Molecular Modeling of Chemosensory Protein 3 from Spodoptera litura and Its Binding Property with Plant Defensive Metabolites

 ,
,  , ,
, ,  ,
,
Abstract
1. Introduction
2. Results
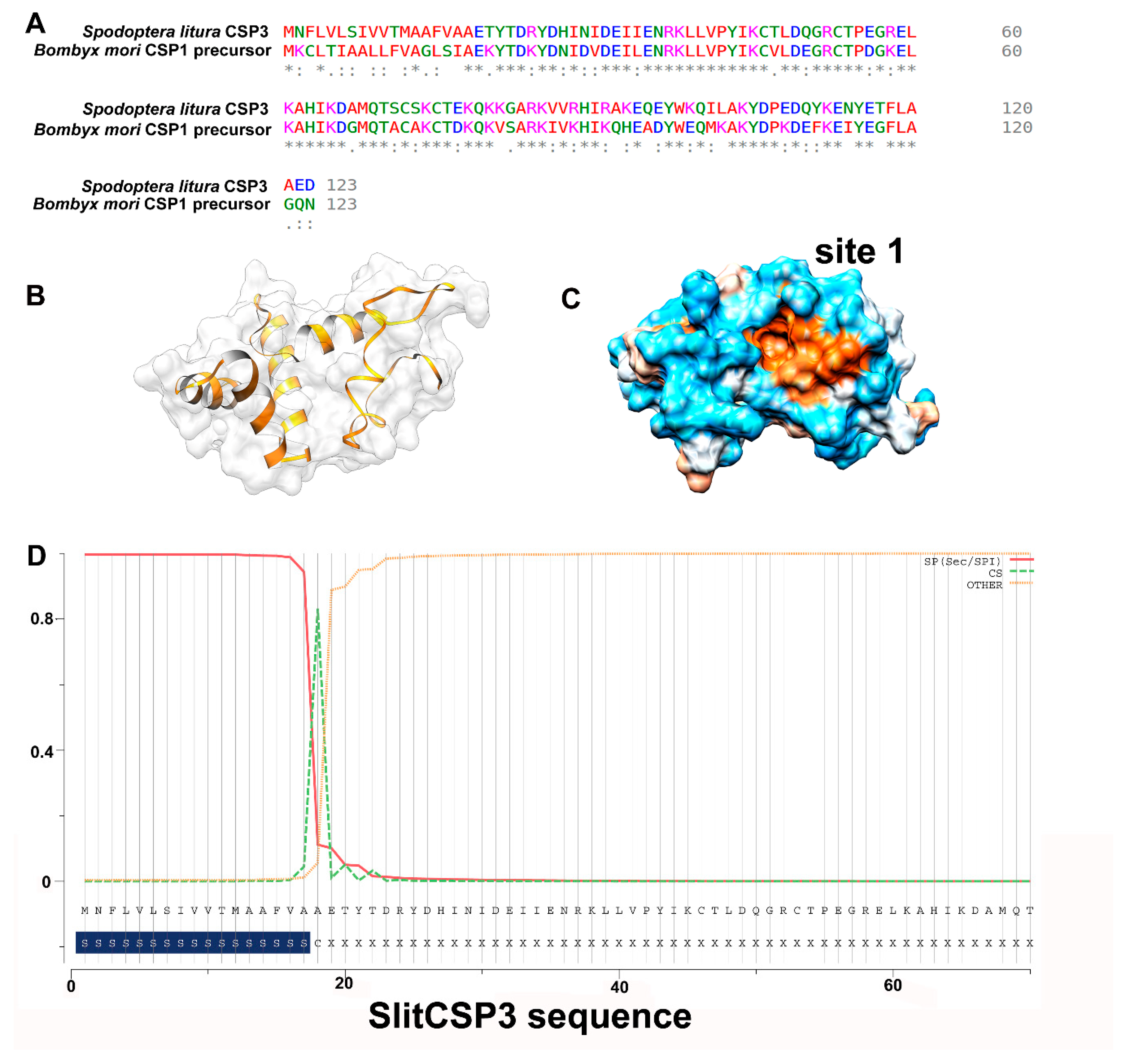
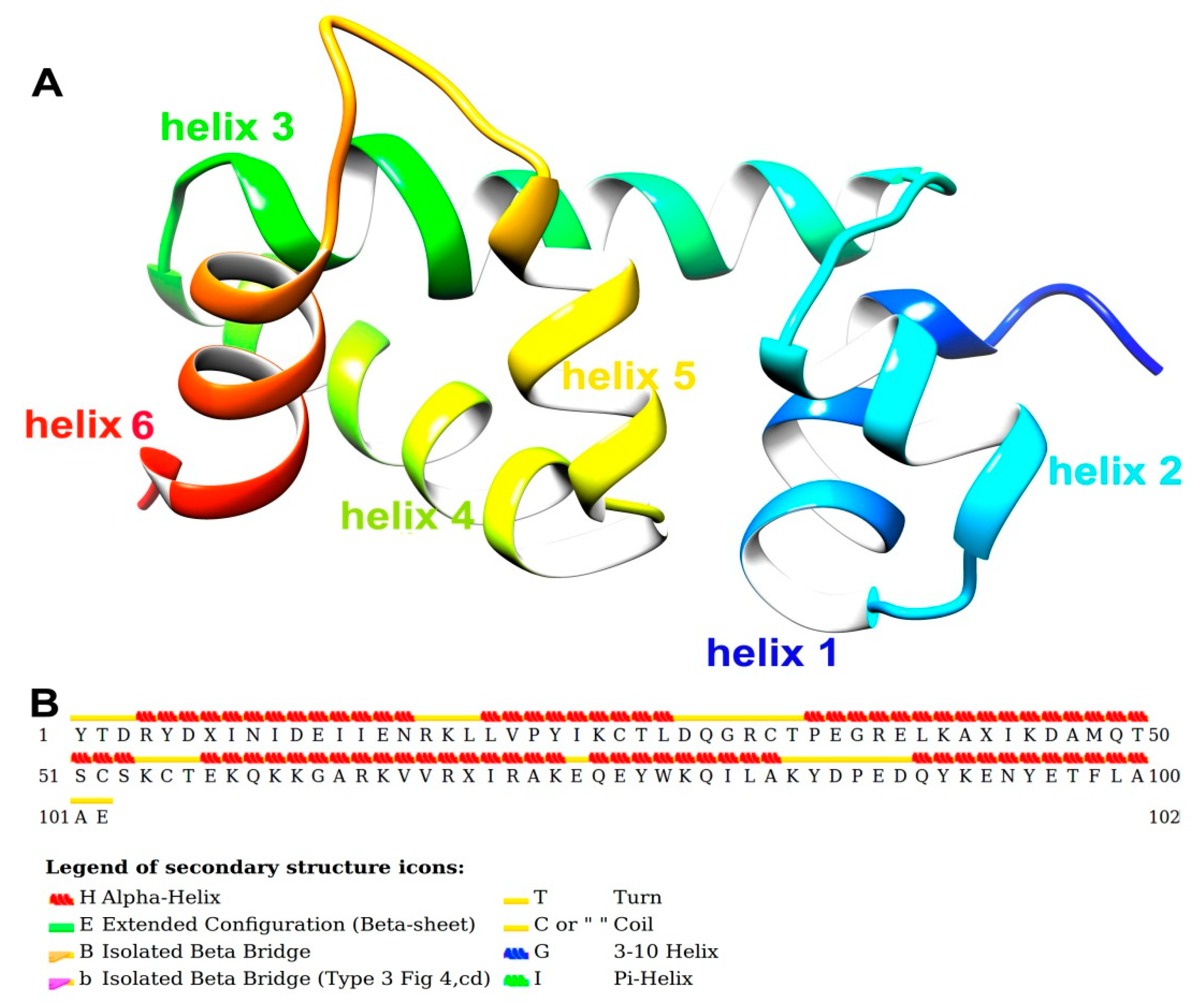
2.1. Sequence Alignment and 3D Modeling of SlitCSP3 Protein
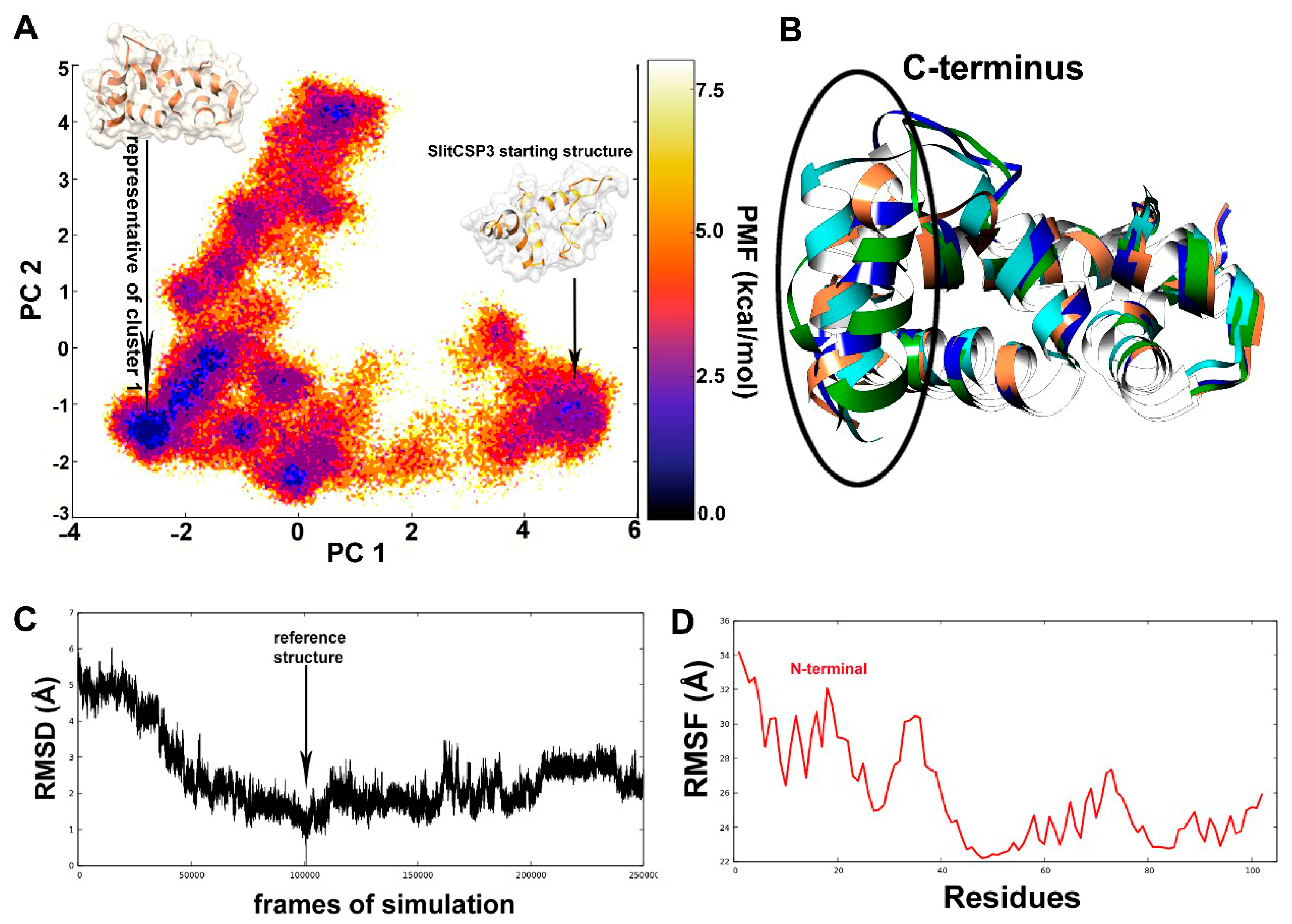
2.2. Accelerated Molecular Dynamics on the Modeled SlitCSP3
2.3. Identification of Binding Sites
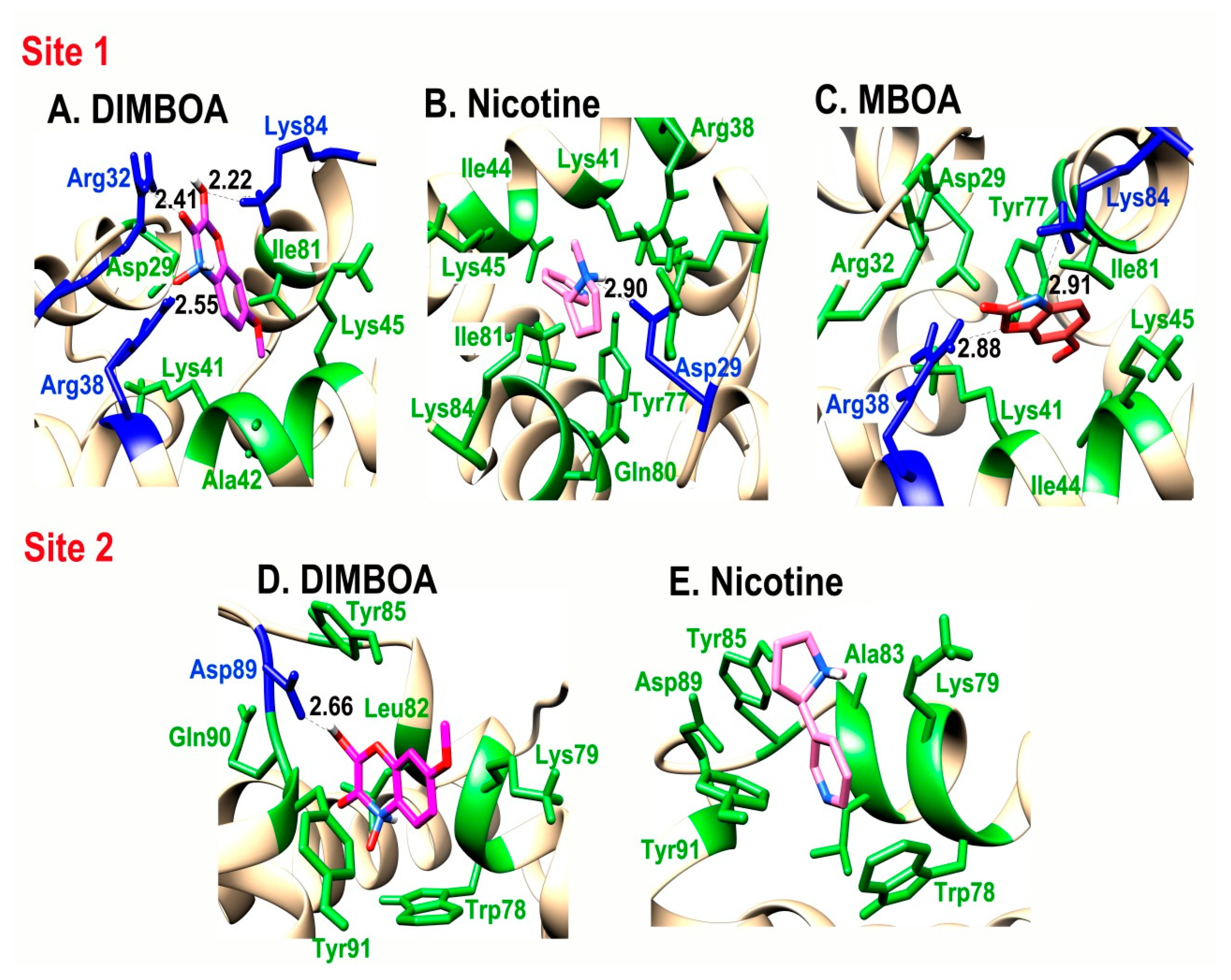
2.4. Interaction of SlitCSP3 with Plant-Defensive Metabolites
2.5. Accelerated MD Simulations of SlitCSP3-Ligand Complexes
3. Discussion
4. Materials and Methods
4.1. Sequence Retrieval and Structure Modeling
4.2. Accelerated Molecular Dynamics
Etotal = Vavg_total + b1 × Natoms, αtotal = b2 × Natoms
4.3. Prediction of SlitCSP3 Binding Sites
4.4. Grid Generation and Docking
4.5. Simulations of Docked Complexes and MM/PBSA Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CSP | Chemosensory proteins |
| (aMD) | Accelerated molecular dynamics |
| SlitCSP3 | Spodoptera litura chemosensory protein 3 |
| PDB | Protein Database |
| SlitCSP3 | Spodoptera litura chemosensory protein 3 |
| RMSD | Root-Mean-Square Deviation |
| RMSF | Root-Mean-Square Atomic Fluctuation |
| DIMBOA | 2-b-D-glucopyranosyloxy-4-hydroxy-7-methoxy-1,4-benzoxazin-3-one |
| HDMBOA | 2-b-D-glucopyranosyloxy-4,7-dimethoxy-1,4-benzoxazin-3-one |
