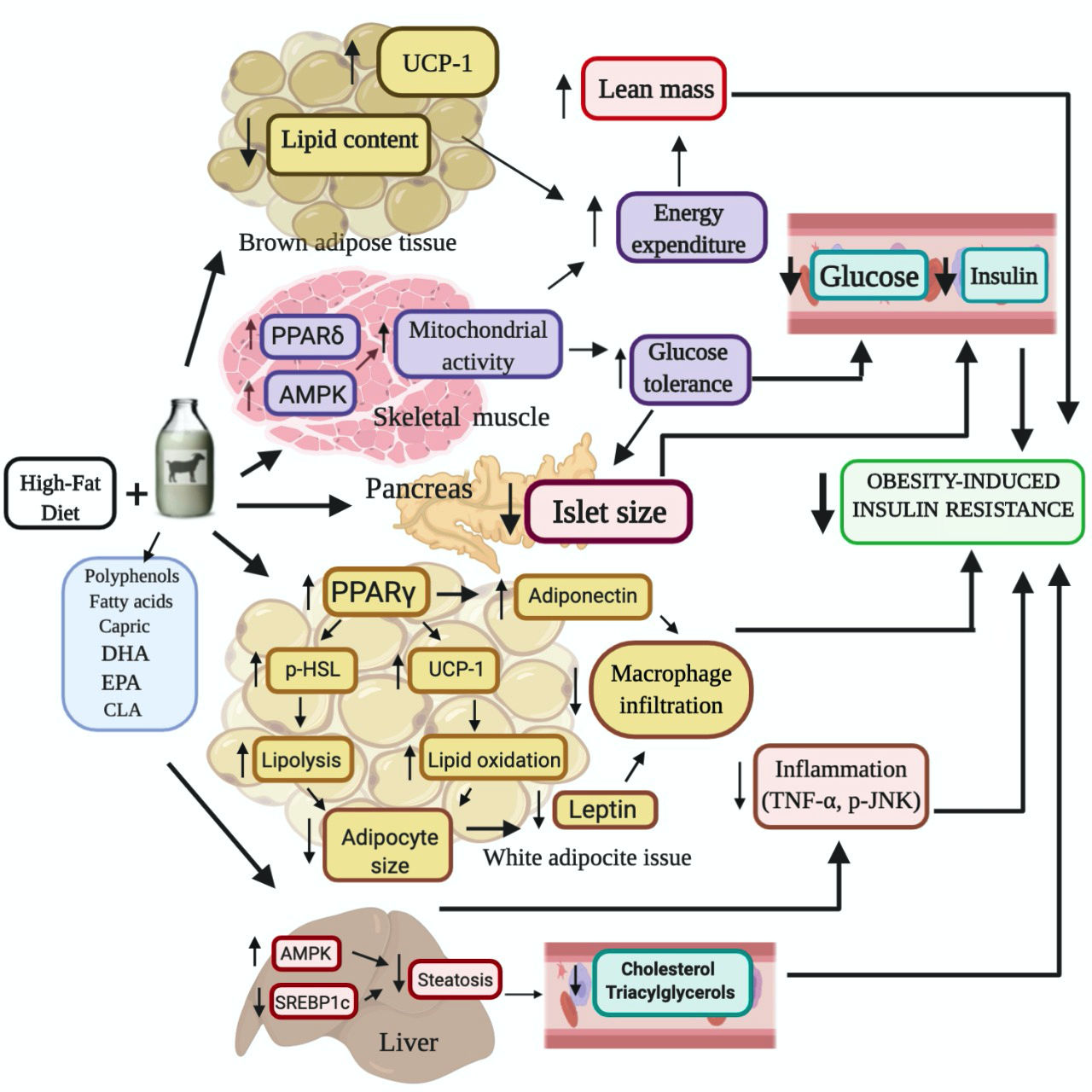
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Experimental Diets
4.3. Total Phenolic Content and Chemical Composition Analysis of Experimental Diets
4.4. Serum Determinations
4.5. Lipid Extraction, Derivatization and Fatty Acids Quantification
4.6. Body Composition and Energy Expenditure Measurement
4.7. Intraperitoneal Glucose and Insulin Tolerance Test
4.8. Histological Analysis of Liver, Pancreas, White and Brown Adipose Tissue
4.9. Mitochondria Abundance and Lipid Content in Skeletal Muscle
4.10. Lipid Content in Liver
4.11. Quantitation of Tumor Necrosis Factor Alpha (TNF-α) in Adipose Tissue
4.12. Immunohistochemistry of UCP-1 in Brown and White Adipose Tissues and Macrophages in Subcutaneous Adipose Tissue
4.13. Liver, Muscle and Adipose Tissue Gene Expression by Quantitative-Polymerase Chain Reaction (PCR)
4.14. Immunoblotting
4.15. Statistical Analyses
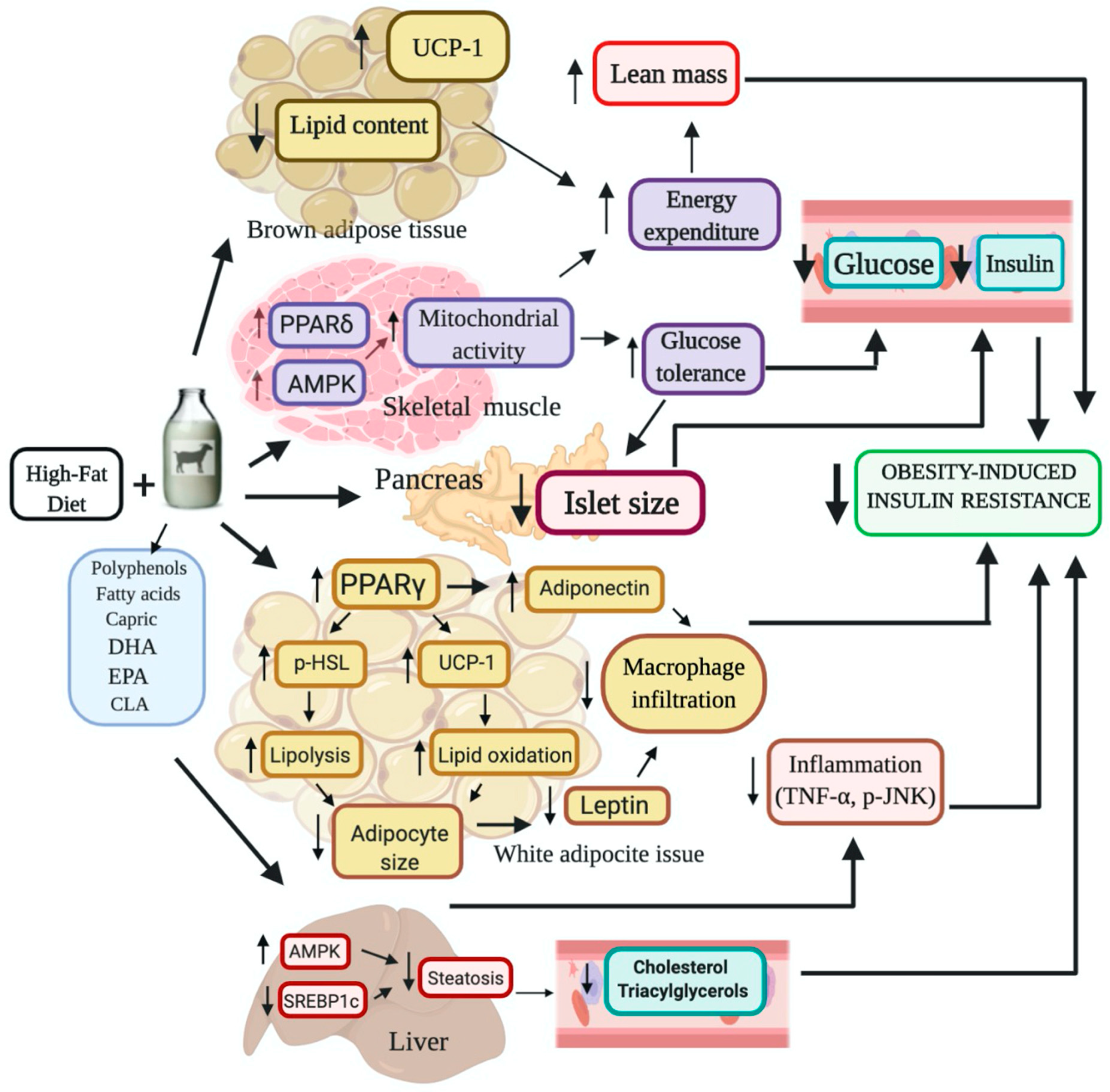
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AF | Acacia farnesiana |
| AMPK | Adenine monophosphate (AMP) activated protein kinase |
| ARA | Arachidonic acid |
| BAT | Brown adipose tissue |
| BODIPY | 4,4-Difluoro-4-bora-3a,4a-diaza-s-indacene |
| CLA | Conjugated linoleic acid |
| CVD | Cardiovascular disease |
| DAPI | 4′,6-Diamidino-2-phenylindole dihydrochloride |
| DHA | Docosahexaenoic acid |
| EPA | Eicosapentaenoic acid |
| GAE | Gallic acid equivalents |
| GC | Gas chromatography |
| H&E | Hematoxylin & eosin |
| HF | High-fat |
| HFAF | High fat Acacia farnesiana |
| HFCD | High fat conventional diet |
| HFG | High fat grazing |
| HmgCOA | 5-Hydroxy-3-methylglutaryl-coenzyme A) reductase |
| HSL | Hormone-sensitive lipase |
| ipGTT | Intraperitoneal Glucose Tolerance Test |
| ipITT | Intraperitoneal Insulin Tolerance Test |
| JNK | c-Jun N-terminal kinase |
| mRNA | Messenger ribonucleic acid |
| MUFA | Monounsaturated fatty acids |
| ORO | Oil Red O solution |
| p-AMPK | Phosphorylated adenine monophosphate (AMP) activated protein kinase |
| p-JNK | Phosphorylated-Jun N-terminal kinase |
| p-HSL | Phosphorylated-Hormone-sensitive lipase |
| PBS | Phosphate buffer saline |
| PCR | Polymerase chain reaction |
| PPARα | Peroxisome proliferator-activated receptor alpha |
| PPARγ | Peroxisome proliferator-activated receptor gamma |
| PPARδ | Peroxisome proliferator-activated receptor delta |
| PUFA | Polyunsaturated fatty acids |
| RER | Respiratory Exchange Ratio |
| SAT | Subcutaneous adipose tissue |
| SCD1 | Stearoyl-CoA desaturase |
| SDH | Succinate Dehydrogenase |
| SREBP-1c | Sterol regulatory element binding protein-1c |
| TNF-α | Tumor necrosis factor alpha |
| UCP-1 | Uncoupling protein one |
| VAT | Visceral adipose tissue |
References
