Abstract
Brain arteriovenous malformation (bAVM) is a congenital defect affecting brain microvasculature, characterized by a direct shunt from arterioles to venules. Germline mutations in several genes related to transforming growth factor beta (TGF-β)/BMP signaling are linked to both sporadic and hereditary phenotypes. However, the low incidence of inherited cases makes the genetic bases of the disease unclear. To increase this knowledge, we performed a whole exome sequencing on five patients, on DNA purified by peripheral blood. Variants were filtered based on frequency and functional class. Those selected were validated by Sanger sequencing. Genes carrying selected variants were prioritized to relate these genes with those already known to be linked to bAVM development. Most of the prioritized genes showed a correlation with the TGF-βNotch signaling and vessel morphogenesis. However, two novel pathways related to cilia morphogenesis and ion homeostasis were enriched in mutated genes. These results suggest novel insights on sporadic bAVM onset and confirm its genetic heterogeneity. The high frequency of germline variants in genes related to TGF-β signaling allows us to hypothesize bAVM as a complex trait resulting from the co-existence of low-penetrance loci. Deeper knowledge on bAVM genetics can improve personalized diagnosis and can be helpful with genotype–phenotype correlations.
1. Introduction
Brain arteriovenous malformations (bAVM, OMIM #108010) are vascular malformations affecting brain vasculature. Lack of a capillary bed and a direct shunt from arterioles to venules, as well as pericyte reduction, are characteristics of the lesions [1]. A vessel tangle is formed, usually called nidus. During transition from the feeding arteries to the nidus and then, to the draining veins, vessels show dysregulated differentiation patterns and severe enlargement. From arteries, blood perfuses to the nidus with high pressure, increasing risk of lesion rupture. Moreover, the mix of arterial and venous circulations within these lesions leads to a deficit in cerebral tissue oxygenation. This complex condition usually results in major clinical manifestations such as intracerebral hemorrhage and epileptic seizures, appearing in almost 50% of patients. Disease incidence is about 0.01% worldwide and usually arises at an early age [2]. Nevertheless, it most often occurs as a sporadic condition and only a few dozen cases are reported as inherited with an autosomal dominant pattern. Hereditary bAVM usually coexists with other vascular syndromes, such as Osler-Weber-Rendu syndrome, also known as hereditary hemorrhagic telangiectasia (HHT). HHT includes different phenotypes caused by mutations in genes related to the transforming growth factor beta 2 (TGF-βII) transduction pathway, such as ENG, ACVRL1, SMAD4, and GDF2 [3,4,5]. Due to the severe remodeling rate and recidivism risk after total surgical resection, bAVMs are considered highly dynamic lesions. Therefore, lesion growth is now thought of as the result of continuous endogenous stimuli due to genetic factors. The low frequency of inherited bAVM makes molecular characterization of the disease difficult. Impaired expression of the ephrin family genes and of other vascular differentiation markers was reported by several authors [6,7]. At the same time, the sporadic nature of the disease can also be considered a result of numerous single nucleotide polymorphisms (SNPs) in genes involved in vasculogenesis and early angiogenesis pathways, as, for instance, in vascular endothelial growth factor (VEGF) and Notch signaling [8]. To improve knowledge on bAVM pathogenesis, we purified DNA from peripheral blood and performed a whole exome sequencing (WES) analysis on a group of five patients affected by sporadic bAVM. Then, we clustered genes carrying the detected mutations, highlighting pathways and prioritizing genes mainly linked to bAVM development.
2. Results
2.1. WES, Bioinformatic Analysis, and Filtering
An average of 81,170,229 million reads were output by the runs. Of these, 92.29% showed a Phred quality score > 30. After duplicate discard, an average of 75,411,435 reads were filtered and mapped to the GRCh38 human reference genome. The percentage of on-target reads was on average 71.57%, calculated on the deduplicated mapped read numbers. The quality report summary of each run is provided in Table S1. Regarding variant calling, a mean of 40,090 variants was annotated for each exome. Following the application of the above-mentioned filtering criteria, about 230 variants were selected for each sample. These included non-synonymous, nonsense, and frameshift mutations, whose reported minor allele frequency (MAF) was estimated to be < 0.01. Full lists are available in Table S2.
2.2. Gene Clustering and Prioritization
The ClueGO (Gene Ontology) enrichment analysis allowed to cluster the mutated genes for each exome in order to highlight the pathways they are involved in. In Table 1 only annotated terms showing the Bonferroni-adjusted p-value ≤ 0.05 were reported, both for each single term and for the entire cluster. The full lists are available in Table S3. Annotations related to cell adhesion (GO:1903392, GO:0051895, R-HSA:382054, GO:0038089, GO:0007157, GO:0120193, GO:1904886), cardiovascular development (WP:3668, GO:0003279, GO:0035904, GO:0060840, GO:0060976, GO:0060411), Notch signaling (GO:0007221, R-HSA:9021450, R-HSA:9013508, GO:0045747, WP:268, R-HSA:9012852), and TGFβR signaling (GO:1903846, GO:0030511) were highly represented in all the AVM samples. Furthermore, ontologies related to microtubule assembly and cell motility (GO:0003341, GO:0001578, GO:0001539, GO:0031122, GO:0060285) were also widely frequent. Interestingly, pathways related to ion conductance (GO:1904063, GO:0034766, GO:2001258, GO:0043267, GO:0016248, GO:0008200) were highly enriched in the AVM5 sample.

Table 1.
ClueGO (Gene Ontology) enrichment analysis results.