| MBOA | 6-Methoxy-2-benzoxazolinone |
| MPQS | ModPipe Quality Score |
| NCBI | National Center for Biotechnology Information |
| CASTp | Computed Atlas of Surface Topography of proteins |
| MM/PBSA | Molecular Mechanics/ Poisson-Boltzmann Surface Area |
| MM/GBSA | Molecular Mechanics/Generalized Born Surface Area |
| PCA | principal component analysis |
| NIIF | National Information Infrastructure Development |
References
- Ahmad, M.; Ghaffar, A.; Rafiq, M. Host plants of leaf worm, Spodoptera litura (Fabricius) (Lepidoptera: Noctuidae) in Pakistan. Asian J. Agric. Biol. 2013, 1, 23–28. [Google Scholar]
- Tuan, S.J.; Li, N.J.; Yeh, C.C. Growth performance and biometric characteristics of Spodoptera litura (Lepidoptera: Noctuidae) reared on different host plants. J. Econ. Entomol. 2015, 108, 2242–2249. [Google Scholar] [CrossRef] [PubMed]
- Yi, X.; Zhao, H.; Dong, X.; Wang, P.; Hu, M.; Zhong, G. BdorCSP2 is important for antifeed and oviposition-deterring activities induced by Rhodojaponin-III against Bactrocera dorsalis. PLoS ONE 2013, 8, e77295. [Google Scholar] [CrossRef]
- Gerofotis, C.D.; Ioannou, C.S.; Nakas, C.T.; Papadopoulos, N.T. The odor of a plant metabolite affects life history traits in dietary restricted adult olive flies. Sci. Rep. 2016, 6, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Shi, Z.; Wang, B.; Lu, M.; Sun, J. Pine defensive monoterpene α-pinene influences the feeding behavior of Dendroctonus valens and its gut bacterial community structure. Int. J. Mol. Sci. 2016, 17, 1734. [Google Scholar] [CrossRef] [PubMed]