- Stergiadis, S.; Nørskov, N.P.; Purup, S.; Givens, I.; Lee, M.R.F. Comparative nutrient profiling of retail goat and cow milk. Nutrients 2019, 11, 2282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, R.; Soares, J.; Garcia, H.; Nascimento, C.; Medeiros, M.; Bomfim, M.; Medeiros, M.C.; Queiroga, R. Goat milk fat naturally enriched with conjugated linoleic acid increased lipoproteins and reduced triacylglycerol in rats. Molecules 2014, 19, 3820–3831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lordan, R.; Tsoupras, A.; Mitra, B.; Zabetakis, I. Dairy fats and cardiovascular disease: Do we really need to be concerned? Foods 2018, 7, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirahatake, K.M.; Astrup, A.; Hill, J.O.; Slavin, J.L.; Allison, D.B.; Maki, K.C. Potential Cardiometabolic Health Benefits of Full-Fat Dairy: The Evidence Base. Adv. Nutr. 2020, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Gijsbers, L.; Ding, E.L.; Malik, V.S.; De Goede, J.; Geleijnse, J.M.; Soedamah-Muthu, S.S. Consumption of dairy foods and diabetes incidence: A dose-response meta-analysis of observational studies. Am. J. Clin. Nutr. 2016, 103, 1111–1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwingshackl, L.; Hoffmann, G.; Schwedhelm, C.; Kalle-Uhlmann, T.; Missbach, B.; Knüppel, S.; Boeing, H. Consumption of dairy products in relation to changes in anthropometric variables in adult populations: A systematic review and meta-analysis of cohort studies. PLoS ONE 2016, 11, e0157461. [Google Scholar] [CrossRef]
- Mena-Sánchez, G.; Becerra-Tomás, N.; Babio, N.; Salas-Salvadó, J. Dairy product consumption in the prevention of metabolic syndrome: A systematic review and meta-analysis of prospective cohort studies. Adv. Nutr. 2019, 10, S144–S153. [Google Scholar] [CrossRef]
- Fernandez, M.A.; Marette, A. Novel perspectives on fermented milks and cardiometabolic health with a focus on type 2 diabetes. Nutr. Rev. 2018, 76, 16–28. [Google Scholar] [CrossRef] [Green Version]
- Zuliani, A.; Esbjerg, L.; Grunert, K.G.; Bovolenta, S. Animal welfare and mountain products from traditional dairy farms: How do consumers perceive complexity? Animals 2018, 8, 207. [Google Scholar] [CrossRef] [Green Version]
- Delgadillo-Puga, C.; Cuchillo-Hilario, M.; León-Ortiz, L.; Ramírez-Rodríguez, A.; Cabiddu, A.; Navarro-Ocaña, A.; Medina-Ocampo, O.N.; Pedraza-Chaverri, J. Goats’ feeding supplementation with Acacia farnesiana pods and their relationship with milk composition: Fatty acids, polyphenols, and antioxidant activity. Animals 2019, 9, 515. [Google Scholar] [CrossRef] [Green Version]
- Haenlein, G.F.W. Goat milk in human nutrition. Small Rumin. Res. 2004, 51, 155–163. [Google Scholar] [CrossRef]