The table reports annotations from ClueGO enrichment analysis. For each sample, the enriched pathways are mentioned as GO Term (3rd column) and the ontology sources (GeneOntology, WikiPathways, Reactome) are reported (4th column), as well as the clustered genes (7th column). Only annotations showing Bonferroni-adjusted p-value ≤ 0.05 for term or group (5th and 6th columns, respectively) are reported. Full results are available in Table S3. ARFGAP: Adenosine diphosphate Ribosylation Factor-GTPase; AVM: Arteriovenous malformation. The number (1–5) indicates the sample; COPI: coating protein 1; GPCR: G protein-coupled receptor; MAP: Mitogen-Activated Protein; MAPK: Mitogen-Activated Protein Kinase; MKK: MAP Kinase Kinase; NCAM: Neural Cell Adhesion Molecule; PDGF: Platelet Derived Growth Factor; PLXND1: Plexin D1; TAK1: TGF-Beta-Activated Kinase 1; TNF: Tumor Necrosis Factor; IRE1: Inositol-Requiring Protein 1; v-SNARE: Vesicle-Soluble NSF (N-Ethylmaleimide-Sensitive Factor) Attachment Protein Receptor.Gene prioritization aimed to relate selected loci with others, already linked to bAVM development. ToppGene output a full list of the Test Gene Set, ordered according to their overall p-value. For each gene, the overall p-value is calculated on the basis of the single p-value for each training parameter considered. Therefore, also in this case, only genes showing an overall p-value ≤ 0.05 were selected for each sample. Together with this criterion, “Gene Ontology (GO) biological process” annotations were considered as prioritized for the five AVM exomes. These are LTBP4, LTBP1, LRP1, and FBN2 for AVM1; TAB1, RELN, and MAP2K3 for AVM2; KDR and EPHA2 for AVM3; NOTCH3, PLXND1, TAB1, CTBP2, SLIT2, RNF111, MAML1, and CHRNB2 for AVM4; and AXIN1, EPHB2, DVL1, and CAMK2B for AVM5 (Table 2). The complete lists with detailed statistical parameters as well as the annotations linking the prioritized genes to the training genes are available in Table S4.
Table 2.
Genes prioritized by ToppGene tool.
Although prioritization analysis was quite exhaustive, further loci were considered. These added loci were selected as they are involved in vasculogenesis and carry missense and nonsense variants. In particular, they were FLT4, NCoR2, CCN1, and GIMAP1 in the AVM2 sample; NOTCH4 in AVM3; and ENG and TGFBR2 in AVM5. In particular, ENG and TGFRB2 are known to be highly linked to brain AVM and HHT development. NCoR2 was affected by the novel nonsense variant c.2078G>T (p.Glu693Ter) (Ensembl Transcript ID: ENST00000405201.5). As a consequence, the mutated protein counts 692 amino acids rather than the 2514 amind acids of the wild-type one. The mutation was detected in a heterozygous condition and resulted as “Damaging” and “Disease causing” in SIFT (https://sift.bii.a-star.edu.sg/) [9] and MutationTaster (http://www.mutationtaster.org/) [10] prediction tools, respectively (not shown).
2.3. Sanger Validation
Table 3 lists the variants detected in the genes previously prioritized. All these variants were confirmed by Sanger sequencing and were not detected in our internal 10 control exomes obtained from healthy subjects. Figure 1 reports only the electropherogram of the novel nonsense variant, c.2078G>T (p.Glu693Ter) affecting the NCoR2 locus.
Table 3.
Variants affecting prioritized genes.
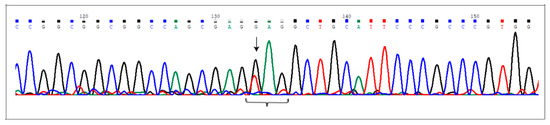
Figure 1.
The c.2078G>T (p.Glu693Ter) at the NCoR2 locus. Electropherogram shows the novel nonsense mutation affecting the NCor2 gene, detected in heterozygous condition in AVM2 sample. Nucleotide substitution is indicated by the arrow. The brace indicates the DNA sequence corresponding to the triplet carrying the mutated nucleotide (the first one).
3. Discussion
The genetic landscape of bAVM is to date quite elusive probably due to the very low frequency of inherited cases. Most patients are affected by sporadic forms whose molecular causes are waiting to be clarified. In many cases, lesions are congenital, increasing in size over the years until they become symptomatic. Due to the dynamic nature of bAVM and its high recidivism rate, it is well accepted that lesions arise as a consequence of continuous endogenous stimuli, as inherited and de novo genetic variants or epigenetic modifications occurring during embryo development [11,12]. Therefore, we performed WES analysis on a group of five patients affected by sporadic bAVM, highlighting the main pathways enriched by germline mutated genes. Despite mutated genes differing among patients, the pathways in which they converge are the same. Firstly, we checked for variants in ENG, NOTCH4, and TGFβR2 that are known to be involved in arteriovenous malformation development [13,14]. Then, we searched for KRAS c.35G>T (p.Gly12Val) that was recurrently detected in bAVM patients [15] and none of them carried the mutation, as well as other variants within the gene. However, this was expected as our analysis was performed on DNA purified by peripheral blood. Mosaic-activating KRAS mutations, indeed, were found in sporadic AVM-derived specimens [16,17].
The ClueGO enrichment analysis revealed pathways related to microtubule formation, cell adhesion, and vascular remodeling as being highly enriched (Table 1). Moreover, prioritization analysis was performed for each sample to detect the main genes involved in TGFβR transduction pathways and, therefore, more likely associated to bAVM onset. As reported in Table S4a,b, several loci are noteworthy of consideration and the most relevant are listed in Table 2. However, among prioritized genes, here we briefly discuss those more likely related to bAVM development, in relation to the single sample. This selection was made considering significant p-values related to phenotypes and pathway, outputted by the ToppGene tool (Table S4a).
3.1. AVM1
Regarding AVM1, we focused on LTBP1, LTBP4, FBN2, and LRP1 loci. FBN2, encoding for Fibrillin 2, and LTBP1 and LTBP4 belonging to the “latent transforming growth factor beta binding proteins” family, are ligands of TGF-β receptors [18,19]. With regard to LRP1, encoding for the LDL receptor related protein 1, expression data showed it is expressed in brain endothelial cells where it contributes to chemotactic cell migration, inducing sphingosine-1-phosphate proangiogenic signaling [20]. Moreover, depletion of LRP1 determines defects of both large and small vessel morphogenesis leading to a lethal phenotype [21].
