Calcium Entry through TRPV1: A Potential Target for the Regulation of Proliferation and Apoptosis in Cancerous and Healthy Cells





Abstract
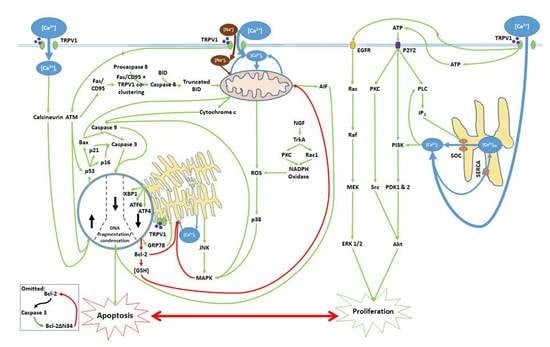
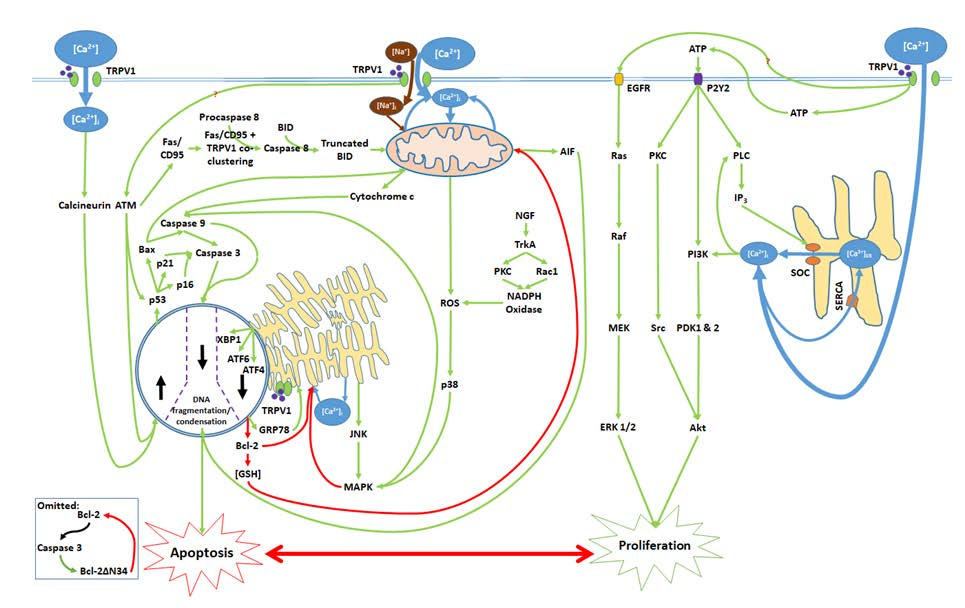
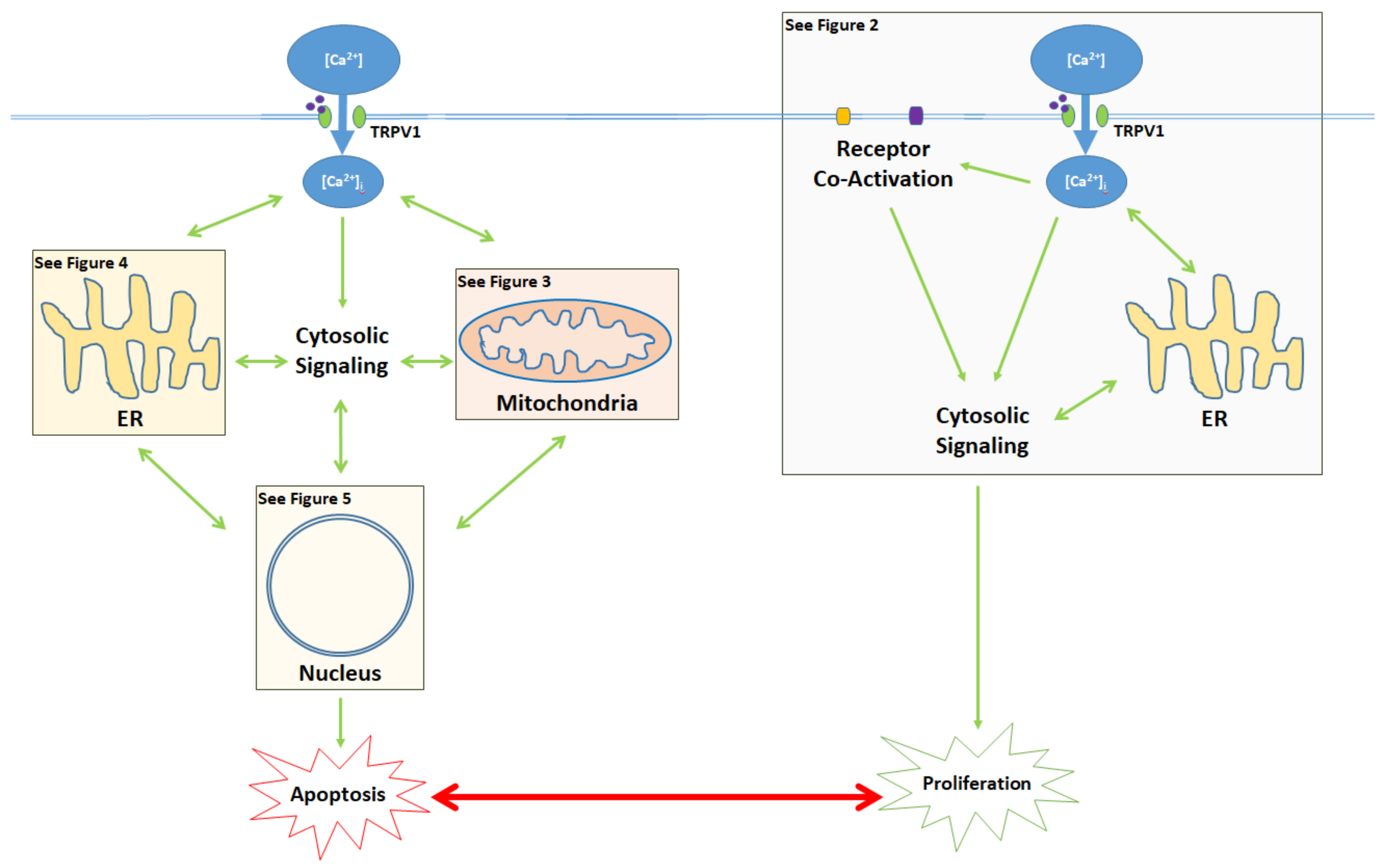
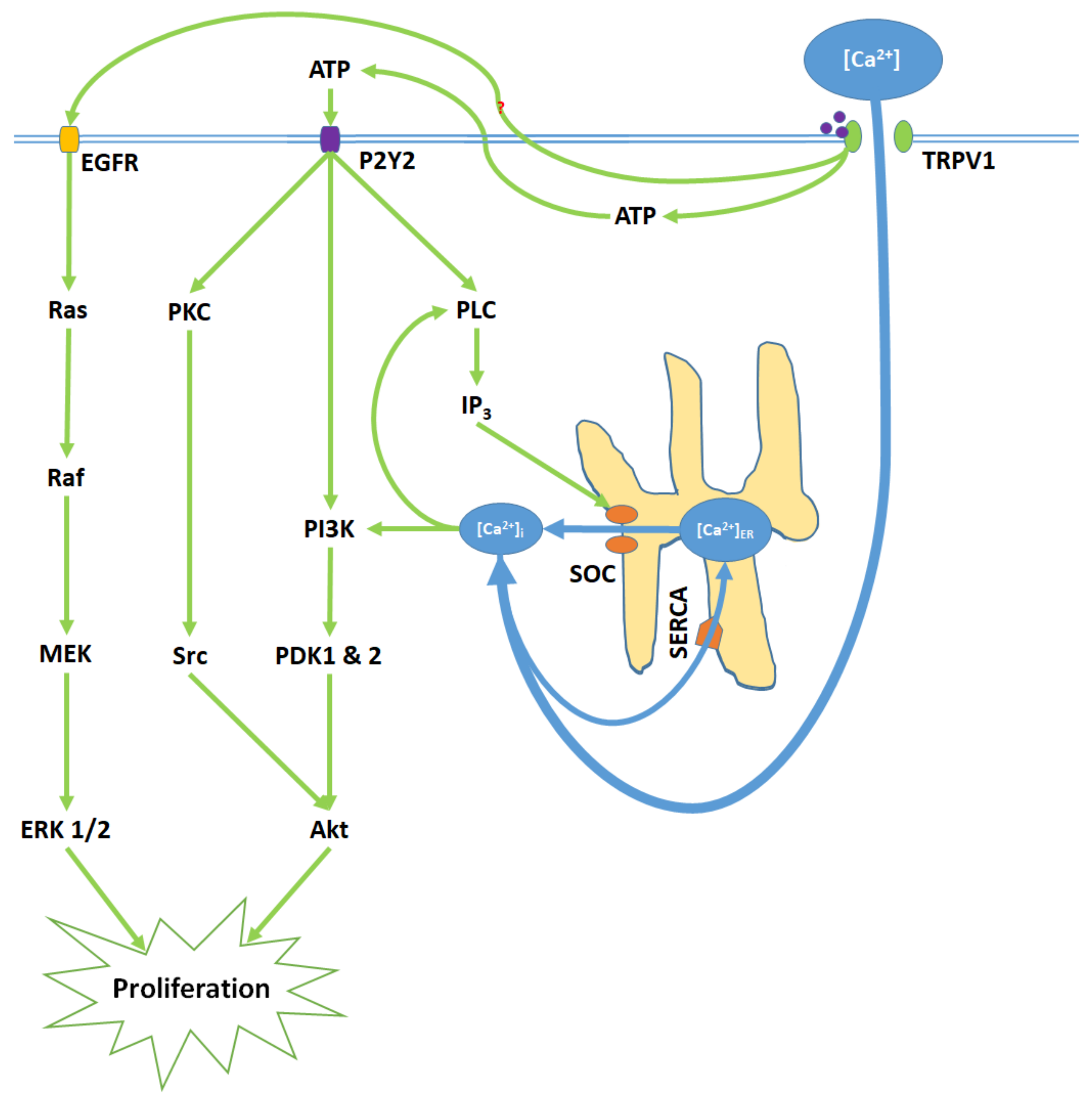
1. [Ca2+]i and the Critical Balance between Apoptosis and Proliferation
2. Expression of TRPV1 in Cancerous and Healthy Tissues
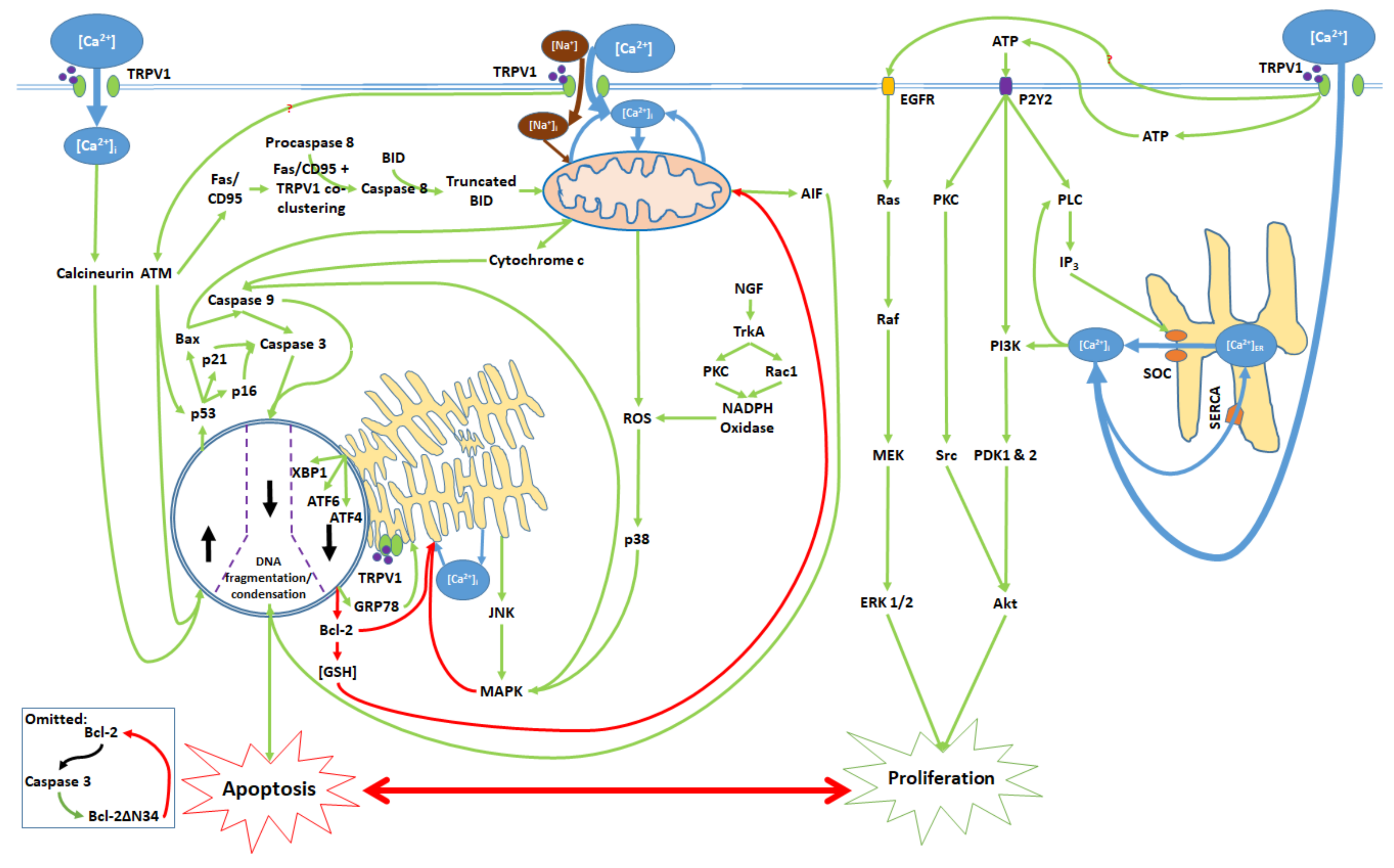
3. Balance Between Apoptosis and Proliferation Mediated by TRPV1
4. TRPV1-Mediated Proliferation
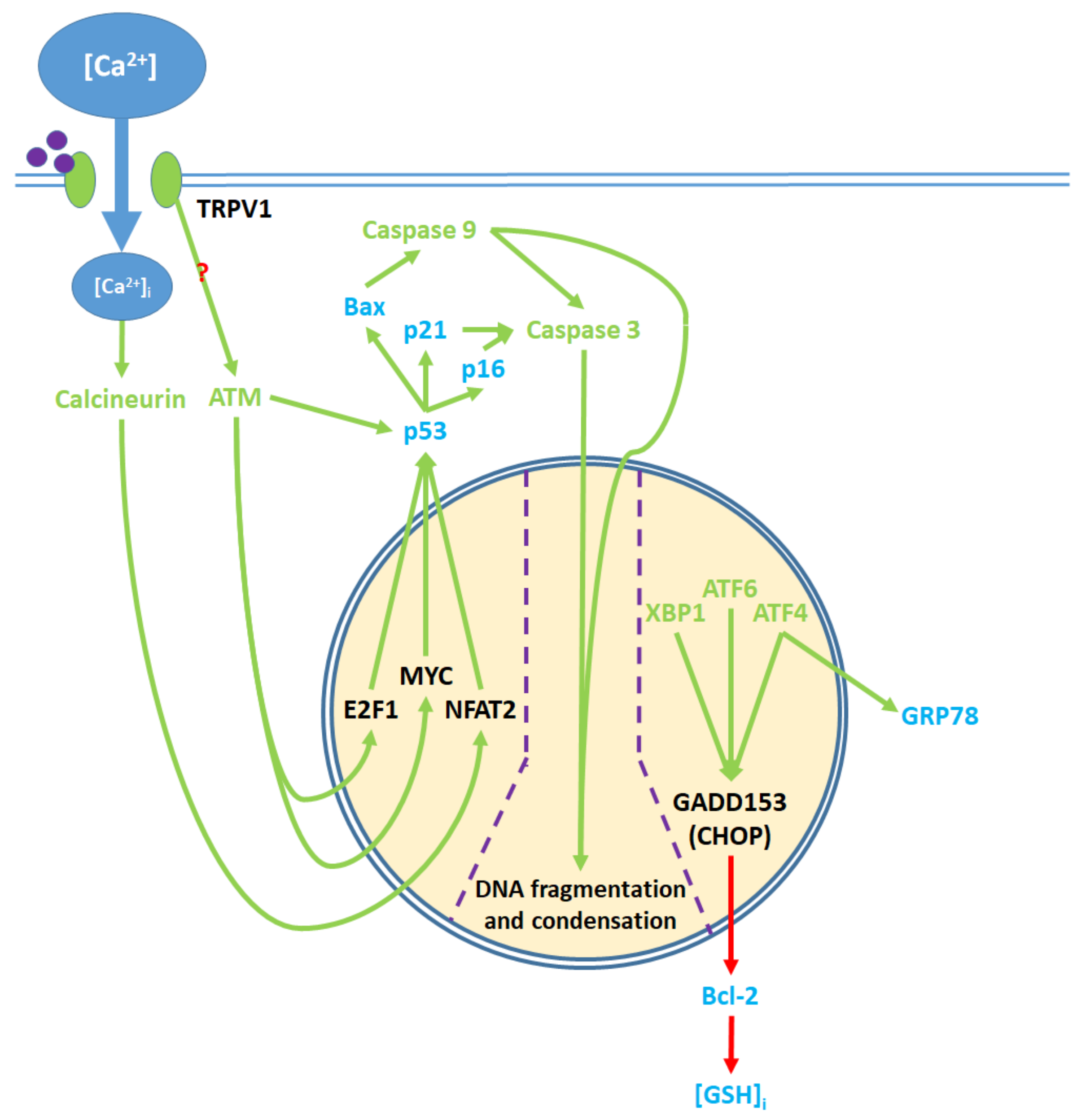
5. The Apoptotic Pathway and Upstream Cytosolic Effects
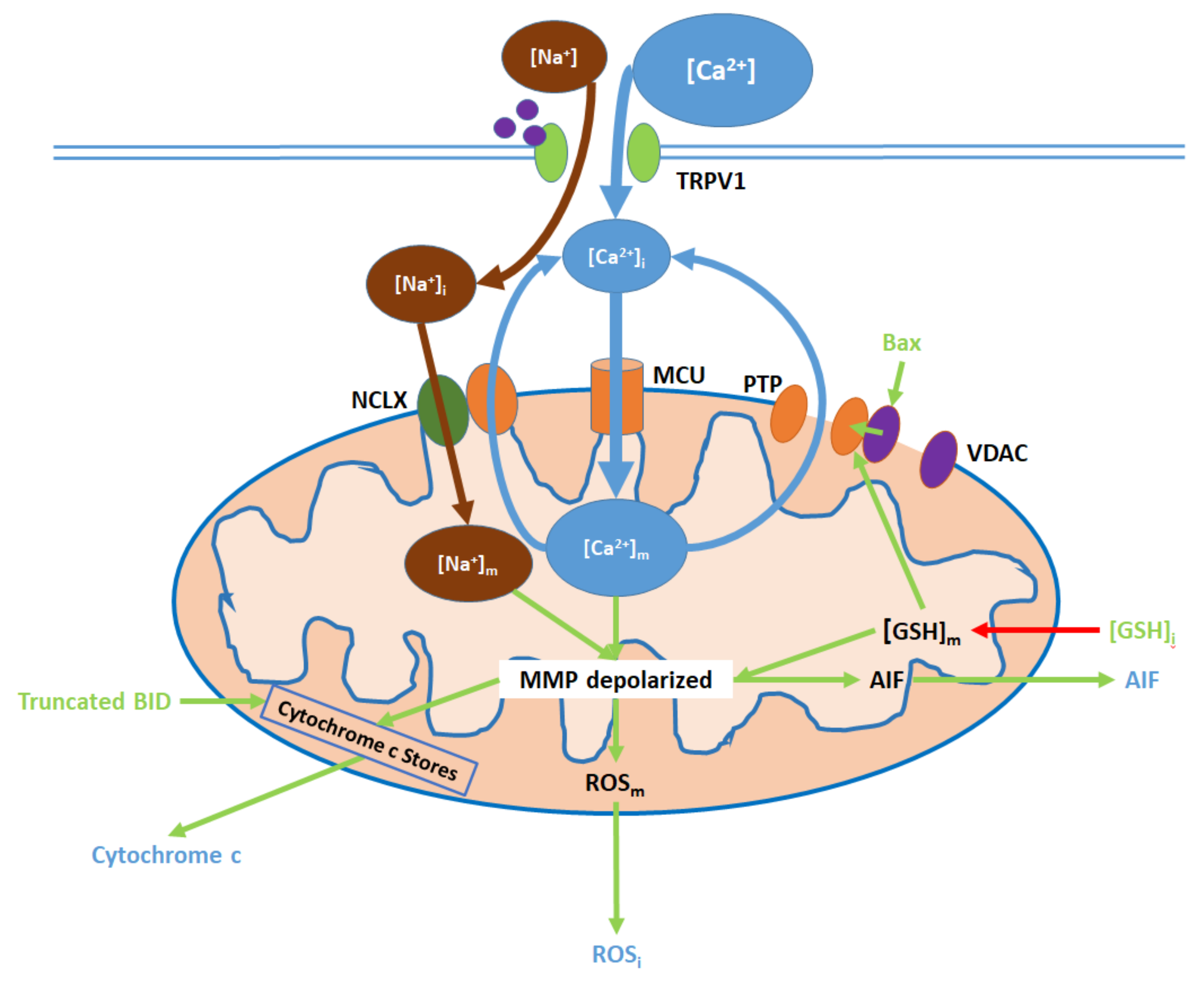
6. Mitochondrial Pathway
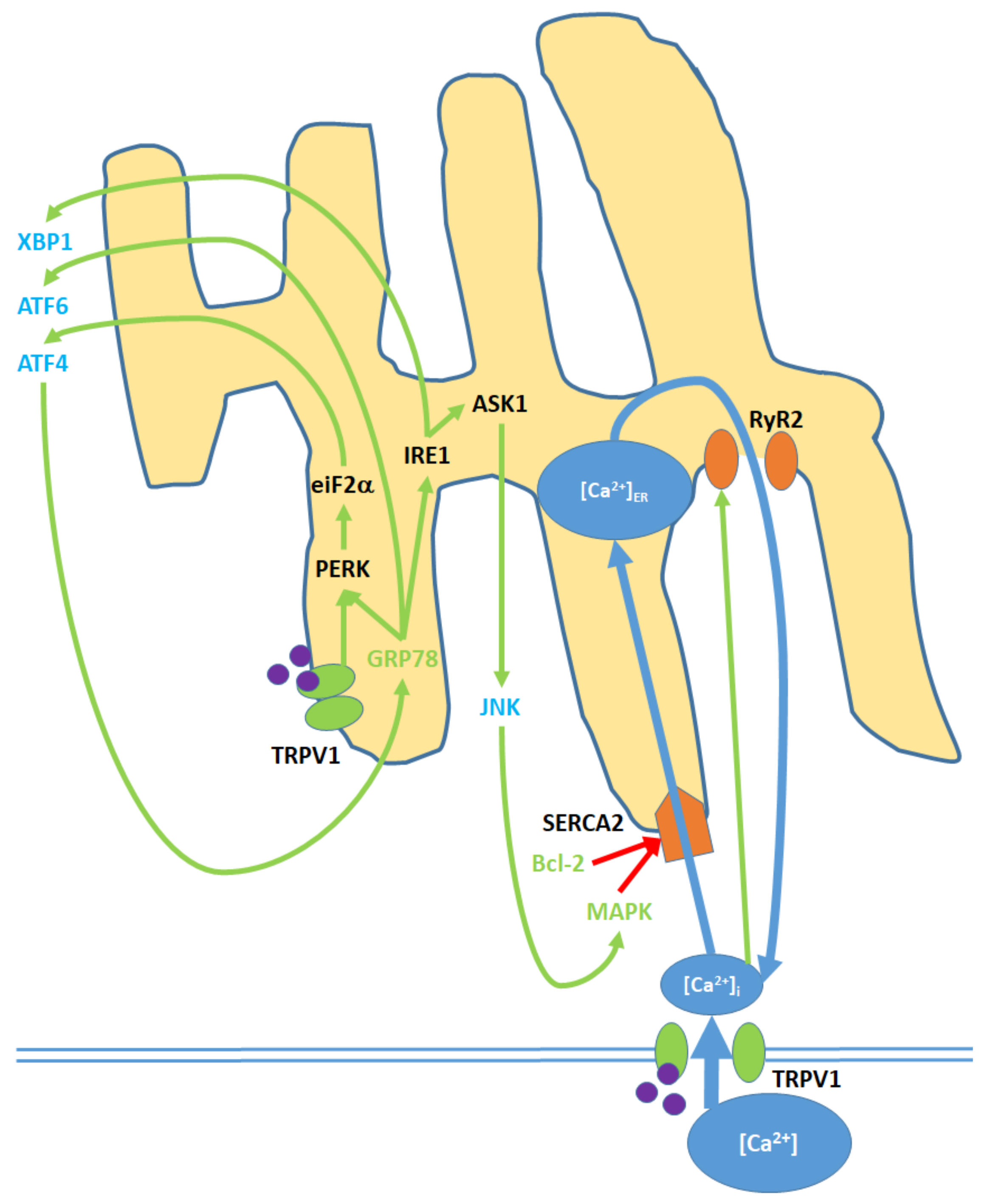
7. Endoplasmic Reticulum (ER) Pathway
8. Nuclear and Downstream Cytosolic Effects
9. TRPV1 as a Potential Target for Anti-Cancer Therapies
Recent Trends and Innovations in Oncologic Approaches Targeting TRPV1
10. Conclusions and Outlook
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 13-HODE | 13-HydroxyOctaDecadEinoic acid |
| 2-AG | 2-ArachidonylGlycerol |
| AEA | Anandamide |
| Akt | Akt serine/threonine protein kinase |
| AIF | Apoptosis Inducing Factor |
| ASMC | Airway Smooth Muscle Cells |
| ATF1 | Activating Transcription Factor 1 |
| ATF3 | Activating Transcription Factor 3 |
| ATF4 | Activating Transcription Factor 4 |
| ATF6 | Activating Transcription Factor 6 |
| ATM | ATM serine-threonine kinase |
| ATP | Adenosine TriPhosphate |
| Bax | Bcl-2 associated X protein |
| Bcl-2 | B-cell lymphoma 2 |
| BID | BH3 Interacting-domain Death agonist |
| BPA | BisPhenol A |
| Ca2+ | Calcium |
| [Ca2+]i | Intracellular Calcium Concentration |
| [Ca2+]m | Mitochondrial Calcium Concentration |
| [Ca2+]ER | Endoplasmic Reticulum Calcium Concentration |
| CaM | CalModulin |
| CB1 | CannaBinoid receptor type 1 |
| CB2 | CannaBinoid receptor type 2 |
| CBD | CannaBiDiol |
| Cdk | Cyclin dependent kinase |
| CGRP | Calcitonin Gene-Related Peptide |
| CGRPR | Calcitonin Gene-Related Peptide Receptor |
| CRC | ColoRectal Cancer |
| DMBA | 7,12-DiMethylBenz[a]Anthracene |
| DNA | DeoxyriboNucleic Acid |
| DRG | Dorsal Root Ganglion |
| E2F1 | E2F transcription factor 1 |
| EA | ElectroAcupuncture |
| ECFC | Endothelial Colony Forming Cells |
| EET | EpoxyEicosaTrienoic acid |
| EGFR | Epidermal Growth Factor Receptor |
| eIF2 | eukaryotic Initiation Factor 2 |
| ER | Endoplasmic Reticulum |
| ERK | Extracellular signal-Regulated Kinase |
| FADD | Fas-Associated protein with Death Domain |
| Fas/CD95 | Fas cell surface death receptor/Cluster of Differentiation 95 |
| GADD153; DDIT3; CHOP | DNA-Damage Inducible Transcript 3; C/EBP HOmologous Protein |
| GSH | Glutathione |
| [GSH]i | Intracellular Glutathione Concentration |
| GRP78; BiP | Binding immunoglobulin Protein |
| HCEC | Human Corneal Epithelial Cells |
| HP | Hypericum perforatum |
| ICR | Institute for Cancer Research |
| IL-1β | InterLeukin 1β |
| IL-6 | InterLeukin 6 |
| IL-8 | InterLeukin 8 |
| IP3 | Inositol triPhosphate |
| IRE1 | Inositol-Requiring Enzyme 1 |
| IRTX | IodoResiniferaToXin |
| JNK | c-Jun N-terminal Kinase |
| MAPK | Mitogen-Activated Protein Kinase |
| MCU | Mitochondrial Calcium Uniporter |
| Mdm2 | Mouse double minute 2 homolog |
| MEK | MAPK-ERK Kinase |
| MET | (R)-METhanandamide |
| mRNA | messenger RiboNucleic Acid |
| MYC | Myc proto-oncogene |
| Na+ | Sodium |
| [Na+]i | Intracellular Sodium Concentration |
| [Na+]m | Mitochondrial Sodium Concentration |
| NADA | N-Arachidonoyl DopAmine |
| NADPH | Nicotinamide Adenine Dinucleotide PHosphate |
| NFAT2 | Nuclear Factor of Activated T-cells 2 |
| NF-κB | Nuclear Factor-Kappa light chain enhancer of activated B cells |
| NGF | Nerve Growth Factor |
| NHA | Normal Human Astrocytes |
| NHBE | Normal Human Bronchial Epithelial cells |
| NHEM | Normal Human Epidermal Melanocytes |
| NHUC | Normal Human Urothelial Cells |
| NIR | Near InfraRed |
| NK1R | NeuroKinin 1 Receptor |
| NPC | Neural Progenitor Cells |
| (m)NPC-CM | (murine) Neural Progenitor Cell-Culture Media |
| p16; CDKN2A | Cyclin-Dependent Kinase Inhibitor 2A |
| p21; CDKN1(A) | Cyclin-Dependent Kinase Inhibitor 1(A) |
| P2Y2 | P2Y purinoceptor 2 |
| p38 (MAPK) | p38 (Mitogen Activated Protein Kinase) |
| p53 | tumor protein p53 |
| PAM | Positive Allosteric Modulator |
| PCOS | PolyCystic Ovary Syndrome |
| PDK1 | Phosphoinositide Dependent Kinase 1 |
| PEG | PolyEthylene Glycol |
| PKC | Protein Kinase C |
| PLC | PhosphoLipase C |
| PI3K | Phosphoinositide-3-Kinase |
| PS | PhosphatidylSerine |
| PTEN | Phosphatase and TENsion homolog |
| PTP | Permeability Transition Pore |
| PTZ | PentyleneTetraZole |
| Rac1 | Ras-related C3 botulinum toxin substrate 1 |
| Raf | Rapidly accelerated fibrosarcoma |
| Ras | Rat sarcoma |
| RCC | Renal Cell Carcinoma |
| RGC | Retinal Ganglion Cells |
| ROS | Reactive Oxygen Species |
| ROSi | Intracellular Reactive Oxygen Species |
| ROSm | Mitochondrial Reactive Oxygen Species |
| RR | Ruthenium Red |
| RTX | ResiniferaToXin |
| RyR2 | Ryanodine Receptor 2 |
| SAEC | Small Airway Epithelial Cells |
| SCID-NOD | Severe Combined ImmunoDeficiency-NonObese Diabetic |
| SERCA | Sarco/Endoplasmic Reticulum Calcium ATPase |
| SGZ | SubGranular Zone |
| SMF | Static Magnetic Field |
| SNI | Sciatic Nerve Injury |
| SNP | Sodium NitroPrusside |
| SOC | Store-Operated Channel |
| SR | Sarcoplasmic Reticulum |
| Src | proto-oncogene tyrosine-protein kinase Src |
| SST | SomatoStaTin |
| sst4 | somatostatin receptor 4 |
| SVZ | SubVentricular Zone |
| T-ALL | T-cell Acute Lymphoblastic Leukemia |
| TG | Trigeminal Ganglia |
| TNF-α | Tumor Necrosis Factor α |
| TrkA | Tropomyosin receptor kinase A |