- Slade, J.D.; Staveley, B.E. Manipulation of components that control feeding behavior in Drosophila melanogaster increases sensitivity to amino acid starvation. Genet. Mol. Res. 2016, 15, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Yi, X.; Qi, J.; Zhou, X.; Hu, M.Y.; Zhong, G.H. Differential expression of chemosensory-protein genes in midguts in response to diet of Spodoptera litura. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Venthur, H.; Zhou, J.J. Odorant receptors and odorant-binding proteins as insect pest control targets: A comparative analysis. Front. Physiol. 2018, 9, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Pelosi, P.; Zhou, J.J.; Ban, L.P.; Calvello, M. Soluble proteins in insect chemical communication. Cell. Mol. Life Sci. 2006, 63, 1658–1676. [Google Scholar] [CrossRef]
- Briand, L.; Swasdipan, N.; Nespoulous, C.; Bézirard, V.; Blon, F.; Huet, J.C.; Ebert, P.; Pernollet, J.C. Characterization of a chemosensory protein (ASP3c) from honeybee (Apis mellifera L.) as a brood pheromone carrier. Eur. J. Biochem. 2002, 269, 4586–4596. [Google Scholar] [CrossRef]
- Picimbon, J.F.; Dietrich, K.; Krieger, J.; Breer, H. Identity and expression pattern of chemosensory proteins in Heliothis virescens (Lepidoptera, Noctuidae). Insect Biochem. Mol. Biol. 2001, 31, 1173–1181. [Google Scholar] [CrossRef]
- Maleszka, J.; Forêt, S.; Saint, R.; Maleszka, R. RNAi-induced phenotypes suggest a novel role for a chemosensory protein CSP5 in the development of embryonic integument in the honeybee (Apis mellifera). Dev. Genes Evol. 2007, 217, 189–196. [Google Scholar] [CrossRef]
- Nomura Kitabayashi, A.; Arai, T.; Kubo, T.; Natori, S. Molecular cloning of cDNA for p10, a novel protein that increases in the regenerating legs of Periplaneta americana (American cockroach). Insect Biochem. Mol. Biol. 1998, 28, 785–790. [Google Scholar] [CrossRef]
- Jacobs, S.P.; Liggins, A.P.; Zhou, J.J.; Pickett, J.A.; Jin, X.; Field, L.M. OS-D-like genes and their expression in aphids (Hemiptera: Aphididae). Insect Mol. Biol. 2005, 14, 423–432. [Google Scholar] [CrossRef]
- Zhou, S.H.; Zhang, J.; Zhang, S.G.; Zhang, L. Expression of chemosensory proteins in hairs on wings of Locusta migratoria (Orthoptera: Acrididae). J. Appl. Entomol. 2008, 132, 439–450. [Google Scholar] [CrossRef]
- Hamelberg, D.; Mongan, J.; McCammon, J.A. Accelerated molecular dynamics: A promising and efficient simulation method for biomolecules. J. Chem. Phys. 2004, 120, 11919–11929. [Google Scholar] [CrossRef] [PubMed]