3.2. AVM2
Regarding data obtained from AVM2 exome, we considered variants affecting TAB1, FLT4, RELN, MAP2K3, CCN1, GIMAP1, and NCoR2 loci. TAB1 encodes for the TGF-β activated kinase 1 binding protein 1 and increases endothelial permeability, mediated by the non-canonical TGF-β pathway following inflammation stimuli [22]. Moreover, TAB1 activates the TAK1 kinase, an upstream modulator of the p38 MAPK signaling. MAP2K3 is also involved [23] in the same pathway and, in particular, a physical interaction between TAK1 and MAP2K3 has been reported [24]. Involvement of inflammatory response in bAVM is, to date, well accepted [25,26] and in this context, we detected a nonsense mutation, the c.699G>A (p.Trp233Ter) in GIMAP1 gene. Together with the TGFβR2 signaling, Notch transduction pathways were also reported as promoting AVM development [27]. Therefore, we also considered prioritizing the NCor2 locus, encoding for nuclear receptor co-repressor. We found the novel nonsense mutation, c.2078G>T (p.Glu693Ter) (Figure 1). FLT4, instead, encodes for the vascular endothelial growth factor receptor 3 (VEGFR3) [28]. Finally, we considered CCN1 locus, encoding for the cellular communication network factor 1. Its expression is increased in the extracellular matrix surrounding microvessels and is growth factor-inducible. The protein promotes integrin-mediated endothelial cell adhesion in response to mechanotransduction signaling [29]. This evidence is well congruent with bAVM pathogenesis due to frequent insult by the high blood pressure within the lesions.
3.3. AVM3
Together with NOTCH4, variants carried by EPHA2, EPHB4, and KDR were detected in sample AVM3. EPHA2 and EPHB2 encode for two proteins belonging to the ephrin family, a subgroup of protein-tyrosine kinase receptors. Ephrins are vessel differentiation markers and their role is pivotal during early vasculogenesis. In particular, mesenchymal cells express EPHB2, a feature of arterial morphogenesis. A model proposed by Adams et al. hypothesizes interaction among type-B ephrins differentially expressed in arteries and veins as the basis of a remodeling process that leads to sprouting and capillary network development [30]. EPHB2 expression is upregulated in capillaries during inflammation. This results in increased endothelial permeability and loss of vessel differentiation [31]. The absence of EphA2, instead, was shown to impair the blood–brain barrier, resulting in inhibition of endothelial cell migration and in enhancement of tight junction formation in human brain microvascular endothelial cells (HBMECs) [32]. KDR encodes for the vascular VEGFR2, essential for the organization of the embryonic vasculature and angiogenic sprouting [33].
3.4. AVM4
The highest number of prioritized genes was in the AVM4 sample. GO annotations for biological process revealed involvement in vasculature morphogenesis for NOTCH3, PLXND1, SLIT2, and MAML1 loci. At the embryo stage, VEGF and Notch transduction signaling modulates PLXND1 expression to guide organ vasculogenesis by promoting endothelial cell migration and proliferation [34]. In adults, instead, PLXND1 expression is physiologically low and limited to a few cell types, such as endothelial cells [35]. Balancing effects on migration were reported for SLIT2 [36]. MAML1 is described as a NOTCH coactivator, even if its role in angiogenesis needs more elucidation [37]. Regarding the TGF-β/BMP pathway, we focused on the RNF111 gene, encoding for the E3 ubiquitin-protein ligase. One of its targets is the SMAD7 protein that acts by inhibiting TGF-β/BMP signaling. Therefore, RNF111 activity is required to promote SMAD7 degradation and to enhance the TGF-β/BMP pathway [38]. TGF-β signaling is also upregulated by increased levels of CTBP2 (C-terminal binding protein 2), driving endothelial-to-mesenchymal transition (EMT) [39]. This gene was also mutated in the AVM4 patient. In the end, we focused our attention on the GO terms regarding the response to hypoxia (GO:0001666, GO:0036293, GO:0070482) with the patient as a carrier of rs55685423 (c.1191G>C, p.Gln397His) in the CHRNB2 locus. This gene encodes for the cholinergic receptor nicotinic beta 2 subunit [40]. Its expression was demonstrated in HBMECs, where it contributes to capillary network formation and to angiogenic response to inflammation [41]. Notably, the same ontologies were also found in the AVM3 patient, annotated by the KDR locus.
3.5. AVM5
Finally, in the AVM5 sample we identified two variants in ENG and TGFBR2 loci, rs139398993 (c.392C>T, p.Pro131Leu) and rs35766612 (c.1159G>T, p.Val387Leu), respectively. Moreover, EPHB2 was also affected by a missense variant. Based on human and mouse phenotype ontologies, DVL1 and AXIN1 were annotated to the “cerebrovascular disease” term and, in particular, with AVM and telangiectasia phenotypes. DVL1 is known to control postnatal angiogenesis [42]. AXIN1 encodes for a negative regulator of the Wnt pathway, also enhancing TGF-β signaling by promoting the degradation of the inhibitory SMAD7, in a RNF111-dependent manner [43]. Surprisingly, it was recently described as an important regulator of embryo central nervous system (CNS) angiogenesis, and overexpression leads to premature vascular regression, followed by progressive dilation and inhibition of vascular maturation [44].
3.6. Novel Insights
Together with TGF-β/Notch signaling, GO annotations derived from the ClueGO enrichment analysis (Table 1) highlight a relevant presence of ontologies related to microtubule and cilia organization (GO:0003341, GO:0001578, GO:0031122, GO:0001539, GO:0060285). A recent study demonstrated that cilia are widely represented in endothelial cells during early vasculogenesis and in the later stages as vessel bifurcation point anastomosis. Zebrafish knock-down for cilia biogenesis gene models showed cilia disassembly following shear stress, resulting in remodeling of endothelial cell architecture and increased permeability and hemorrhagic events. Moreover, hemorrhages were only observed in head vasculature and were not observed in the trunk or caudal vessels [45]. Based on this evidence, germline defects in genes controlling cilia assembly might also contribute to brain AVM development as the result of mechanical stress induced by high blood flow and pressure. Clearly, this hypothesis needs to be adequately validated.