| TRPV1 | Transient Receptor Potential Vanilloid 1 |
| VDAC | Voltage Dependent Anion Channel |
| VGCC | Voltage Gated Calcium Channel |
| XBP1 | X-box Binding Protein 1 |
References
- Cvejic, D.; Selemetjev, S.; Savin, S.; Paunovic, I.; Tatic, S. Changes in the balance between proliferation and apoptosis during the progression of malignancy in thyroid tumours. Eur. J. Histochem. 2009, 53, e8. [Google Scholar] [CrossRef] [PubMed]
- Hao, X.; Du, M.; Bishop, A.E.; Talbot, I.C. Imbalance between proliferation and apoptosis in the development of colorectal carcinoma. Virchows Arch. 1998, 433, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Afanas’ev, V.N.; Korol, B.A.; Mantsygin Yu, A.; Nelipovich, P.A.; Pechatnikov, V.A.; Umansky, S.R. Flow cytometry and biochemical analysis of DNA degradation characteristic of two types of cell death. FEBS Lett. 1986, 194, 347–350. [Google Scholar] [CrossRef]
- Reed, J.C. Mechanisms of apoptosis. Am. J. Pathol. 2000, 157, 1415–1430. [Google Scholar] [CrossRef]
- Green, D.R.; Reed, J.C. Mitochondria and apoptosis. Science 1998, 281, 1309–1312. [Google Scholar] [CrossRef]
- Kuwana, T.; Newmeyer, D.D. Bcl-2-family proteins and the role of mitochondria in apoptosis. Curr. Opin. Cell Biol. 2003, 15, 691–699. [Google Scholar] [CrossRef]
- Orrenius, S.; Zhivotovsky, B.; Nicotera, P. Regulation of cell death: The calcium-apoptosis link. Nat. Rev. Mol. Cell Biol. 2003, 4, 552–565. [Google Scholar] [CrossRef]
- Varghese, E.; Samuel, S.M.; Sadiq, Z.; Kubatka, P.; Liskova, A.; Benacka, J.; Pazinka, P.; Kruzliak, P.; Busselberg, D. Anti-Cancer Agents in Proliferation and Cell Death: The Calcium Connection. Int. J. Mol. Sci. 2019, 20, 17. [Google Scholar] [CrossRef]
- Santulli, G.; Lewis, D.; des Georges, A.; Marks, A.R.; Frank, J. Ryanodine Receptor Structure and Function in Health and Disease. Subcell. Biochem. 2018, 87, 329–352. [Google Scholar] [CrossRef]
- Gambardella, J.; Lombardi, A.; Morelli, M.B.; Ferrara, J.; Santulli, G. Inositol 1,4,5-Trisphosphate Receptors in Human Disease: A Comprehensive Update. J. Clin. Med. 2020, 9, 96. [Google Scholar] [CrossRef]
- Santulli, G.; Nakashima, R.; Yuan, Q.; Marks, A.R. Intracellular calcium release channels: An update. J. Physiol. 2017, 595, 3041–3051. [Google Scholar] [CrossRef]
- Kania, E.; Roest, G.; Vervliet, T.; Parys, J.B.; Bultynck, G. IP3 Receptor-Mediated Calcium Signaling and Its Role in Autophagy in Cancer. Front. Oncol. 2017, 7, 140. [Google Scholar] [CrossRef]
- Shin, D.-H.; Leem, D.-G.; Shin, J.-S.; Kim, J.-I.; Kim, K.-T.; Choi, S.Y.; Lee, M.-H.; Choi, J.-H.; Lee, K.-T. Compound K induced apoptosis via endoplasmic reticulum Ca2+ release through ryanodine receptor in human lung cancer cells. J. Ginseng Res. 2018, 42, 165–174. [Google Scholar] [CrossRef]
- Panner, A.; Wurster, R.D. T-type calcium channels and tumor proliferation. Cell Calcium 2006, 40, 253–259. [Google Scholar] [CrossRef]
- Valerie, N.C.; Dziegielewska, B.; Hosing, A.S.; Augustin, E.; Gray, L.S.; Brautigan, D.L.; Larner, J.M.; Dziegielewski, J. Inhibition of T-type calcium channels disrupts Akt signaling and promotes apoptosis in glioblastoma cells. Biochem. Pharm. 2013, 85, 888–897. [Google Scholar] [CrossRef]
- Cano-Abad, M.A.F.; Villarroya, M.; Garcı‘a, A.G.; Gabilan, N.H.; Lo´pez, M.G. Calcium entry through L-type calcium channels causes mitochondrial disruption and chromaffin cell death. J. Biol. Chem. 2001, 276, 39695–39704. [Google Scholar] [CrossRef]
- Nicotera, P.; Orrenius, S. The role of calcium in apoptosis. Cell Calcium. 1998, 23, 173–180. [Google Scholar] [CrossRef]
- Stewart, T.A.; Yapa, K.T.; Monteith, G.R. Altered calcium signaling in cancer cells. Biochim. Biophys. Acta 2015, 1848, 2502–2511. [Google Scholar] [CrossRef]
- Florea, A.M.; Busselberg, D. Anti-cancer drugs interfere with intracellular calcium signaling. Neurotoxicology 2009, 30, 803–810. [Google Scholar] [CrossRef]
- Raynal, N.J.; Lee, J.T.; Wang, Y.; Beaudry, A.; Madireddi, P.; Garriga, J.; Malouf, G.G.; Dumont, S.; Dettman, E.J.; Gharibyan, V.; et al. Targeting Calcium Signaling Induces Epigenetic Reactivation of Tumor Suppressor Genes in Cancer. Cancer Res. 2016, 76, 1494–1505. [Google Scholar] [CrossRef]
- Clapham, D.E. TRP channels as cellular sensors. Nature 2003, 426, 517–524. [Google Scholar] [CrossRef]
- Huang, J.; Liu, J.; Qiu, L. Transient receptor potential vanilloid 1 promotes EGFR ubiquitination and modulates EGFR/MAPK signalling in pancreatic cancer cells. Cell Biochem. Funct. 2020. [Google Scholar] [CrossRef]
- So, C.L.; Milevskiy, M.J.G.; Monteith, G.R. Transient receptor potential cation channel subfamily V and breast cancer. Lab. Investig. 2020, 100, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Zygmunt, P.M.; Petersson, J.; Andersson, D.A.; Chuang, H.; Sorgard, M.; Di Marzo, V.; Julius, D.; Hogestatt, E.D. Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature 1999, 400, 452–457. [Google Scholar] [CrossRef] [PubMed]
- Caterina, M.J.; Schumacher, M.A.; Tominaga, M.; Rosen, T.A.; Levine, J.D.; Julius, D. The capsaicin receptor: A heat-activated ion channel in the pain pathway. Nature 1997, 389, 816–824. [Google Scholar] [CrossRef] [PubMed]
- Bautista, D.; Julius, D. Fire in the hole: Pore dilation of the capsaicin receptor TRPV1. Nat. Neurosci. 2008, 11, 528–529. [Google Scholar] [CrossRef]
- Satheesh, N.J.; Uehara, Y.; Fedotova, J.; Pohanka, M.; Büsselberg, D.; Kruzliak, P. TRPV currents and their role in the nociception and neuroplasticity. Neuropeptides 2016, 57, 1–8. [Google Scholar] [CrossRef]
- Szallasi, A.; Cruz, F.; Geppetti, P. TRPV1: A therapeutic target for novel analgesic drugs? Trends Mol. Med. 2006, 12, 545–554. [Google Scholar] [CrossRef]
- Christie, S.; Wittert, G.A.; Li, H.; Page, A.J. Involvement of TRPV1 Channels in Energy Homeostasis. Front. Endocrinol. (Lausanne) 2018, 9, 420. [Google Scholar] [CrossRef]