- Hamelberg, D.; De Oliveira, C.A.F.; McCammon, J.A. Sampling of slow diffusive conformational transitions with accelerated molecular dynamics. J. Chem. Phys. 2007, 127, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Jansen, S.; Chmelík, J.; Žídek, L.; Padrta, P.; Novák, P.; Zdráhal, Z.; Picimbon, J.F.; Löfstedt, C.; Sklenář, V. Structure of Bombyx mori chemosensory protein 1 in solution. Arch. Insect Biochem. Physiol. 2007, 66, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Lartigue, A.; Campanacci, V.; Roussel, A.; Larsson, A.M.; Alwyn Jones, T.; Tegoni, M.; Cambillau, C. X-ray structure and ligand binding study of a moth chemosensory protein. J. Biol. Chem. 2002, 277, 32094–32098. [Google Scholar] [CrossRef]
- Campanacci, V.; Lartigue, A.; Hällberg, B.M.; Jones, T.A.; Giudici-Orticoni, M.T.; Tegoni, M.; Cambillau, C. Moth chemosensory protein exhibits drastic conformational changes and cooperativity on ligand binding. Proc. Natl. Acad. Sci. USA 2003, 100, 5069–5074. [Google Scholar] [CrossRef]
- Tomaselli, S.; Crescenzi, O.; Sanfelice, D.; Eiso, A.B.; Wechselberger, R.; Angeli, S.; Scaloni, A.; Boelens, R.; Tancredi, T.; Pelosi, P.; et al. Solution structure of a chemosensory protein from the desert locust Schistocerca gregaria. Biochemistry 2006, 45, 10606–10613. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Dong, X.; Liu, J.; Hu, M.; Zhong, G.; Geng, P.; Yi, X. Molecular cloning, expression and molecular modeling of chemosensory protein from Spodoptera litura and its binding properties with Rhodojaponin III. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the MM_PBSA and MM_GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model 2011, 51, 69–82. [Google Scholar] [CrossRef]
- Eswar, N.; John, B.; Mirkovic, N.; Fiser, A.; Ilyin, V.A.; Pieper, U.; Stuart, A.C.; Marti-Renom, M.A.; Madhusudhan, M.S.; Yerkovich, B.; et al. Tools for comparative protein structure modeling and analysis. Nucleic Acids Res. 2003, 31, 3375–3380. [Google Scholar] [CrossRef] [PubMed]
- Karolewski, Z.; Fitt, B.D.L.; Latunde-Dada, A.O.; Foster, S.J.; Todd, A.D.; Downes, K.; Evans, N. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Plant Pathol. 2006, 55, 3389–3402. [Google Scholar] [CrossRef]
- Johnson, M.; Zaretskaya, I.; Raytselis, Y.; Merezhuk, Y.; McGinnis, S.; Madden, T.L. NCBI BLAST: A better web interface. Nucleic Acids Res. 2008, 36, 5–9. [Google Scholar] [CrossRef]
- Almagro Armenteros, J.J.; Tsirigos, K.D.; Sønderby, C.K.; Petersen, T.N.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019, 37, 420–423. [Google Scholar] [CrossRef]
- Tyagi, C.; Marik, T.; Szekeres, A.; Vágvölgyi, C.; Kredics, L.; Ötvös, F. Tripleurin XIIc: Peptide folding dynamics in aqueous and hydrophobic environment mimic using accelerated molecular dynamics. Molecules 2019, 24, 358. [Google Scholar] [CrossRef]