- Cabiddu, A.; Delgadillo-Puga, C.; Decandia, M.; Molle, A.G. Extensive ruminant production systems and milk quality with emphasis on unsaturated fatty acids, volatile compounds, antioxidant protection degree and phenol content. Animals 2019, 9, 771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, J.M. Leptin and the regulation of body weight. Keio J. Med. 2011, 60, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, W.; Carballo-Jane, E.; McLaren, D.G.; Mendoza, V.H.; Gagen, K.; Geoghagen, N.S.; McNamara, L.A.; Gorski, J.N.; Eiermann, G.J.; Petrov, A.; et al. Plasma lipid profiling across species for the identification of optimal animal models of human dyslipidemia. J. Lipid Res. 2012, 53, 51–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bn, S.; D’Souza, S.S.; Abraham, A. Insulin resistance precedes glucose intolerance and hyperleptinaemia in high-fat simple carbohydrate-fed C57BL/6J mice. Endokrynol. Pol. 2016, 67, 592–598. [Google Scholar] [CrossRef] [Green Version]
- Okada, T.; Chong, W.L.; Hu, J.; Hinault, C.; Michael, M.D.; Krützfeldt, J.; Yin, K.; Holzenberger, M.; Stoffel, M.; Kulkarni, R.N. Insulin receptors in β-cells are critical for islet compensatory growth response to insulin resistance. Proc. Natl. Acad. Sci. USA 2007, 104, 8977–8982. [Google Scholar] [CrossRef] [Green Version]
- Bal, N.C.; Singh, S.; Reis, F.C.G.; Maurya, S.K.; Pani, S.; Rowland, L.A.; Periasamy, M. Both brown adipose tissue and skeletal muscle thermogenesis processes are activated during mild to severe cold adaptation in mice. J. Biol. Chem. 2017, 292, 16616–16625. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, I.; Walsh, K. The whitening of brown fat and its implications for weight management in obesity. Curr. Obes. Rep. 2015, 4, 224–229. [Google Scholar] [CrossRef]
- Manio, M.C.C.; Inoue, K.; Fujitani, M.; Matsumura, S.; Fushiki, T. Combined pharmacological activation of AMPK and PPARδ potentiates the effects of exercise in trained mice. Phys. Rep. 2016, 4, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Thomas, D.; Apovian, C. Macrophage functions in lean and obese adipose tissue. Metab. Clin. Exp. 2017, 72, 120–143. [Google Scholar] [CrossRef]
- Braun, K.; Oeckl, J.; Westermeier, J.; Li, Y.; Klingenspor, M. Non-adrenergic control of lipolysis and thermogenesis in adipose tissues. J. Exp. Biol. 2018, 221, jeb.165381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medina-Gomez, G.; Gray, S.L.; Yetukuri, L.; Shimomura, K.; Virtue, S.; Campbell, M.; Curtis, R.K.; Jimenez-Linan, M.; Blount, M.; Yeo, G.S.H.; et al. PPAR gamma 2 prevents lipotoxicity by controlling adipose tissue expandability and peripheral lipid metabolism. PLoS Gen. 2007, 3, 634–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.J.; Huang, C.J.; Xie, D. Anti-obesity effects of conjugated linoleic acid, docosahexaenoic acid, and eicosapentaenoic acid. Mol. Nutr. Food Res. 2008, 52, 631–645. [Google Scholar] [CrossRef] [PubMed]