- Jeong, K.Y. Changes in TRPV1-Mediated Physiological Function in Rats Systemically Treated With Capsaicin on the Neonate. Int. J. Mol. Sci. 2020, 21, 3143. [Google Scholar] [CrossRef]
- Inprasit, C.; Lin, Y.-W. TRPV1 Responses in the Cerebellum Lobules V, VIa and VII Using Electroacupuncture Treatment for Inflammatory Hyperalgesia in Murine Model. Int. J. Mol. Sci. 2020, 21, 3312. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.; Carver, C.M.; Mullen, P.; Milne, S.; Lukacs, V.; Shapiro, M.S.; Gamper, N. Local Ca(2+) signals couple activation of TRPV1 and ANO1 sensory ion channels. Sci. Signal. 2020, 13. [Google Scholar] [CrossRef]
- Buch, T.R.H.; Buch, E.A.M.; Boekhoff, I.; Steinritz, D.; Aigner, A. Role of Chemosensory TRP Channels in Lung Cancer. Pharmaceuticals 2018, 11, 90. [Google Scholar] [CrossRef]
- Sappington, R.M.; Sidorova, T.; Long, D.J.; Calkins, D.J. TRPV1: Contribution to retinal ganglion cell apoptosis and increased intracellular Ca2+ with exposure to hydrostatic pressure. Invest. Ophthalmol. Vis. Sci. 2009, 50, 717–728. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zhang, X.; Kuang, H.; Wu, J.; Guo, Y.; Ma, L. Effect of TRPV1 channel on the proliferation and apoptosis in asthmatic rat airway smooth muscle cells. Exp. Lung Res. 2013, 39, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Shirakawa, H.; Yamaoka, T.; Sanpei, K.; Sasaoka, H.; Nakagawa, T.; Kaneko, S. TRPV1 stimulation triggers apoptotic cell death of rat cortical neurons. Biochem. Biophys. Res. Commun. 2008, 377, 1211–1215. [Google Scholar] [CrossRef] [PubMed]
- Stock, K.; Garthe, A.; de Almeida Sassi, F.; Glass, R.; Wolf, S.A.; Kettenmann, H. The capsaicin receptor TRPV1 as a novel modulator of neural precursor cell proliferation. Stem. Cells 2014, 32, 3183–3195. [Google Scholar] [CrossRef]
- Sun, Z.; Han, J.; Zhao, W.; Zhang, Y.; Wang, S.; Ye, L.; Liu, T.; Zheng, L. TRPV1 activation exacerbates hypoxia/reoxygenation-induced apoptosis in H9C2 cells via calcium overload and mitochondrial dysfunction. Int. J. Mol. Sci. 2014, 15, 18362–18380. [Google Scholar] [CrossRef]
- Hu, F.; Sun, W.W.; Zhao, X.T.; Cui, Z.J.; Yang, W.X. TRPV1 mediates cell death in rat synovial fibroblasts through calcium entry-dependent ROS production and mitochondrial depolarization. Biochem. Biophys. Res. Commun. 2008, 369, 989–993. [Google Scholar] [CrossRef]
- Denda, S.; Denda, M.; Inoue, K.; Hibino, T. Glycolic acid induces keratinocyte proliferation in a skin equivalent model via TRPV1 activation. J. Derm. Sci. 2010, 57, 108–113. [Google Scholar] [CrossRef]
- Hofmann, N.A.; Barth, S.; Waldeck-Weiermair, M.; Klec, C.; Strunk, D.; Malli, R.; Graier, W.F. TRPV1 mediates cellular uptake of anandamide and thus promotes endothelial cell proliferation and network-formation. Biol. Open 2014, 3, 1164–1172. [Google Scholar] [CrossRef] [PubMed]
- Vercelli, C.; Barbero, R.; Cuniberti, B.; Odore, R.; Re, G. Expression and functionality of TRPV1 receptor in human MCF-7 and canine CF.41 cells. Vet. Comp. Oncol. 2015, 13, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Stock, K.; Kumar, J.; Synowitz, M.; Petrosino, S.; Imperatore, R.; Smith, E.S.J.; Wend, P.; Purfürst, B.; Nuber, U.A.; Gurok, U.; et al. Neural precursor cells induce cell death of high-grade astrocytomas through stimulation of TRPV1. Nat. Med. 2012, 18, 1232. [Google Scholar] [CrossRef]
- Jambrina, E.; Alonso, R.; Alcalde, M.; del Carmen Rodrı́guez, M.; Serrano, A.; Martı́nez, A.C.; Garcı́a-Sancho, J.; Izquierdo, M. Calcium influx through receptor-operated channel induces mitochondria-triggered paraptotic cell death. J. Biol. Chem. 2003, 278, 14134–14145. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Wang, G.; Tao, H.; Yang, Z.; Wang, Y.; Meng, Z.; Zhou, J. Capsaicin mediates caspases activation and induces apoptosis through P38 and JNK MAPK pathways in human renal carcinoma. BMC Cancer 2016, 16, 790. [Google Scholar] [CrossRef]
- Sanchez, M.G.; Sanchez, A.M.; Collado, B.; Malagarie-Cazenave, S.; Olea, N.; Carmena, M.J.; Prieto, J.C.; Diaz-Laviada, I.I. Expression of the transient receptor potential vanilloid 1 (TRPV1) in LNCaP and PC-3 prostate cancer cells and in human prostate tissue. Eur. J. Pharm. 2005, 515, 20–27. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Basu, S. Fas-associated factor 1 is a negative regulator in capsaicin induced cancer cell apoptosis. Cancer Lett. 2010, 287, 142–149. [Google Scholar] [CrossRef]
- Hou, N.; He, X.; Yang, Y.; Fu, J.; Zhang, W.; Guo, Z.; Hu, Y.; Liang, L.; Xie, W.; Xiong, H.; et al. TRPV1 Induced Apoptosis of Colorectal Cancer Cells by Activating Calcineurin-NFAT2-p53 Signaling Pathway. Biomed. Res. Int. 2019, 2019, 6712536. [Google Scholar] [CrossRef]
- Amantini, C.; Mosca, M.; Nabissi, M.; Lucciarini, R.; Caprodossi, S.; Arcella, A.; Santoni, G. Capsaicin_induced apoptosis of glioma cells is mediated by TRPV1 vanilloid receptor and requires p38 MAPK activation. J. Neurochem. 2007, 102, 977–990. [Google Scholar] [CrossRef]
- Fonseca, B.M.; Correia-da-Silva, G.; Teixeira, N.A. Cannabinoid-induced cell death in endometrial cancer cells: Involvement of TRPV1 receptors in apoptosis. J. Physiol. Biochem. 2018, 74, 261–272. [Google Scholar] [CrossRef]
- Wu, Y.Y.; Liu, X.Y.; Zhuo, D.X.; Huang, H.B.; Zhang, F.B.; Liao, S.F. Decreased expression of TRPV1 in renal cell carcinoma: Association with tumor Fuhrman grades and histopathological subtypes. Cancer Manag. Res. 2018, 10, 1647–1655. [Google Scholar] [CrossRef] [PubMed]
- Amantini, C.; Ballarini, P.; Caprodossi, S.; Nabissi, M.; Morelli, M.B.; Lucciarini, R.; Cardarelli, M.A.; Mammana, G.; Santoni, G. Triggering of transient receptor potential vanilloid type 1 (TRPV1) by capsaicin induces Fas/CD95-mediated apoptosis of urothelial cancer cells in an ATM-dependent manner. Carcinogenesis 2009, 30, 1320–1329. [Google Scholar] [CrossRef] [PubMed]