Another important property of the blood–brain barrier is the highly selective control of solute transport which is maintained by the exact spatial distribution of membrane transporters and ion channels. Polarization is a key factor for morpho-functional homeostasis of endothelial cells and was shown to be driven by VEGF via Ca2+ specific signaling pathways [46]. Moreover, dysregulation of K+ ion influx in non-excitable cells was shown to lead to hyperpolarization of membrane potential with consequent increased intracellular Ca2+. This results in enhancement of cell proliferation and was also demonstrated in brain capillary endothelial cells [47]. However, if physiological Ca2+ concentration is abnormally excessive, endothelial cells undergo apoptosis [48]. Therefore, we focused attention on GO terms from the ClueGO analysis of sample AVM5. As shown in Table 1, GO terms related to transmembrane ion transport (GO:1904063, GO:0034766, GO:2001258, GO:0043267, GO:0016248, GO:0008200) are largely enriched, as well as those related to membrane biogenesis (GO:0044091, GO:0071709, GO:1904886) making the hypothesis of misregulation in bAVM onset conceivable.
Although these are preliminary findings, they are in accordance with what was recently published by Hauer and colleagues. They describe dysregulated expression of genes also involving cytoskeleton network and transmembrane transport in bAVM-derived specimens, when compared to intracranial control arteries [49]. Therefore, these results allow to elicit other mechanisms in pathogenesis of bAVM not only confined to the canonical TGFβR2 pathway.
3.7. Final Considerations
We discussed loci affected by germline variants in five bAVM samples. Despite these loci differing among the samples, they converged in regulation of the same cellular signaling pathways. This interconnection is represented in Figure 2. The image was obtained by STRING tool Version 11.0 (https://string-db.org/) [50]. Details on nodes and edges are supplied in Table S5.
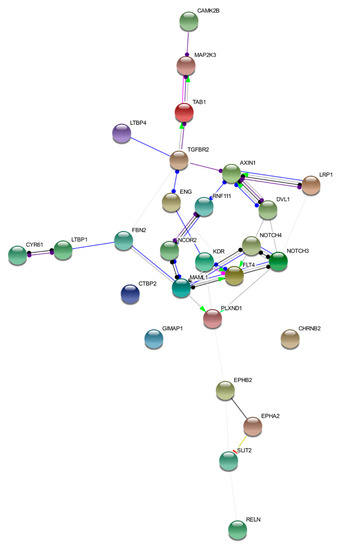
Figure 2.
Functional network of prioritized genes. The image describes observed and inferred functional interactions linking prioritized genes. Nodes represent input proteins while edges represent protein–protein associations. Each edge color indicates a specific annotation. Green: activation; red: inhibition; blue: binding; light blue: phenotype; black: reaction; violet: catalysis; pink: posttranslational modification; yellow: transcriptional regulation.
According to prioritized genes, our data support findings previously reported [51,52] and, in particular, the genetic heterogeneity of the disease. These results suggest that sporadic bAVM is probably not a monogenic condition, rather it arises during early vasculogenesis at the embryo stage. In particular, following fertilization, the combination of both inherited and eventually de novo genetic variants in numerous loci controlling vessel development could result in early vasculogenesis impairment and lesion onset. However, the evidence that these patients develop lesions only in the CNS underlines the importance of the cross-talk between glial cells and endothelium during neurodevelopment and blood–brain barrier morphogenesis. Most genes considered here show an early peculiar expression in neural progenitor cells that contributes to correct vasculogenesis and angiogenetic processes. In this context, proteins related to axon guidance such as Slits, plexins, and ephrins are exhaustive examples [53,54]. However, a last consideration regards genes involved in DNA repair such as WRN, FANCC, BRCA2, TP53BP1, and others carrying rare missense variants. These variants might cause protein functional alteration and, subsequently, DNA repair impairment. At the embryo stage, this might trigger DNA errors resulting in somatic mutations. Clearly, as our study focused on germline variants, the role of somatic mutations is not evaluated here.
Germline genetic variants are endogenous and permanent factors affecting both early vasculogenesis and late angiogenesis. Endothelial remodeling is a continuous phenomenon, and this can explain the increased recidivism rate of bAVM. Therefore, our hypothesis regards the possibility of considering bAVM as the result of the co-existence of numerous low-penetrance loci controlling different processes during endothelial cell differentiation and maturation. This idea is supported by two observations. We selected only loci affected by rare variants (MAF < 0.01) and then, those most likely related to rare disease onset. Rare variants at the same loci were searched for in our internal 10 control exomes obtained from healthy subjects and none was detected.
Clearly, the main limitation of the study is related to the few samples considered and, certainly, results require further validations on a larger patient cohort.
Despite this being a preliminary investigation, the possibility of detecting novel loci and germline mutations potentially involved in bAVM onset will allow to hypothesize a strategy for molecular diagnosis, preferably based on a panel of selected genes.
4. Materials and Methods
4.1. Patient Recruitment and WES Analysis
The study was performed on a group of five Italian patients (AVM1–5) diagnosed with bAVM following cerebral angiography investigation (Figure 3). A severity lesion score was assigned to each patient based on the Spetzler–Martin grading system [55]. Anamnestic data are presented in Table 4. No familiar history of bAVM was reported for the patients and they were classified as sporadic. Patients were fully informed on their enrolment in the study and informed consent was obtained, and for underaged patients as well. DNA samples were collected from peripheral blood and purified by the QIAamp DNA Blood Mini Kit (Qiagen). Qualitative and quantitative measurements of the samples were performed by NanoDrop spectrophotometer (Thermo Fisher Scientific) and by a Qubit fluorometer (Thermo Fisher Scientific). Paired-end libraries were obtained by the SureSelect Human All Exon V7 (Agilent) kit and sequenced on a HiSeq 2500 Illumina platform.
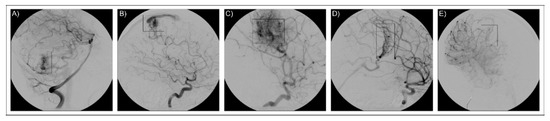
Figure 3.
Imaging for neuroradiological diagnosis. The figure reports cerebral angiographies that confirm the presence of bAVM lesions. The five images refer to the five patients as reported in the text: (A) AVM1, (B) AVM2, (C) AVM3, (D) AVM4, (E) AVM5. AVM lesions are framed in the rectangular box.
Table 4.
Anamnesis data of the patients enrolled in the study.