- Moslehi, A.; Hamidi-zad, Z. Role of SREBPs in liver diseases: A Mini-review. J. Clin. Transl. Hepatol. 2018, 6, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, Z.; Bond, N.J.; Ashmore, T.; Sanders, F.; Ament, Z.; Wang, X.; Murray, A.J.; Bellafante, E.; Virtue, S.; Vidal-Puig, A.; et al. Lipid zonation and phospholipid remodeling in nonalcoholic fatty liver disease. Hepatology 2017, 65, 1165–1180. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Kang, M.S.; Nam, M.; Kim, S.A.; Hwang, G.S.; Kim, H.S. Eicosapentaenoic acid (EPA) modulates glucose metabolism by targeting AMP-Activated Protein Kinase (AMPK) pathway. Int. J. Mol. Sci. 2019, 20, 4751. [Google Scholar] [CrossRef] [Green Version]
- González-Granillo, M.; Steffensen, K.R.; Granados, O.; Torres, N.; Korach-André, M.; Ortíz, V.; Aguilar-Salinas, C.; Jakobsson, T.; Díaz-Villaseñor, A.; Loza-Valdes, A.; et al. Soy protein isoflavones differentially regulate liver X receptor isoforms to modulate lipid metabolism and cholesterol transport in the liver and intestine in mice. Diabetologia 2012, 55, 2469–2478. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, H.; Umemura, A.; Taniguchi, K.; Font-Burgada, J.; Dhar, D.; Ogata, H.; Zhong, Z.; Valasek, M.A.; Seki, E.; Hidalgo, J.; et al. ER Stress cooperates with hypernutrition to trigger TNF-Dependent spontaneous HCC development. Cancer Cell. 2014, 26, 331–343. [Google Scholar] [CrossRef] [Green Version]
- Imamura, S.; Morioka, T.; Yamazaki, Y.; Numaguchi, R.; Urata, H.; Motoyama, K.; Mori, K.; Fukumoto, S.; Shoji, T.; Emoto, M.; et al. Plasma polyunsaturated fatty acid profile and delta-5 desaturase activity are altered in patients with type 2 diabetes. Metab. Clin. Exp. 2014, 63, 1432–1438. [Google Scholar] [CrossRef]
- Vázquez, C.V.; Rojas, M.G.V.; Ramírez, C.A.; Chávez-Servín, J.L.; García-Gasca, T.; Ferriz Martínez, R.A.; García, O.P.; Rosado, J.L.; López-Sabater, C.M.; Castellote, A.I.; et al. Total phenolic compounds in milk from different species. Design of an extraction technique for quantification using the Folin-Ciocalteu method. Food Chem. 2015, 176, 480–486. [Google Scholar] [CrossRef]
- Hilario, M.C.; Puga, C.D.; Ocaña, A.N.; Romo, F.P.