- Puntambekar, P.; Mukherjea, D.; Jajoo, S.; Ramkumar, V. Essential role of Rac1/NADPH oxidase in nerve growth factor induction of TRPV1 expression. J. Neurochem. 2005, 95, 1689–1703. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Guo, W.; Ma, J.; Xu, P.; Zhang, W.; Guo, S.; Liu, L.; Ma, J.; Shi, Q.; Jian, Z.; et al. Downregulated TRPV1 Expression Contributes to Melanoma Growth via the Calcineurin-ATF3-p53 Pathway. J. Invest. Derm. 2018, 138, 2205–2215. [Google Scholar] [CrossRef] [PubMed]
- Nazıroğlu, M.; Çiğ, B.; Blum, W.; Vizler, C.; Buhala, A.; Marton, A.; Katona, R.; Jósvay, K.; Schwaller, B.; Oláh, Z.; et al. Targeting breast cancer cells by MRS1477, a positive allosteric modulator of TRPV1 channels. PLoS ONE 2017, 12, e0179950. [Google Scholar]
- Xie, R.; Xu, J.; Wen, G.; Jin, H.; Liu, X.; Yang, Y.; Ji, B.; Jiang, Y.; Song, P.; Dong, H.; et al. The P2Y2 nucleotide receptor mediates the proliferation and migration of human hepatocellular carcinoma cells induced by ATP. J. Biol. Chem. 2014, 289, 19137–19149. [Google Scholar] [CrossRef]
- Sabala, P.; Czajkowski, R.; Przybylek, K.; Kalita, K.; Kaczmarek, L.; Baranska, J. Two subtypes of G protein-coupled nucleotide receptors, P2Y1 and P2Y2 are involved in calcium signalling in glioma C6 cells. Br. J. Pharm. 2001, 132, 393–402. [Google Scholar] [CrossRef][Green Version]
- Liu, L.; Yudin, Y.; Rohacs, T. Diacylglycerol kinases regulate TRPV1 channel activity. J. Biol. Chem. 2020. [Google Scholar] [CrossRef]
- Heo, J.S.; Han, H.J. ATP stimulates mouse embryonic stem cell proliferation via protein kinase C, phosphatidylinositol 3-kinase/Akt, and mitogen-activated protein kinase signaling pathwats. Stem. Cells 2006, 24, 2637–2648. [Google Scholar] [CrossRef]
- Danciu, T.E.; Adam, R.M.; Naruse, K.; Freeman, M.R.; Hauschka, P.V. Calcium regulates the PI3K-Akt pathway in stretched osteoblasts. FEBS Lett. 2003, 536, 193–197. [Google Scholar] [CrossRef]
- Katz, S.; Ayala, V.; Santillán, G.; Boland, R. Activation of the PI3K/Akt signaling pathway through P2Y2 receptors by extracellular ATP is involved in osteoblastic cell proliferation. Arch. Biochem. Biophys. 2011, 513, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, Z.; Capó-Aponte, J.E.; Zhang, F.; Pan, Z.; Reinach, P.S. Epidermal growth factor recept or transactivation by the cannabinoid receptor (CB1) and transient receptor potential vanilloid 1 (TRPV1) induces differential responses in corneal epithelial cells. Exp. Eye Res. 2010, 91, 462–471. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Bode, A.M.; Zhu, F.; Liu, K.; Zhang, J.; Kim, M.O.; Langfald, A.K. TRPV1-antagonist AMG9810 promotes mouse skin tumorigenesis through EGFR/Akt signaling. Carcinogenesis 2011, 32, 779–785. [Google Scholar] [CrossRef] [PubMed]
- Roberts, P.J.; Der, C.J. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 2007, 26, 3291–3310. [Google Scholar] [CrossRef]
- Uslusoy, F.; Nazıroğlu, M.; Çiğ, B. Inhibition of the TRPM2 and TRPV1 channels through Hypericum perforatum in sciatic nerve injury-induced rats demonstrates their key role in apoptosis and mitochondrial oxidative stress of sciatic nerve and dorsal root ganglion. Front. Physiol. 2017, 8, 335. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Wang, F.; Yang, Y.; Ma, W.; Lin, Z.; Cheng, N.; Long, Y.; Deng, S.; Li, Z. Recurrent activations of transient receptor potential vanilloid-1 and vanilloid-4 promote cellular proliferation and migration in esophageal squamous cell carcinoma cells. FEBS Open. Biol. 2019, 9, 206–225. [Google Scholar] [CrossRef]
- Chung, M.K.; Güler, A.D.; Caterina, M.J. TRPV1 shows dynamic ionic selectivity during agonist stimulation. Nat. Neurosci 2008, 11, 555. [Google Scholar] [CrossRef]
- Pereira, G.J.V.; Tavares, M.T.; Azevedo, R.A.; Martins, B.B.; Cunha, M.R.; Bhardwaj, R.; Cury, Y.; Zambelli, V.O.; Barbosa, E.G.; Hediger, M.A.; et al. Capsaicin-like analogue induced selective apoptosis in A2058 melanoma cells: Design, synthesis and molecular modeling. Bioorg Med. Chem. 2019, 27, 2893–2904. [Google Scholar] [CrossRef]
- Defo Deeh, P.B.; Watcho, P.; Wankeu_Nya, M.; Ngadjui, E.; Usman, U.Z. The methanolic extract of Guibourtia tessmannii (caesalpiniaceae) and selenium modulate cytosolic calcium accumulation, apoptosis and oxidative stress in R2C tumour Leydig cells: Involvement of TRPV 1 channels. Andrologia 2019, 51, e13216. [Google Scholar] [CrossRef]
- Köse, S.A.; Nazırolu, M. N-acetyl cysteine reduces oxidative toxicity, apoptosis, and calcium entry through TRPV1 channels in the neutrophils of patients with polycystic ovary syndrome. Free Radic Res. 2015, 49, 338–346. [Google Scholar] [CrossRef]
- Chen, W.T.; Lin, G.B.; Lin, S.H.; Lu, C.H.; Hsieh, C.H.; Ma, B.L.; Chao, C.Y. Static magnetic field enhances the anticancer efficacy of capsaicin on HepG2 cells via capsaicin receptor TRPV1. PLoS ONE 2018, 13, e0191078. [Google Scholar] [CrossRef]
- Pan, L.; Song, K.; Hu, F.; Sun, W.; Lee, I. Nitric oxide induces apoptosis associated with TRPV1 channel-mediated Ca2+ entry via S-nitrosylation in osteoblasts. Eur. J. Pharm. 2013, 715, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Lakshmi, S.; Joshi, P.G. Co-activation of P2Y2 receptor and TRPV channel by ATP: Implications for ATP induced pain. Cell Mol. Neurobiol. 2005, 25, 819–832. [Google Scholar] [CrossRef] [PubMed]
- Nita, I.I.; Caspi, Y.; Gudes, S.; Fishman, D.; Lev, S.; Hersfinkel, M.; Binshtok, A.M. Privileged crosstalk between TRPV1 channels and mitochondrial calcium shuttling machinery controls nociception. Bba-Mol. Cell Res. 2016, 1863, 2868–2880. [Google Scholar] [CrossRef]