- Pierce, L.C.T.; Salomon-Ferrer, R.; Augusto, F.; De Oliveira, C.; McCammon, J.A.; Walker, R.C. Routine access to millisecond time scale events with accelerated molecular dynamics. J. Chem. Theory Comput. 2012, 8, 2997–3002. [Google Scholar] [CrossRef]
- Koukos, P.I.; Glykos, N.M. Grcarma: A fully automated task-oriented interface for the analysis of molecular dynamics trajectories. J. Comput. Chem. 2013, 34, 2310–2312. [Google Scholar] [CrossRef]
- Miao, Y.; Sinko, W.; Pierce, L.; Bucher, D.; Walker, R.C.; McCammon, J.A. Improved reweighting of accelerated molecular dynamics simulations for free energy calculation. J. Chem. Theory Comput. 2014, 10, 2677–2689. [Google Scholar] [CrossRef] [PubMed]
- Jing, Z.; Sun, H. A comment on the reweighting method for accelerated molecular dynamics simulations. J. Chem. Theory Comput. 2015, 11, 2395–2397. [Google Scholar] [CrossRef]
- Tian, W.; Chen, C.; Lei, X.; Zhao, J.; Liang, J. CASTp 3.0: Computed atlas of surface topography of proteins. Nucleic Acids Res. 2018, 46, W363–W367. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef]
- Release, S. 2: LigPrep. Schrödinger, LLC: New York, NY, USA, 2017. [Google Scholar]
- Wang, J.; Wang, W.; Kollman, P.; Case, D. Antechamber, an accessory software package for molecular mechanical calculations. J. Am. Chem. Soc. 2001, 222, U403. [Google Scholar]