-G. Antioxidant activity, bioactive polyphenols in Mexican goats’ milk cheeses on summer grazing. J. Dairy Res. 2010, 77, 20–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dludla, P.V.; Nkambule, B.B.; Jack, B.; Mkandla, Z.; Mutize, T.; Silvestri, S.; Orlando, P.; Tiano, L.; Johan Louw Mazibuko-Mbeje, S.E. Inflammation and oxidative stress in an obese state and the protective effects of gallic acid. Nutrients 2019, 11, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Y.; Wen, L.; Sun, J.; Bai, W.; Jiao, R.; Hu, Y.; He, P.; Ou, S. Cytoprotective mechanism of ferulic acid against high glucose-induced oxidative stress in cardiomyocytes and hepatocytes. Food Nut. Res. 2016, 60, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Jeong, S.J.; Seo, C.S.; Lim, H.S.; Sohn, E.; Yun, J.; Kim, B.Y. Simultaneous determination of the traditional herbal formula Ukgansan and the in vitro antioxidant activity of ferulic acid as an active compound. Molecules 2018, 23, 1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Mao, L.; Xu, P.; Wang, Y. Effects of (-)-Epigallocatechin Gallate (EGCG) on Energy Expenditure and Microglia-Mediated Hypothalamic Inflammation in Mice Fed a High-Fat Diet. Nutrients 2018, 10, 1681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hursel, R.; Westerterp-Plantenga, M.S. Catechin- and caffeine-rich teas for control of body weight in humans 1-4. Am. J. Clin. Nutr. 2013, 98, 1682–1693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahkeshani, N.; Farzaei, F.; Fotouhi, M.; Alavi, S.S.; Bahramsoltani, R.; Naseri, R.; Momtaz, S.; Abbasabadi, Z.; Rahimi, J.; Farzaei, M.H.; et al. Pharmacological effects of gallic acid in health and disease: A mechanistic review. Iran. J. Basic Med. Sci. 2019, 22, 225–237. [Google Scholar] [CrossRef]
- Palacios-González, B.; Vargas-Castillo, A.; Velázquez-Villegas, L.A.; Vasquez-Reyes, S.; López, P.; Noriega, L.G.; Aleman, G.; Tovar-Palacio, C.; Torre-Villalvazo, I.; Yang, L.-J.; et al. Genistein increases the thermogenic program of subcutaneous WAT and increases energy expenditure in mice. J. Nut. Biochem. 2019, 68, 59–68. [Google Scholar] [CrossRef]
- Avior, Y.; Bomze, D.; Ramon, O.; Nahmias, Y. Flavonoids as dietary regulators of nuclear receptor activity. Food Funct. 2013, 4, 831–844. [Google Scholar] [CrossRef] [Green Version]
- Lefterova, M.I.; Steger, D.J.; Zhuo, D.; Qatanani, M.; Mullican, S.E.; Tuteja, G.; Manduchi, E.; Grant, G.R.; Lazar, M.A. Cell-Specific Determinants of peroxisome proliferator-activated receptor γ function in adipocytes and macrophages. Mol. Cell. Biol. 2010, 30, 2078–2089. [Google Scholar] [CrossRef] [Green Version]
- Luquet, S.; Lopez-Soriano, J.; Holst, D.; Gaudel, C.; Jehl-Pietri, C.; Fredenrich, A.; Grimaldi, P.A. Roles of peroxisome proliferator-activated receptor delta (PPARδ) in the control of fatty acid catabolism. A new target for the treatment of metabolic syndrome. Biochimie 2004, 86, 833–837. [Google Scholar] [CrossRef] [PubMed]
- Abete, I.; Goyenechea, E.; Zulet, M.A.; Martínez, J.A. Obesity and metabolic syndrome: Potential benefit from specific nutritional components. Nut. Met. Cardiovasc. Dis. 2011, 21. [Google Scholar] [CrossRef] [PubMed]