- Nazırolu, M.; Övey, I.S. Involvement of apoptosis and calcium accumulation through TRPV1 channels in neurobiology of epilepsy. Neuroscience 2015, 293, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Vercelli, C.; Barbero, R.; Cuniberti, B.; Racca, S.; Abbadessa, G.; Piccione, F.; Re, G. Transient receptor potential vanilloid 1 expression and functionality in mcf-7 cells: A preliminary investigation. J. Breast Cancer 2014, 17, 332–338. [Google Scholar] [CrossRef]
- Ghazizadeh, V.; Nazırolu, M. Electromagnetic radiation (Wi-Fi) and epilepsy induce calcium entry and apoptosis through activation of TRPV1 channel in hippocampus and dorsal root ganglion of rats. Metab. Brain Dis. 2014, 29, 787–799. [Google Scholar] [CrossRef]
- Thomas, K.C.; Sabnis, A.S.; Johansen, M.E.; Lanza, D.L.; Moos, P.J.; Yost, G.S.; Reilly, C.A. Transient receptor potential vanilloid 1 agonists cause endoplasmic reticulum stress and cell death in human lung cells. J. Pharm. Exp. 2007, 321, 830–838. [Google Scholar] [CrossRef]
- Agopyan, N.; Head, J.; Yu, S.; Simon, S.A. TRPV1 receptors mediate particulate matter-induced apoptosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2004, 286, L563–L572. [Google Scholar] [CrossRef]
- He, L.; Poblenz, A.T.; Medrano, C.J.; Fox, D.A. Lead and calcium produce rod photoreceptor cell apoptosis by opening the mitochondrial permeability transition pore. J. Biol. Chem. 2000, 275, 12175–12184. [Google Scholar] [CrossRef]
- Narita, M.; Shimizu, S.; Ito, T.; Chittenden, T.; Lutz, R.J.; Matsuda, H.; Tsujimoto, Y. Bax interacts with the permeability transition pore to induce permeability transition and cytochrome c release in isolated mitochondria. Proc. Natl. Acad. Sci. USA 1998, 95, 14681–14686. [Google Scholar] [CrossRef]
- Armstrong, J.S.; Jones, D.P. Glutathione depletion enforces the mitochondrial permeability transition and causes cell death in Bcl-2 overexpressing HL60 cells. Faseb J. 2002, 16, 1263–1265. [Google Scholar] [CrossRef] [PubMed]
- Douglas, M.G.; Cockrell, R.S. Mitochondrial cation-hydrogen ion exchange. Sodium selective transport by mitochondria and submitochondrial particles. J. Biol. Chem. 1974, 249, 5464–5471. [Google Scholar] [PubMed]
- Numata, M.; Petrecca, K.; Lake, N.; Orlowski, J. Identification of a mitochondrial Na+/H+ exchanger. J. Biol. Chem. 1998, 273, 6951–6959. [Google Scholar] [CrossRef]
- Smaili, S.S.; Russell, J.T. Permeability transition pore regulates both mitochondrial membrane potential and agonist-evoked Ca2+ signals in oligodendrocyte progenitors. Cell Calcium. 1999, 26, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, J.S.; Steinauer, K.K.; Hornung, B.; Irish, J.M.; Lecane, P.; Birrell, G.W.; Peehl, D.M.; Knox, S.J. Role of glutathione depletion and reactive oxygen species generation in apoptotic signaling in a human B lymphoma cell line. Cell Death Differ. 2002, 9, 252–263. [Google Scholar] [CrossRef] [PubMed]
- Ip, S.W.; Lan, S.H.; Lu, H.F.; Huang, A.C.; Yang, J.S.; Lin, J.P.; Wood, W.G. Capsaicin mediates apoptosis in human nasopharyngeal carcinoma NPC-TW 039 cells through mitochondrial depolarization and endoplasmic reticulum stress. Hum. Exp. Toxicol. 2012, 31, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Cande, C.; Stephanou, N.C.; Jiang, S.; Gurbuxani, S.; Larochette, N.; Daugas, E.; Garrido, C.; Kroemer, G.; Wu, H. DNA binding is required for the apoptogenic action of apoptosis inducing factor. Nat. Struct. Biol. 2002, 9, 680–684. [Google Scholar] [CrossRef]
- Krizanova, O.; Steliarova, I.; Csaderova, L.; Pastorek, M.; Hudecova, S. Capsaicin induces apoptosis in PC12 cells through ER stress. Oncol. Rep. 2014, 31, 581–588. [Google Scholar] [CrossRef]
- Dremina, E.S.; Sharov, V.S.; Kumar, K.; Zaidi, A.; Michaelis, E.K.; Schoneich, C. Anti-apoptotic protein Bcl-2 interacts with and destabilizes the sarcoplasmic/endoplasmic reticulum Ca2+-ATPase (SERCA). Biochem. J. 2004, 383, 361–370. [Google Scholar] [CrossRef]
- Laver, D.R.; Lamb, G.D. Inactivation of Ca2+ release channels (ryanodine receptors RyR1 and RyR2) with rapid steps in [Ca2+] and voltage. Biophys. J. 1998, 74, 2352–2364. [Google Scholar] [CrossRef]
- Andrews, C.; Ho, P.D.; Dillmann, W.H.; Glembotski, C.C.; McDonough, P.M. The MKK6-p38 MAPK pathway prolongs the cardiac contractile calcium transient, downregulates SERCA2, and activates NF-AT. Cardiovasc. Res. 2003, 59, 46–56. [Google Scholar] [CrossRef]
- Luo, S.; Baumeister, P.; Yang, S.; Abcouwer, S.F.; Lee, A.S. Induction of GRP78/BiP by translational block Activation of the Grp78 promoter by ATF4 through an upstream ATF/CRE site independent of the endoplasmic reticulum stress elements. J. Biol. Chem. 2003, 278, 37375–37385. [Google Scholar] [CrossRef]
- Lim, M.P.; Devi, L.A.; Rozenfeld, R. Cannabidiol causes activated hepatic stellate cell death through a mechanism of endoplasmic reticulum stress-induced apoptosis. Cell Death Dis. 2011, 2, e170. [Google Scholar] [CrossRef] [PubMed]
- Barlow, C.; Brown, K.D.; Deng, C.X.; Tagle, D.A.; Wynshaw-Boris, A. Atm selectively regulates distinct p53-dependent cell-cycle checkpoint and apoptotic pathways. Nat. Genet. 1997, 17, 453–456. [Google Scholar] [CrossRef] [PubMed]
- Canman, C.E.; Lim, D.S.; Cimprich, K.A.; Taya, Y.; Tamai, K.; Sakaguchi, K.; Appella, E.; Kastan, M.B.; Siliciano, J.D. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science 1998, 281, 1677–1679. [Google Scholar] [CrossRef]
- Katsuda, K.; Kataoka, M.; Uno, F.; Murakami, T.; Kondo, T.; Roth, J.A.; Tanaka, N.; Fujiwara, T. Activation of caspase-3 and cleavage of Rb are associated with p16-mediated apoptosis in human non-small cell lung cancer cells. Oncogene 2002, 21, 2108–2113. [Google Scholar] [CrossRef]
- Hernandez, A.M.; Colvin, E.S.; Chen, Y.C.; Geiss, S.L.; Eller, L.E.; Fueger, P.T. Upregulation of p21 activates the intrinsic apoptotic pathway in beta-cells. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E1281–E1290. [Google Scholar] [CrossRef][Green Version]