4.2. Bioinformatic Analysis
FastQ data obtained from sequencing runs were quality checked by the FastQC (v.0.11.7) tool (http://www.bioinformatics.babraham.ac.uk/projects/fastqc). Only reads presenting a Phred score ≥ 28 were selected after trimming and aligned to the GRCh38 Human Reference Genome by the Burrows–Wheeler Aligner (BWA) algorithm [56]. Duplicate reads were removed by the MarkDuplicate tool provided by Picard toolkit (v.2.18.23) (“Picard Toolkit.” 2019. Broad Institute, GitHub Repository. http://broadinstitute.github.io/picard/; Broad Institute). Then, the Indel realignment and the base recalibration were performed by the Genome Analysis Toolkit (GATK) (v.4.1.3.0) (https://software.broadinstitute.org/gatk/). Variant calling was executed by FreeBayes [57], while ANNOVAR v.2018Apr16 [58] was used for variant annotation.
4.3. Variant Filtering Criteria
Before proceeding with downstream analysis, annotated genes and variants were filtered on the basis of several criteria. Variants showing quality score < 150 were discarded. This threshold value was established by the observation that several variants with depth lower than 150 were not confirmed by the following Sanger sequencing validation. Filtered variants were classified by functional class and intronic, synonymous, non-coding RNA, and untranslated regions affecting variants were discarded. Missense, nonsense, frameshift, and short indels presenting an MAF < 0.01 were selected. Rare variants were preferred based on the low worldwide incidence of bAVM. For the MAF-based filtering, the values reported in the Genome Aggregation Database (https://gnomad.broadinstitute.org/) [59] and in the 1000 Genomes phase 3 project [60] were considered.
4.4. Gene Clustering and Prioritization
To visualize and functionally group genes carrying filtered variants, the ClueGO plug-in of Cytoscape software was used for each sample [61]. Clustering was performed on the basis of the GO Biological Process, REACTOME and WikiPathways ontologies. Groups showing Bonferroni step down corrected p-value ≤ 0.05 were considered significant and therefore selected for the purpose. Genes within the chosen groups were added to the Test Gene Set in ToppGene (https://toppgene.cchmc.org/) [62], a web-based tool for prioritization of candidate genes based on functional similarity to a training gene list. The Training Gene Set group was made up of ENG, ACVRL1, TGFBR2, SMAD4, and GDF2 genes, already known to be causative of HHT and of a few familiar bAVM cases without HHT. The training parameters selected were “GO:Biological Process”, “Human Phenotype”, “Mouse Phenotype”, “Pathway”, “PubMed”, “Interaction”, and “Disease”. Five different analyses were run, one for each exome data. Statistical parameters were calculated applying the Bonferroni correction, and p-values ≤ 0.05 were considered significant.
4.5. Sanger Validation
Variants carried by prioritized genes were validated by Sanger sequencing, next to polymerase chain reaction (PCR) amplification. Primer sequences and PCR conditions are available upon request. Sanger sequencing was carried out using the BigDyeTerminator© v3.1 Cycle Sequencing Kit chemistry and run on a 3130xl Genetic Analyzer (Applied Biosystems, Thermo Fisher Scientific). Moreover, all variants here considered were further searched in an in-house exome-control dataset obtained by WES data, collected on a cohort of 10 Caucasian healthy subjects, heterogeneous for sex and age. The healthy condition was confirmed by computed tomography.
Patients agreed to be enrolled in the study and their informed consents were obtained. The manuscript does not contain information attributable to their identity. The study involves human participants and was approved by the local Ethics Committee “A.O.U. G. Martino”, N.11/2011 date of approval: 14 December.2011.
5. Conclusions
As knowledge on bAVM is still very elusive, we recruited a group of patients affected by sporadic bAVM and performed WES analysis. Clustering of genes which were affected by rare variants highlighted cytoskeleton impairment as well as defective ion conduction in endothelial cells. Therefore, we hypothesize perturbations at these pathways as possible mechanisms involved in bAVM pathogenesis. We prioritized genes more likely linked to lesion development as FBN2, TAB1, NCoR2, SLIT2, RNF111, CAMK2B, EPHA2, and EPHB2. Although to date no correlation has been reported between gene mutations and clinical phenotype, further characterization of pathways involved in bAVM development could provide a valid criterion to relate molecular features to clinical presentation. In particular, lesion site, bleeding risk, and patient outcome could represent valid prognostic factors linked to patient genotype.
Supplementary Materials
Supplementary materials are available at https://www.mdpi.com/1422-0067/21/12/4321/s1. Table S1: AVM Sample Reports of WES Analysis, Table S2: Selected Variants, Table S3: ClueGO Pathways, Table S4: ToppGene Results. Table S5.: STRING Results.