- Julibert, A.; del Mar Bibiloni, M.; Tur, J.A. Dietary fat intake and metabolic syndrome in adults: A systematic review. Nut. Met. Cardiovasc. Dis. 2019, 29, 887–905. [Google Scholar] [CrossRef] [PubMed]
- Dinicolantonio, J.J.; O’Keefe, J.H. Importance of maintaining a low omega-6/omega-3 ratio for reducing inflammation. Open Heart 2018, 5, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Ralston, J.C.; Lyons, C.L.; Cooke, A.A.; Aoife, M.; Falvey, A.; Finucane, O.M.; McGillicuddy, F.C.; Rutter, G.A.; Roche, H.M. Dietary substitution of saturated with monounsaturated fatty acids within high-fat diets attenuates hyperinsulinemia and pancreatic islet dysfunction. Br. J. Nutr. 2020, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, N.; Matsuzaka, T.; Suzuki, M.; Nakano, Y.; Zao, H.; Yokoo, T.; Suzuki-Kemuriyama, N.; Kuba, M.; Okajima, Y.; Takeuchi, Y.; et al. Ablation of Elovl6 protects pancreatic islets from high-fat diet-induced impairment of insulin secretion. Biochem. Biophys. Res. Commun. 2014, 450, 318–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhaswant, M.; Poudyal, H.; Brown, L. Mechanisms of enhanced insulin secretion and sensitivity with n-3 unsaturated fatty acids. J. Nutr. Biochem. 2015, 26, 571–584. [Google Scholar] [CrossRef]
- Pinel, A.; Morio-Liondore, B.; Capel, F. n-3 polyunsaturated fatty acids modulate metabolism of insulin-sensitive tissues: Implication for the prevention of type 2 diabetes. J. Phys. Biochem. 2013, 70, 647–658. [Google Scholar] [CrossRef]
- Albert, B.B.; Derraik, J.G.; Brennan, C.M.; Biggs, J.B.; Smith, G.C.; Garg, M.L.; Cameron-Smith, D.; Hofman, P.L.; Cutfield, W.S. Higher omega-3 index is associated with increased insulin sensitivity and more favorable metabolic profile in middle-aged overweight men. Sci Rep. 2014, 4, 6697. [Google Scholar] [CrossRef] [Green Version]
- Golan-Gerstl, R.; Shiff, Y.E.; Moshayoff, V.; Schecter, D.; Leshkowitz, D.; Reif, S. Characterization and biological function of milk-derived miRNAs. Mol. Nutr. Food Res. 2017, 61, 1–26. [Google Scholar] [CrossRef]
- Mohanty, D.P.; Mohapatra, S.; Misra, S.; Sahu, P.S. Milk derived bioactive peptides and their impact on human health—A review. Saudi J. Biol. Sci. 2016, 23, 577–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reagan-Shaw, S.; Nihal, M.; Ahmad, N. Dose translation from animal to human studies revisted. FASEB J. 2007, 22, 659–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Center for Drug Evaluation and Research; Center for Biologics Evaluation and Research. Estimating the Safe Starting Dose in Clinical Trials for Therapeutics in Adult Healthy Volunteers; U.S. Food and Drug Administration: Rockville, MD, USA, 2002.