- Cheng, E.H.; Kirsch, D.G.; Clem, R.J.; Ravi, R.; Kastan, M.B.; Bedi, A.; Ueno, K.; Hardwick, J.M. Conversion of Bcl-2 to a Bax-like death effector by caspases. Science 1997, 278, 1966–1968. [Google Scholar] [CrossRef]
- Gil, Y.G.; Kang, M.K. Capsaicin induces apoptosis and terminal differentiation in human glioma A172 cells. Life Sci. 2008, 82, 997–1003. [Google Scholar] [CrossRef]
- Song, J.; Lee, J.H.; Lee, S.H.; Park, K.A.; Lee, W.T.; Lee, J.E. TRPV1 Activation in Primary Cortical Neurons Induces Calcium-Dependent Programmed Cell Death. Exp. Neurobiol. 2013, 22, 51–57. [Google Scholar] [CrossRef]
- Wu, T.T.; Peters, A.A.; Tan, P.T.; Roberts-Thomson, S.J.; Monteith, G.R. Consequences of activating the calcium-permeable ion channel TRPV1 in breast cancer cells with regulated TRPV1 expression. Cell Calcium. 2014, 56, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Blagosklonny, M.V. P53: An ubiquitous target of anticancer drugs. Int. J. Cancer 2002, 98, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Issaeva, N.; Bozko, P.; Enge, M.; Protopopova, M.; Verhoef, L.G.; Masucci, M.; Pramanik, A.; Selivanova, G. Small molecule RITA binds to p53, blocks p53-HDM-2 interaction and activates p53 function in tumors. Nat. Med. 2004, 10, 1321–1328. [Google Scholar] [CrossRef] [PubMed]
- Park, G.Y.; Wilson, J.J.; Song, Y.; Lippard, S.J. Phenanthriplatin, a monofunctional DNA-binding platinum anticancer drug candidate with unusual potency and cellular activity profile. Proc. Natl. Acad. Sci. USA 2012, 109, 11987–11992. [Google Scholar] [CrossRef]
- Satheesh, N.J.; Busselberg, D. The role of intracellular calcium for the development and treatment of neuroblastoma. Cancers 2015, 7, 823–848. [Google Scholar] [CrossRef]
- Roderick, H.L.; Cook, S.J. Ca2+ signalling checkpoints in cancer: Remodelling Ca2+ for cancer cell proliferation and survival. Nat. Rev. Cancer 2008, 8, 361–375. [Google Scholar] [CrossRef]
- Al-Taweel, N.; Varghese, E.; Florea, A.M.; Büsselberg, D. Cisplatin (CDDP) triggers cell death of MCF-7 cells following disruption of intracellular calcium ([Ca2+] i) homeostasis. J. Toxicol. Sci. 2014, 39, 765–774. [Google Scholar] [CrossRef]
- Varghese, E.; Busselberg, D. Auranofin, an anti-rheumatic gold compound, modulates apoptosis by elevating the intracellular calcium concentration ([ca2+]I) in mcf-7 breast cancer cells. Cancers 2014, 6, 2243–2258. [Google Scholar] [CrossRef]
- Günes, D.A.; Florea, A.-M.; Splettstoesser, F.; Büsselberg, D. Co-application of arsenic trioxide (As2O3) and cisplatin (CDDP) on human SY-5Y neuroblastoma cells has differential effects on the intracellular calcium concentration ([Ca2+]i) and cytotoxicity. NeuroToxicology 2009, 30, 194–202. [Google Scholar] [CrossRef]
- Aoki, M.; Batista, O.; Bellacosa, A.; Tsichlis, P.; Vogt, P.K. The akt kinase: Molecular determinants of oncogenicity. Proc. Natl. Acad. Sci. USA 1998, 95, 14950–14955. [Google Scholar] [CrossRef]
- Wu, Y.-R.; Qi, H.-J.; Deng, D.-F.; Luo, Y.-Y.; Yang, S.-L. MicroRNA-21 promotes cell proliferation, migration, and resistance to apoptosis through PTEN/PI3K/AKT signaling pathway in esophageal cancer. Tumor. Biol. 2016, 37, 12061–12070. [Google Scholar] [CrossRef]
- Jin, Y.; Feng, S.J.; Qiu, S.; Shao, N.; Zheng, J.H. LncRNA MALAT1 promotes proliferation and metastasis in epithelial ovarian cancer via the PI3K-AKT pathway. Eur. Rev. Med. Pharm. Sci. 2017, 21, 3176–3184. [Google Scholar]
- Gao, N.; Zhang, Z.; Jiang, B.H.; Shi, X. Role of PI3K/AKT/mTOR signaling in the cell cycle progression of human prostate cancer. Biochem. Biophys. Res. Commun. 2003, 310, 1124–1132. [Google Scholar] [CrossRef]
- Berns, K.; Horlings, H.M.; Hennessy, B.T.; Madiredjo, M.; Hijmans, E.M.; Beelen, K.; Linn, S.C.; Gonzalez-Angulo, A.M.; Stemke-Hale, K.; Hauptmann, M.; et al. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell.
- Abrams, S.L.; Steelman, L.S.; Shelton, J.G.; Wong, E.W.T.; Chappell, W.H.; Bäsecke, J.; Stivala, F.; Donia, M.; Nicoletti, F.; Libra, M.; et al. The Raf/MEK/ERK pathway can govern drug resistance, apoptosis and sensitivity to targeted therapy. Cell Cycle 2010, 9, 1781–1791. [Google Scholar] [CrossRef]
- Bon, G.; Loria, R.; Amoreo, C.A.; Verdina, A.; Sperduti, I.; Mastrofrancesco, A.; Soddu, S.; Diodoro, M.G.; Mottolese, M.; Todaro, M.; et al. Dual targeting of HER3 and MEK may overcome HER3-dependent drug-resistance of colon cancers. Oncotarget 2017, 8, 108463–108479. [Google Scholar] [CrossRef]
- Dekker, L.V.; Leitges, M.; Altschuler, G.; Mistry, N.; McDermott, A.; Roes, J.; Segal, A.W. Protein kinase C-β contributes to NADPH oxidase activation in neutrophils. Biochem. J. 2000, 347, 285–289. [Google Scholar] [CrossRef]
- Descamps, S.; Toillon, R.-A.; Adriaenssens, E.; Pawlowski, V.R.; Cool, S.M.; Nurcombe, V.; Bourhis, X.L.; Boilly, B.n.; Peyrati, J.-P.; Hondermarck, H. Nerve Growth Factor stimulates proliferation and survival of human breast cancer cells through two distinct signaling pathways. J. Biol. Chem. 2001, 276, 17864–17870. [Google Scholar] [CrossRef]
- Bujak, J.K.; Kosmala, D.; Szopa, I.M.; Majchrzak, K.; Bednarczyk, P. Inflammation, Cancer and Immunity-Implication of TRPV1 Channel. Front. Oncol. 2019, 9, 1087. [Google Scholar] [CrossRef]
- Afroz, S.; Arakaki, R.; Iwasa, T.; Oshima, M.; Hosoki, M.; Inoue, M.; Baba, O.; Okayama, Y.; Matsuka, Y. CGRP Induces Differential Regulation of Cytokines from Satellite Glial Cells in Trigeminal Ganglia and Orofacial Nociception. Int. J. Mol. Sci. 2019, 20, 711. [Google Scholar] [CrossRef] [PubMed]