Author Contributions
Conceptualization, C.S.; methodology, C.S., L.D. and C.T.; investigation: M.L. and F.G.; resources: E.M. and A.A.C.; data curation: L.D.; writing—original draft preparation, C.S.; writing—review and editing, A.S.; supervision, R.D. All authors have read and agreed the final version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Winkler, E.A.; Birk, H.; Burkhardt, J.K.; Chen, X.; Yue, J.K.; Guo, D.; Rutledge, W.C.; Lasker, G.F.; Partow, C.; Tihan, T.; et al. Reductions in Brain Pericytes Are Associated With Arteriovenous Malformation Vascular Instability. J. Neurosurg. 2018, 129, 1464–1474. [Google Scholar] [CrossRef] [PubMed]
- Ozpinar, A.; Mendez, G.; Abla, A.A. Epidemiology, genetics, pathophysiology, and prognostic classifications of cerebral arteriovenous malformations. Handb. Clin. Neurol. 2017, 143, 5–13. [Google Scholar] [PubMed]
- Zhang, D.; Zhou, F.; Zhao, X.; Bao, B.; Chen, J.; Yang, J. Endoglin is a conserved regulator of vasculogenesis in zebrafish—Implications for hereditary haemorrhagic telangiectasia. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef] [PubMed]
- Crist, A.M.; Lee, A.R.; Patel, N.R.; Westhoff, D.E.; Meadows, S.M. Vascular Deficiency of Smad4 Causes Arteriovenous Malformations: A Mouse Model of Hereditary Hemorrhagic Telangiectasia. Angiogenesis 2018, 21, 363–380. [Google Scholar] [CrossRef]
- Wooderchak-Donahue, W.L.; McDonald, J.; O’Fallon, B.; Upton, P.D.; Li, W.; Roman, B.L.; Young, S.; Plant, P.; Fülöp, G.T.; Langa, C.; et al. BMP9 Mutations Cause a Vascular-Anomaly Syndrome With Phenotypic Overlap With Hereditary Hemorrhagic Telangiectasia. Am. J. Hum. Genet. 2013, 93, 530–537. [Google Scholar] [CrossRef]
- Sasahara, A.; Kasuya, H.; Akagawa, H.; Ujiie, H.; Kubo, O.; Sasaki, T.; Onda, H.; Sakamoto, Y.; Krischek, B.; Hori, T.; et al. Increased expression of ephrin A1 in brain arteriovenous malformation: DNA microarray analysis. Neurosurg. Rev. 2007, 30, 299–305. [Google Scholar] [CrossRef]
- Bai, J.; Wang, Y.J.; Liu, L.; Zhao, Y.L. Ephrin B2 and EphB4 selectively mark arterial and venous vessels in cerebral arteriovenous malformation. J. Int. Med. Res. 2014, 42, 405–415. [Google Scholar] [CrossRef]
- Fang, J.; Hirschi, K. Molecular regulation of arteriovenous endothelial cell specification. F1000Research 2019, 8, F1000 Faculty Rev-1208. [Google Scholar] [CrossRef]
- Vaser, R.; Adusumalli, S.; Leng, S.N.; Sikic, M.; Ng, P.C. SIFT missense predictions for genomes. Nat. Protoc. 2016, 11, 1–9. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef]
- Chen, X.; Liu, Y.; Zhou, S.; Nie, S.; Lin, Z.; Zhou, C.; Sun, J.; Gao, X.; Huang, Y. Methylation of the CDKN2A Gene Increases the Risk of Brain Arteriovenous Malformations. J. Mol. Neurosci. 2019, 69, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.M.; Surendran, S.; Abraham, M.; Rajavelu, A.; Kartha, C.C. Genetic and epigenetic mechanisms in the development of arteriovenous malformations in the brain. Clin. Epigenetics 2016, 8, 78. [Google Scholar] [CrossRef] [PubMed]
- Tual-Chalot, S.; Garcia-Collado, M.; Redgrave, R.E.; Singh, E.; Davison, B.; Park, C.; Lin, H.; Luli, S.; Jin, Y.; Wang, Y.; et al. Loss of Endothelial Endoglin Promotes High-Output Heart Failure Through Peripheral Arteriovenous Shunting Driven by VEGF Signaling. Circ. Res. 2020, 126, 243–257. [Google Scholar] [CrossRef] [PubMed]
- Delev, D.; Pavlova, A.; Grote, A.; Boström, A.; Höllig, A.; Schramm, J.; Fimmers, R.; Oldenburg, J.; Simon, M. NOTCH4 gene polymorphisms as potential risk factors for brain arteriovenous malformation development and hemorrhagic presentation. J. Neurosurg. 2017, 126, 1552–1559. [Google Scholar] [CrossRef]
- Goss, J.A.; Huang, A.Y.; Smith, E.; Konczyk, D.J.; Smits, P.J.; Sudduth, C.L.; Stapleton, C.; Patel, A.; Alexandrescu, S.; Warman, M.L.; et al. Somatic mutations in intracranial arteriovenous malformations. PLoS ONE 2019, 14, e0226852. [Google Scholar] [CrossRef]
- Al-Olabi, L.; Polubothu, S.; Dowsett, K.; Andrews, K.A.; Stadnik, P.; Joseph, A.P.; Knox, R.; Pittman, A.; Clark, G.; Baird, W.; et al. Mosaic RAS/MAPK Variants Cause Sporadic Vascular Malformations Which Respond to Targeted Therapy. J. Clin. Investig. 2018, 128, 1496–1508. [Google Scholar] [CrossRef]
- Priemer, D.S.; Vortmeyer, A.O.; Zhang, S.; Chang, H.Y.; Curless, K.L.; Cheng, L. Activating KRAS mutations in arteriovenous malformations of the brain: Frequency and clinicopathologic correlation. Hum. Pathol. 2019, 89, 33–39. [Google Scholar] [CrossRef]
- Davis, M.R.; Andersson, R.; Severin, J.; de Hoon, M.; Bertin, N.; Baillie, J.K.; Kawaji, H.; Sandelin, A.; Forrest, A.R.; Summers, K.M. FANTOM Consortium: Transcriptional profiling of the human fibrillin/LTBP gene family, key regulators of mesenchymal cell functions. Mol. Genet. Metab. 2014, 112, 73–83. [Google Scholar] [CrossRef]
- Doetschman, T.; Barnett, J.V.; Runyan, R.B.; Camenisch, T.D.; Heimark, R.L.; Granzier, H.L.; Conway, S.J.; Azhar, M. Transforming growth factor beta signaling in adult cardiovascular diseases and repair. Cell Tissue Res. 2012, 347, 203–223. [Google Scholar] [CrossRef]