- Derrell, C.J.; Gebhart, G.F.; Gonder, J.C.; Keeling, M.E.; Kohn, D.F. The 1996 guide for the care and use of laboratory animals. ILAR 1997, 38, 41–48. [Google Scholar]
- Reeves, P.G.; Forrest, H.N.; Fahey, G.C. AIN-93 purified diets for laboratory rodents: Final report of the American Institute of Nutrition ad hoc writing committee on the reformulation of the AIN-76A rodent diet. J. Nutr. 1993, 123, 1939–1951. [Google Scholar] [CrossRef]
- Singleton, V.L.; Orthofer, R.; Lamuela-Raventós, R.M. Analysis of total phenols and other oxidation substrates and antioxidants by means of Folin-Ciocalteu reagent. Methods Enzymol. 1999, 299, 152–178. [Google Scholar] [CrossRef]
- Folch, J.; Lees, M.; Sloane Stanley, G.H. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar]
- Barakat, H.A.; Dohm, G.L.; Loesche, P.; Tapscott, E.B.; Smith, C. Lipid content and fatty acid composition of heart and muscle of the BIO 82.62 cardiomyopathic hamster. Lipids 1976, 11, 747–751. [Google Scholar] [CrossRef]
- Bowe, J.E.; Franklin, Z.J.; Hauge-Evans, A.C.; King, A.J.; Persaud, S.J.; Jones, P.M. Assessing glucose homeostasis in rodent models. J. Endocrinol. 2014, 222, 13–25. [Google Scholar] [CrossRef]
- Galarraga, M.; Campión, J.; Muñoz-Barrutia, A.; Boqué, N.; Moreno, H.; Martínez, J.A.; Milagro, F.; Ortiz-de-Solórzano, C. Adiposoft: Automated software for the analysis of white adipose tissue cellularity in histological sections. J. Lipid Res. 2012, 53, 2791–2796. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, H.; Williams, E.G.; Mouchiroud, L.; Cantó, C.; Fan, W.; Downes, M.; Héligon, C.; Barish, G.D.; Desvergne, B.; Evans, R.M.; et al. NCoR1 is a conserved physiological modulator of muscle mass and oxidative function. Cell 2011, 147, 827–839. [Google Scholar] [CrossRef] [Green Version]
- Kalmar, B.; Blanco, G.; Greensmith, L. Determination of muscle fiber type in rodents. Curr. Protoc. Mouse Biol. 2012, 2, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Leal-Díaz, A.M.; Noriega, L.G.; Torre-Villalvazo, I.; Torres, N.; Alemán-Escondrillas, G.; López-Romero, P.; Sánchez-Tapia, M.; Aguilar-López, M.; Furuzawa-Carballeda, J.; Velázquez-Villegas, L.A.; et al. Aguamiel concentrate from Agave salmiana and its extracted saponins attenuated obesity and hepatic steatosis and increased Akkermansia muciniphila in C57BL6 mice. Sci. Rep. 2016, 28, 34242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehlem, A.; Hagberg, C.E.; Muhl, L.; Eriksson, U.; Falkevall, A. Imaging of neutral lipids by oil red O for analyzing the metabolic status in health and disease. Nat. Protoc. 2013, 8, 1149–1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Méndez-Flores, S.; Hernández-Molina, G.; Enríquez, A.B.; Faz-Muñoz, D.; Esquivel, Y.; Pacheco-Molina, C.; Furuzawa-Carballeda, J. Cytokines and Effector/Regulatory Cells Characterization in the Physiopathology of Cutaneous Lupus Erythematosus: A Cross-Sectional Study. Mediat. Inflamm. 2016, 7074829. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ingredient (%) | Control | HF | HFCD | HFG | HFAF |
|---|---|---|---|---|---|
| Dry whole goat milk | -- | -- | 60.0 | 60.0 | 60.0 |
| Casein a | 20.0 | 20.0 | 6.0 | 1.0 | -- |
| Sucrose b | 10.0 | 23.9 | 12 | 15.0 | 14.0 |
| Corn starch c | 40.0 | -- | -- | -- | -- |
| Maltodextrin d | 13.0 | 13.0 | -- | -- | -- |