- Mashaghi, A.; Marmalidou, A.; Tehrani, M.; Grace, P.M.; Pothoulakis, C.; Dana, R. Neuropeptide substance P and the immune response. Cell Mol. Life Sci. 2016, 73, 4249–4264. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Georgescu, S.R.; Sarbu, M.I.; Matei, C.; Ilie, M.A.; Caruntu, C.; Constantin, C.; Neagu, M.; Tampa, M. Capsaicin: Friend or Foe in Skin Cancer and Other Related Malignancies? Nutrients 2017, 9, 1365. [Google Scholar] [CrossRef]
- Erin, N. Role of sensory neurons, neuroimmune pathways, and transient receptor potential vanilloid 1 (TRPV1) channels in a murine model of breast cancer metastasis. Cancer Immunol. Immunother. 2020, 69, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Peluso, G.; Petillo, O.; Melone, M.A.; Mazzarella, G.; Ranieri, M.; Tajana, G.F. Modulation of cytokine production in activated human monocytes by somatostatin. Neuropeptides 1996, 30, 443–451. [Google Scholar] [CrossRef]
- Pinter, E.; Helyes, Z.; Szolcsanyi, J. Inhibitory effect of somatostatin on inflammation and nociception. Pharm. Ther. 2006, 112, 440–456. [Google Scholar] [CrossRef]
- Li, Y.R.; Gupta, P. Immune aspects of the bi-directional neuroimmune facilitator TRPV1. Mol. Biol. Rep. 2019, 46, 1499–1510. [Google Scholar] [CrossRef]
- Fernandes, E.S.; Cerqueira, A.R.A.; Soares, A.G.; Costa, S.K.P. Capsaicin and Its Role in Chronic Diseases. In Drug Discovery from Mother Nature; Gupta, S.C., Prasad, S., Aggarwal, B.B., Eds.; Springer: Cham, Switzerland, 2016; Volume 929. [Google Scholar]
- Walker, J.; Ley, J.P.; Schwerzler, J.; Lieder, B.; Beltran, L.; Ziemba, P.M.; Hatt, H.; Hans, J.; Widder, S.; Krammer, G.E.; et al. Nonivamide, a capsaicin analogue, exhibits anti-inflammatory properties in peripheral blood mononuclear cells and U-937 macrophages. Mol. Nutr. Food Res. 2017, 61. [Google Scholar] [CrossRef]
- Gao, H.; Yang, B.J.; Li, N.; Feng, L.M.; Shi, X.Y.; Zhao, W.H.; Liu, S.J. Bisphenol A and hormone-associated cancers: Current progress and perspectives. Medicine (Baltimore) 2015, 94, e211. [Google Scholar] [CrossRef]
- Guzel, K.G.U.; Naziroglu, M.; Ceyhan, D. Bisphenol A-Induced Cell Proliferation and Mitochondrial Oxidative Stress Are Diminished via Modulation of TRPV1 Channel in Estrogen Positive Breast Cancer Cell by Selenium Treatment. Biol. Trace Elem. Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Geng, S.; Zheng, Y.; Meng, M.; Guo, Z.; Cao, N.; Ma, X.; Du, Z.; Li, J.; Duan, Y.; Du, G. Gingerol Reverses the Cancer-Promoting Effect of Capsaicin by Increased TRPV1 Level in a Urethane-Induced Lung Carcinogenic Model. J. Agric. Food Chem. 2016, 64, 6203–6211. [Google Scholar] [CrossRef]
- Nur, G.; Naziroglu, M.; Deveci, H.A. Synergic prooxidant, apoptotic and TRPV1 channel activator effects of alpha-lipoic acid and cisplatin in MCF-7 breast cancer cells. J. Recept Signal. Transduct. Res. 2017, 37, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Çetin, E.S.; Nazıroğlu, M.; Çiğ, B.; Övey, İ.S.; Koşar, P.A. Selenium potentiates the anticancer effect of cisplatin against oxidative stress and calcium ion signaling-induced intracellular toxicity in MCF-7 breast cancer cells: Involvement of the TRPV1 channel. J. Recept. Sig. Transd. 2017, 37, 84–93. [Google Scholar] [CrossRef]
- Zheng, L.; Chen, J.; Ma, Z.; Liu, W.; Yang, F.; Yang, Z.; Wang, K.; Wang, X.; He, D.; Li, L.; et al. Capsaicin enhances anti-proliferation efficacy of pirarubicin via activating TRPV1 and inhibiting PCNA nuclear translocation in 5637 cells. Mol. Med. Rep. 2016, 13, 881–887. [Google Scholar] [CrossRef] [PubMed]
- Kosar, P.A.; Naziroglu, M.; Ovey, I.S.; Cig, B. Synergic Effects of Doxorubicin and Melatonin on Apoptosis and Mitochondrial Oxidative Stress in MCF-7 Breast Cancer Cells: Involvement of TRPV1 Channels. J. Membr. Biol. 2016, 249, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Amgalan, D.; Garner, T.P.; Kitsis, R.N. A small-molecule allosteric inhibitor of BAX protects against doxorubicin-induced cardiomyopathy. Nat. Cancer 2020, 1, 315–328. [Google Scholar] [CrossRef]
- Deveci, H.A.; Naziroglu, M.; Nur, G. 5-Fluorouracil-induced mitochondrial oxidative cytotoxicity and apoptosis are increased in MCF-7 human breast cancer cells by TRPV1 channel activation but not Hypericum perforatum treatment. Mol. Cell Biochem. 2018, 439, 189–198. [Google Scholar] [CrossRef]
- Chen, Y.; Li, J.; Jin, L.; Lei, K.; Liu, H.; Yang, Y. Fibulin-5 contributes to colorectal cancer cell apoptosis via the ROS/MAPK and Akt signal pathways by downregulating transient receptor potential cation channel subfamily V member 1. J. Cell Biochem. 2019, 120, 17838–17846. [Google Scholar] [CrossRef]
- Punzo, F.; Manzo, I.; Tortora, C.; Pota, E.; Angelo, V.; Bellini, G.; Di Paola, A.; Verace, F.; Casale, F.; Rossi, F. Effects of CB2 and TRPV1 receptors’ stimulation in pediatric acute T-lymphoblastic leukemia. Oncotarget 2018, 9, 21244–21258. [Google Scholar] [CrossRef][Green Version]
- Mao, Y.; Liu, X. Bioresponsive Nanomedicine: The Next Step of Deadliest Cancers’ Theranostics. Front. Chem. 2020, 8, 257. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Zhang, J.; Zhang, W.; Foda, M.F.; Zhang, Y.; Ge, L.; Han, H. Intracellular Ca(2+) Cascade Guided by NIR-II Photothermal Switch for Specific Tumor Therapy. iScience 2020, 23, 101049. [Google Scholar] [CrossRef] [PubMed]
- Zhen, X.; Xie, C.; Jiang, Y.; Ai, X.; Xing, B.; Pu, K. Semiconducting Photothermal Nanoagonist for Remote-Controlled Specific Cancer Therapy. Nano Lett. 2018, 18, 1498–1505. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Guerrero, A.; Espinosa-Duran, J.M.; Velasco-Medina, J. TRPV1 channel as a target for cancer therapy using CNT-based drug delivery systems. Eur. Biophys. J. 2016, 45, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Baker, C.; Rodrigues, T.; de Almeida, B.P.; Barbosa-Morais, N.L.; Bernardes, G.J.L. Natural product-drug conjugates for modulation of TRPV1-expressing tumors. Bioorg. Med. Chem. 2019, 27, 2531–2536. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Pan, J.B.; Zhao, W.; Chen, H.Y.; Xu, J.J. Gold nanorod-assisted near-infrared light-mediated regulation of membrane ion channels activates apoptotic pathways. Chem. Commun. 2020. [Google Scholar] [CrossRef]