- Vézina, A.; Charfi, C.; Zgheib, A.; Annabi, B. Cerebrovascular Angiogenic Reprogramming upon LRP1 Repression: Impact on Sphingosine-1-Phosphate-Mediated Signaling in Brain Endothelial Cell Chemotactism. Mol. Neurobiol. 2018, 55, 3551–3563. [Google Scholar] [CrossRef]
- Nakajima, C.; Haffner, P.; Goerke, S.M.; Zurhove, K.; Adelmann, G.; Frotscher, M.; Herz, J.; Bock, H.H.; May, P. The lipoprotein receptor LRP1 modulates sphingosine-1-phosphate signaling and is essential for vascular development. Development 2014, 141, 4513–4525. [Google Scholar] [CrossRef] [PubMed]
- Grimsey, N.J.; Lin, Y.; Narala, R.; Rada, C.C.; Mejia-Pena, H.; Trejo, J. G protein-coupled receptors activate p38 MAPK via a non-canonical TAB1-TAB2- and TAB1-TAB3-dependent pathway in endothelial cells. J. Biol. Chem. 2019, 294, 5867–5878. [Google Scholar] [CrossRef] [PubMed]
- Sylvain-Prévost, S.; Ear, T.; Simard, F.A.; Fortin, C.F.; Dubois, C.M.; Flamand, N.; McDonald, P.P. Activation of TAK1 by Chemotactic and Growth Factors, and Its Impact on Human Neutrophil Signaling and Functional Responses. J. Immunol. 2015, 195, 5393–5403. [Google Scholar] [CrossRef] [PubMed]
- Di, Y.; Li, S.; Wang, L.; Zhang, Y.; Dorf, M.E. Homeostatic interactions between MEKK3 and TAK1 involved in NF-kappaB signaling. Cell. Signal. 2008, 20, 705–713. [Google Scholar] [CrossRef]
- Noshiro, S.; Mikami, T.; Kataoka-Sasaki, Y.; Sasaki, M.; Hashi, K.; Ohtaki, S.; Wanibuchi, M.; Mikuni, N.; Kocsis, J.D.; Honmou, O. Biological relevance of tissue factor and IL-6 in arteriovenous malformations. Neurosurg. Rev. 2017, 40, 359–367. [Google Scholar] [CrossRef]
- Shoemaker, L.D.; Fuentes, L.F.; Santiago, S.M.; Allen, B.M.; Cook, D.J.; Steinberg, G.K.; Chang, S.D. Human brain arteriovenous malformations express lymphatic-associated genes. Ann. Clin. Transl. Neurol. 2014, 1, 982–995. [Google Scholar] [CrossRef]
- Murphy, P.A.; Kim, T.N.; Huang, L.; Nielsen, C.M.; Lawton, M.T.; Adams, R.H.; Schaffer, C.B.; Wang, R.A. Constitutively active Notch4 receptor elicits brain arteriovenous malformations through enlargement of capillary-like vessels. Proc. Natl. Acad. Sci. USA 2014, 111, 18007–18012. [Google Scholar] [CrossRef]
- Hou, Y.; Shin, Y.J.; Han, E.J.; Choi, J.S.; Park, J.M.; Cha, J.H.; Choi, J.Y.; Lee, M.Y. Distribution of vascular endothelial growth factor receptor-3/Flt4 mRNA in adult rat central nervous system. J. Chem. Neuroanat. 2011, 42, 56–64. [Google Scholar] [CrossRef]
- Chaqour, B. Caught between a “Rho” and a hard place: Are CCN1/CYR61 and CCN2/CTGF the arbiters of microvascular stiffness? J. Cell Commun. Signal. 2019. [Google Scholar] [CrossRef]
- Adams, R.H.; Wilkinson, G.A.; Weiss, C.; Diella, F.; Gale, N.W.; Deutsch, U.; Risau, W.; Klein, R. Roles of ephrinB ligands and EphB receptors in cardiovascular development: Demarcation of arterial/venous domains, vascular morphogenesis, and sprouting angiogenesis. Genes Dev. 1999, 13, 295–306. [Google Scholar] [CrossRef]
- Liu, H.; Devraj, K.; Möller, K.; Liebner, S.; Hecker, M.; Korff, T. EphrinB-mediated reverse signalling controls junctional integrity and pro-inflammatory differentiation of endothelial cells. Thromb. Haemost. 2014, 112, 151–163. [Google Scholar] [CrossRef] [PubMed]
- Zhou, N.; Zhao, W.D.; Liu, D.X.; Liang, Y.; Fang, W.G.; Li, B.; Chen, Y.H. Inactivation of EphA2 promotes tight junction formation and impairs angiogenesis in brain endothelial cells. Microvasc. Res. 2011, 82, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Doronzo, G.; Astanina, E.; Corà, D.; Chiabotto, G.; Comunanza, V.; Noghero, A.; Neri, F.; Puliafito, A.; Primo, L.; Spampanato, C.; et al. TFEB controls vascular development by regulating the proliferation of endothelial cells. EMBO J. 2019, 38, e98250. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Oh, W.J.; Gaiano, N.; Yoshida, Y.; Gu, C. Semaphorin 3E-Plexin-D1 signaling regulates VEGF function in developmental angiogenesis via a feedback mechanism. Genes Dev. 2011, 25, 1399–1411. [Google Scholar] [CrossRef] [PubMed]
- Gu, C.; Yoshida, Y.; Livet, J.; Reimert, D.V.; Mann, F.; Merte, J.; Henderson, C.E.; Jessell, T.M.; Kolodkin, A.L.; Ginty, D.D. Semaphorin 3E and plexin-D1 control vascular pattern independently of neuropilins. Science 2005, 307, 265–268. [Google Scholar] [CrossRef]
- Li, G.J.; Yang, Y.; Yang, G.K.; Wan, J.; Cui, D.L.; Ma, Z.H.; Du, L.J.; Zhang, G.M. Slit2 suppresses endothelial cell proliferation and migration by inhibiting the VEGF-Notch signaling pathway. Mol. Med. Rep. 2017, 15, 1981–1988. [Google Scholar] [CrossRef]
- Zhang, S.; Kim, J.Y.; Xu, S.; Liu, H.; Yin, M.; Koroleva, M.; Guo, J.; Pei, X.; Jin, Z.G. Endothelial-specific YY1 governs sprouting angiogenesis through directly interacting with RBPJ. Proc. Natl. Acad. Sci. USA 2020, 117, 4792–4801. [Google Scholar] [CrossRef]
- Koinuma, D.; Shinozaki, M.; Komuro, A.; Goto, K.; Saitoh, M.; Hanyu, A.; Ebina, M.; Nukiwa, T.; Miyazawa, K.; Imamura, T.; et al. Arkadia amplifies TGF-beta superfamily signalling through degradation of Smad7. EMBO J. 2003, 22, 6458–6470. [Google Scholar] [CrossRef]
- Zhou, P.; Wan, X.; Zou, Y.; Chen, Z.; Zhong, A. Transforming growth factor beta (TGF-β) is activated by the CtBP2-p300-AP1 transcriptional complex in chronic renal failure. Int. J. Biol. Sci. 2020, 16, 204–215. [Google Scholar] [CrossRef]