| Lard | -- | 28.0 | 7.0 | 9.0 | 11.0 |
| Soy oil e | 7.0 | 5.0 | 5.0 | 5.0 | 5.0 |
| Cellulose f | 5.0 | 5.0 | 5.0 | 5.0 | 5.0 |
| Vitamin Mix g | 1.0 | 1.1 | 1.1 | 1.1 | 1.1 |
| Mineral Mix h | 3.5 | 3.5 | 3.5 | 3.5 | 3.5 |
| L-Cysteine i | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 |
| Choline j | 0.25 | 0.25 | 0.25 | 0.25 | 0.25 |
| Total | 100 | 100 | 100 | 100 | 100 |
| % Energy (kcal) from: | |||||
| Protein | 20.6 | 20.6 | 20.6 | 20.8 | 20.8 |
| Fat | 7.0 | 32.0 | 32.4 | 32.0 | 32.4 |
| Carbohydrates | 63.0 | 36.9 | 36.5 | 36.5 | 36.1 |
| Polyphenol content (mg/GAE per 100 g of diet) k | 1.68 | 1.65 | 16.1 | 17.4 | 17.8 |
| Protein | Gene (Mouse) | Forward Sequence (5′-3′) | Reverse Sequence (3′-5′) |
|---|---|---|---|
| SREBP-1c | Srebf1 | AGACAAACTGCCCATCCACC | AAGCGGATGTAGTCGATGGC |
| PPARδ | Ppard | CTCTTCATCGCGGCCATCATTCT | TCTGCCATCTTCTGCAGCAGCTT |
| AMPKα | PRKAA2 | ACCTGAGAACGTCCTGCTTG | GGCCTGCGTACAATCTTCCT |
| PPARγ2 | Pparg2 | CTCCTGTTGACCCAGAGCAT | GAAGTTGGTGGGCCAGAA |
| adiponectin | Adipoq | CTGACGACACCAAAAGGGCT | CCAACCTGCACAAGTTCCCT |
| leptin | Lep | CAAGCAGTGCCTATCCAGA | AAGCCCAGGAATGAAGTCCA |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Delgadillo-Puga, C.; Noriega, L.G.; Morales-Romero, A.M.; Nieto-Camacho, A.; Granados-Portillo, O.; Rodríguez-López, L.A.; Alemán, G.; Furuzawa-Carballeda, J.; Tovar, A.R.; Cisneros-Zevallos, L.; et al. Goat’s Milk Intake Prevents Obesity, Hepatic Steatosis and Insulin Resistance in Mice Fed A High-Fat Diet by Reducing Inflammatory Markers and Increasing Energy Expenditure and Mitochondrial Content in Skeletal Muscle. Int. J. Mol. Sci. 2020, 21, 5530. https://doi.org/10.3390/ijms21155530
Delgadillo-Puga C, Noriega LG, Morales-Romero AM, Nieto-Camacho A, Granados-Portillo O, Rodríguez-López LA, Alemán G, Furuzawa-Carballeda J, Tovar AR, Cisneros-Zevallos L, et al. Goat’s Milk Intake Prevents Obesity, Hepatic Steatosis and Insulin Resistance in Mice Fed A High-Fat Diet by Reducing Inflammatory Markers and Increasing Energy Expenditure and Mitochondrial Content in Skeletal Muscle. International Journal of Molecular Sciences. 2020; 21(15):5530. https://doi.org/10.3390/ijms21155530
Chicago/Turabian StyleDelgadillo-Puga, Claudia, Lilia G. Noriega, Aurora M. Morales-Romero, Antonio Nieto-Camacho, Omar Granados-Portillo, Leonardo A. Rodríguez-López, Gabriela Alemán, Janette Furuzawa-Carballeda, Armando R. Tovar, Luis Cisneros-Zevallos, and et al. 2020. "Goat’s Milk Intake Prevents Obesity, Hepatic Steatosis and Insulin Resistance in Mice Fed A High-Fat Diet by Reducing Inflammatory Markers and Increasing Energy Expenditure and Mitochondrial Content in Skeletal Muscle" International Journal of Molecular Sciences 21, no. 15: 5530. https://doi.org/10.3390/ijms21155530
APA StyleDelgadillo-Puga, C., Noriega, L. G., Morales-Romero, A. M., Nieto-Camacho, A., Granados-Portillo, O., Rodríguez-López, L. A., Alemán, G., Furuzawa-Carballeda, J., Tovar, A. R., Cisneros-Zevallos, L., & Torre-Villalvazo, I. (2020). Goat’s Milk Intake Prevents Obesity, Hepatic Steatosis and Insulin Resistance in Mice Fed A High-Fat Diet by Reducing Inflammatory Markers and Increasing Energy Expenditure and Mitochondrial Content in Skeletal Muscle. International Journal of Molecular Sciences, 21(15), 5530. https://doi.org/10.3390/ijms21155530