- Lozano, C.; Cordova, C.; Marchant, I.; Zuniga, R.; Ochova, P.; Ramirez-Barrantes, R.; Gonzalez-Arriagada, W.A.; Rodriguez, B.; Olivero, P. Intracellular aggregated TRPV1 is associated with lower survival in breast cancer patients. Breast Cancer 2018, 10, 161–168. [Google Scholar] [CrossRef]
- Han, G.H.; Chay, D.B.; Nam, S.; Cho, H.; Chung, J.Y.; Kim, J.H. Prognostic Significance of Transient Receptor Potential Vanilloid Type 1 (TRPV1) and Phosphatase and Tension Homolog (PTEN) in Epithelial Ovarian Cancer. Cancer Genom. Proteom. 2020, 17, 309–319. [Google Scholar] [CrossRef]
- Han, G.H.; Chay, D.B.; Nam, S.; Cho, H.; Chung, J.Y.; Kim, J.H. The Combination of Transient Receptor Potential Vanilloid Type 1 (TRPV1) and Phosphatase and Tension Homolog (PTEN) is an Effective Prognostic Biomarker in Cervical Cancer. Int. J. Gynecol. Pathol. 2020. [Google Scholar] [CrossRef]
- Sharma, S.K.; Vijay, S.; Gore, S.; Dore, T.M.; Jagannathan, R. Measuring Cellular Ion Transport by Magnetoencephalography. ACS Omega 2020, 5, 4024–4031. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Localization/Cancer Type | Cell Line/Source | TRPV1 mRNA Expression | TRPV1 Protein Expression | Source |
|---|---|---|---|---|
| Non-Cancer | ||||
| Eyes | Whole retina, Sprague-Dewley rats | Yes | Yes | [34] |
| Retinal RGC, Sprague-Dewley rat | Yes | Yes | [34] | |
| Primary retinal RGC, Sprague-Dewley rat | Yes | Yes | [34] | |
| Whole retina, DBA/2 mice | -- | Yes | [34] | |
| Whole retina, C57 mice | -- | Yes | [34] | |
| Lung | ASMC, Sprague-Dewley rats | Yes | Yes | [35] |
| ASMC, chronic asthmatic Sprague-Dewley rats | Yes | Yes | [35] | |
| Nervous System | Cortical neuron, Wistar rat | Yes | Yes | [36] |
| Brain, Sprague-Dewley rat | -- | Yes | [34] | |
| Brain, C57 mouse | -- | Yes | [34] | |
| Type 1 SGZ NPC, p7-21, murine | Yes | Yes | [37] | |
| Type B SVZ NPC, p7-p21, murine | Yes | Yes | [37] | |
| Heart | H9C2 | Yes | Yes | [38] |
| Joints | Synoviocytes, Wistar rat | Yes | -- | [39] |
| Skin | Epidermis, human skin | -- | Yes | [40] |
| In-vitro Reconstructed Skin Equivalent Model | -- | Yes | [40] | |
| Circulatory/Endothelium | ECFC | -- | Yes | [41] |
| EA.hy926 | -- | Yes | [41] | |
| Cancer | ||||
| Breast Cancer | MCF-7 | -- | Yes | [42] |
| CF.41 | -- | Yes | [42] | |
| Nervous System Cancer | GL261 | -- | Yes | [43] |
| Leukemia | Jurkat | -- | Yes | [44] |
| Renal Cell Carcinoma | 786-O | Yes | Yes | [45] |
| Bladder Cancer | T24 | Yes | Yes | [45] |
| 5637 | Yes | Yes | [45] | |
| Prostate Cancer | LNCaP | Yes | Yes | [46] |
| PC-3 | Yes | Yes | [46] | |
| Sarcoma | Meth A | Yes | Yes | [47] |
| CMS5 | Yes | Yes | [47] |
| Cancer Type | Cell Line/Source | TRPV1 mRNA vs. Normal | TRPV1 Protein vs. Normal | Normal Comparison | Source |
|---|---|---|---|---|---|
| Colorectal | Human CRC | -- | Decreased | Human Colorectal Sample | [48] |
| Nervous System | U87 | Decreased | Decreased | NHA | [49] |
| U373 | Increased | Increased | NHA | [49] | |
| FLS | Decreased | -- | NHA | [49] | |
| FC1 | Decreased | -- | NHA | [49] | |
| High Grade Astrocyte | Increased | -- | Low Grade Astrocyte | [43] | |
| “Brain Tumors” | Increased | -- | “Tumor Free Brain” | [43] | |
| Endometrial | Ishikawa | NC | Decreased | HFF-1 | [50] |
| Hec50co | NC | Decreased | HFF-1 | [50] | |
| Renal | Human RCC | Decreased | Decreased | Human Renal Sample | [51] |
| RT4 | Increased | Increased | NHUC | [52] | |
| TCCSUP | Decreased | Decreased | NHUC | [52] | |
| J82 | Decreased | Decreased | NHUC | [52] | |
| EJ | Decreased | Decreased | NHUC | [52] | |
| Pheochromocytoma | PC12 | -- | Decreased | Rat DRG | [53] |
| Melanoma | WM793B | NC | NC | NHEM | [54] |
| WM35 | Decreased | Decreased | NHEM | [54] | |
| 1205Lu | Decreased | Decreased | NHEM | [54] | |
| 451Lu | Decreased | Decreased | NHEM | [54] | |
| UACC 62 | Decreased | Decreased | NHEM | [54] | |
| UACC 257 | Decreased | Decreased | NHEM | [54] | |
| Hs 294T | Decreased | Decreased | NHEM | [54] | |
| A375 | Decreased | Decreased | NHEM | [54] | |
| A2058 | Decreased | Decreased | NHEM | [54] | |
| Sk-mel-5 | Decreased | Decreased | NHEM | [54] | |
| Primary human melanoma | Decreased | Decreased | Human melanocytic nevus tissues | [54] | |
| Metastatic human melanoma | Decreased | Decreased | Human melanocytic nevus tissues | [54] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhai, K.; Liskova, A.; Kubatka, P.; Büsselberg, D. Calcium Entry through TRPV1: A Potential Target for the Regulation of Proliferation and Apoptosis in Cancerous and Healthy Cells. Int. J. Mol. Sci. 2020, 21, 4177. https://doi.org/10.3390/ijms21114177
Zhai K, Liskova A, Kubatka P, Büsselberg D. Calcium Entry through TRPV1: A Potential Target for the Regulation of Proliferation and Apoptosis in Cancerous and Healthy Cells. International Journal of Molecular Sciences. 2020; 21(11):4177. https://doi.org/10.3390/ijms21114177
Chicago/Turabian StyleZhai, Kevin, Alena Liskova, Peter Kubatka, and Dietrich Büsselberg. 2020. "Calcium Entry through TRPV1: A Potential Target for the Regulation of Proliferation and Apoptosis in Cancerous and Healthy Cells" International Journal of Molecular Sciences 21, no. 11: 4177. https://doi.org/10.3390/ijms21114177
APA StyleZhai, K., Liskova, A., Kubatka, P., & Büsselberg, D. (2020). Calcium Entry through TRPV1: A Potential Target for the Regulation of Proliferation and Apoptosis in Cancerous and Healthy Cells. International Journal of Molecular Sciences, 21(11), 4177. https://doi.org/10.3390/ijms21114177