- Villa, C.; Colombo, G.; Meneghini, S.; Gotti, C.; Moretti, M.; Ferini-Strambi, L.; Chisci, E.; Giovannoni, R.; Becchetti, A.; Combi, R. CHRNA2 and Nocturnal Frontal Lobe Epilepsy: Identification and Characterization of a Novel Loss of Function Mutation. Front. Mol. Neurosci. 2019, 12, 17. [Google Scholar] [CrossRef]
- Heeschen, C.; Weis, M.; Aicher, A.; Dimmeler, S.; Cooke, J.P. A novel angiogenic pathway mediated by non-neuronal nicotinic acetylcholine receptors. J. Clin. Investig. 2002, 110, 527–536. [Google Scholar] [CrossRef]
- Peghaire, C.; Bats, M.L.; Sewduth, R.; Jeanningros, S.; Jaspard, B.; Couffinhal, T.; Duplàa, C.; Dufourcq, P. Fzd7 (Frizzled-7) Expressed by Endothelial Cells Controls Blood Vessel Formation Through Wnt/β-Catenin Canonical Signaling. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2369–2380. [Google Scholar] [CrossRef]
- Liu, W.; Rui, H.; Wang, J.; Lin, S.; He, Y.; Chen, M.; Li, Q.; Ye, Z.; Zhang, S.; Chan, S.C.; et al. Axin is a scaffold protein in TGF-beta signaling that promotes degradation of Smad7 by Arkadia. EMBO J. 2006, 25, 1646–1658. [Google Scholar] [CrossRef] [PubMed]
- Jensen, L.D.; Hot, B.; Ramsköld, D.; Germano, R.F.V.; Yokota, C.; Giatrellis, S.; Lauschke, V.M.; Hubmacher, D.; Li, M.X.; Hupe, M.; et al. Disruption of the Extracellular Matrix Progressively Impairs Central Nervous System Vascular Maturation Downstream of β-Catenin Signaling. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1432–1447. [Google Scholar] [CrossRef] [PubMed]
- Eisa-Beygi, S.; Benslimane, F.M.; El-Rass, S.; Prabhudesai, S.; Abdelrasoul, M.K.A.; Simpson, P.M.; Yalcin, H.C.; Burrows, P.E.; Ramchandran, R. Characterization of Endothelial Cilia Distribution During Cerebral-Vascular Development in Zebrafish (Danio rerio). Arterioscler. Thromb. Vasc. Biol. 2018, 38, 2806–2818. [Google Scholar] [CrossRef] [PubMed]
- Noren, D.P.; Chou, W.H.; Lee, S.H.; Qutub, A.A.; Warmflash, A.; Wagner, D.S.; Popel, A.S.; Levchenko, A. Endothelial cells decode VEGF-mediated Ca2+ signaling patterns to produce distinct functional responses. Sci. Signal. 2016, 9, ra20. [Google Scholar] [CrossRef] [PubMed]
- Yamamura, H.; Suzuki, Y.; Yamamura, H.; Asai, K.; Imaizumi, Y. Hypoxic stress up-regulates Kir2.1 expression and facilitates cell proliferation in brain capillary endothelial cells. Biochem. Biophys. Res. Commun. 2016, 476, 386–392. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, D.; Kito, H.; Yamamoto, S.; Ohya, S.; Yamamura, H.; Asai, K.; Imaizumi, Y. Contribution of K(ir)2 potassium channels to ATP-induced cell death in brain capillary endothelial cells and reconstructed HEK293 cell model. Am. J. Physiol. Cell. Physiol. 2011, 300, C75–C86. [Google Scholar] [CrossRef]
- Hauer, A.J.; Kleinloog, R.; Giuliani, F.; Rinkel, G.J.E.; de Kort, G.A.; Berkelbach van der Sprenkel, J.W.; van der Zwan, A.; Gosselaar, P.H.; van Rijen, P.C.; de Boer-Bergsma, J.J.; et al. RNA-Sequencing Highlights Inflammation and Impaired Integrity of the Vascular Wall in Brain Arteriovenous Malformations. Stroke 2020, 51, 268–274. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Wang, K.; Zhao, S.; Liu, B.; Zhang, Q.; Li, Y.; Liu, J.; Shen, Y.; Ding, X.; Lin, J.; Wu, Y.; et al. Perturbations of BMP/TGF-β and VEGF/VEGFR signalling pathways in non-syndromic sporadic brain arteriovenous malformations (BAVM). J. Med. Genet. 2018, 55, 675–684. [Google Scholar] [CrossRef] [PubMed]
- Scimone, C.; Donato, L.; Alafaci, C.; Granata, F.; Rinaldi, C.; Longo, M.; D’Angelo, R.; Sidoti, A. High-Throughput Sequencing to Detect Novel Likely Gene-Disrupting Variants in Pathogenesis of Sporadic Brain Arteriovenous Malformations. Front. Genet. 2020, 11, 146. [Google Scholar] [CrossRef] [PubMed]
- Guijarro-Muñoz, I.; Cuesta, A.M.; Alvarez-Cienfuegos, A.; Geng, J.G.; Alvarez-Vallina, L.; Sanz, L. The axonal repellent Slit2 inhibits pericyte migration: Potential implications in angiogenesis. Exp. Cell Res. 2012, 318, 371–378. [Google Scholar] [CrossRef]
- Harde, E.; Nicholson, L.; Furones Cuadrado, B.; Bissen, D.; Wigge, S.; Urban, S.; Segarra, M.; Ruiz de Almodóvar, C.; Acker-Palmer, A. EphrinB2 regulates VEGFR2 during dendritogenesis and hippocampal circuitry development. Elife 2019, 8, e49819. [Google Scholar] [CrossRef]
- Spetzler, R.F.; Martin, N.A. A Proposed Grading System for Arteriovenous Malformations. J. Neurosurg. 1986, 65, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Garrison, E.; Marth, G. Haplotype-based variant detection from short-read sequencing. arXiv 2012, arXiv:1207.3907v2. [Google Scholar]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. BioRxiv 2019, 531210. [Google Scholar] [CrossRef]
- 1000 Genomes Project Consortium; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.H.; Pagès, F.; Trajanoski, Z.; Galon, J. ClueGO: A cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Bardes, E.E.; Aronow, B.J.; Jegga, A.G. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009, 37, W305–W311. [Google Scholar] [CrossRef] [PubMed]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).