- Case, D.; Ben-Shalom, I.; Brozell, S.; Cerutti, D.; Cheatham, T., III; Cruzeiro, V.; Gohlke, H.; Goetz, A.W.; Greene, D.; Harris, R.; et al. Amber 18. Univ. Calif. San Fr. 2018. [Google Scholar]
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Greene, D.; Xiao, L.; Qi, R.; Luo, R. Recent developments and applications of the MM/PBSA method. Front. Mol. Biosci. 2018, 4, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Pelosi, P.; Iovinella, I.; Zhu, J.; Wang, G.; Dani, F.R. Beyond chemoreception: Diverse tasks of soluble olfactory proteins in insects. Biol. Rev. 2018, 93, 184–200. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Mao, Y.; Zhang, L. Binding properties of four antennae-expressed chemosensory proteins (CSPs) with insecticides indicates the adaption of Spodoptera litura to environment. Pestic. Biochem. Physiol. 2018, 146, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Arnaud, P.; Offmann, B.; Picimbon, J.F. Genotyping and bio-sensing chemosensory proteins in insects. Sensors 2017, 17, 1801. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
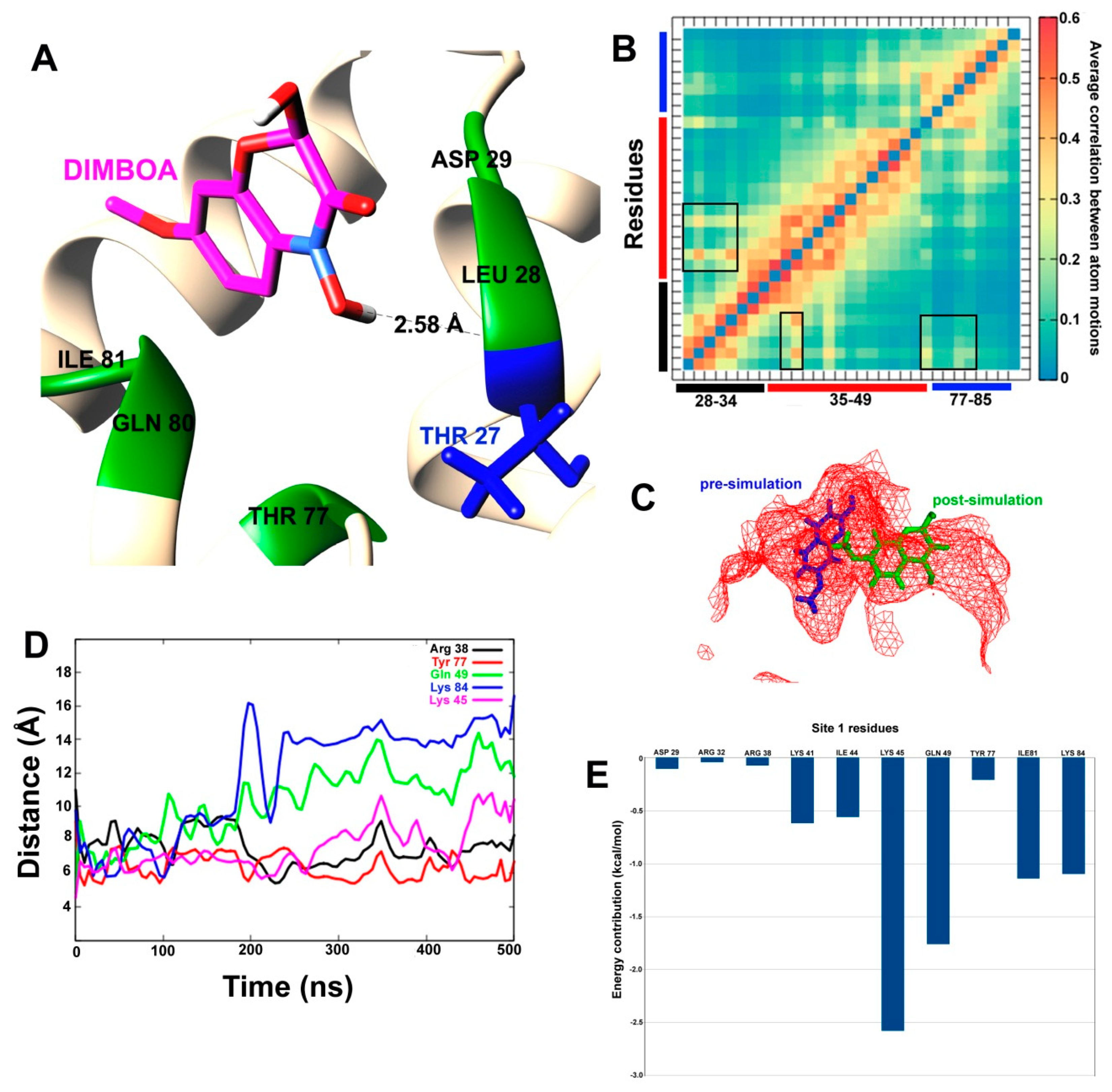{kind=link}
{kind=link}
| Site 1 | Residues Forming Hydrogen Bonds | Residues Involved in Hydrophobic Interactions |
|---|---|---|
| DIMBOA | Arg32, Arg38, Lys84 | Asp29, Lys41, Ala42, Lys45, Ile81 |
| MBOA | Arg38, Lys84 | Asp29, Arg32, Lys41, Ile44, Lys45, Tyr77, Ile81 |
| Nicotine | Asp29 | Arg38, Lys41, Ile44, Lys45, Tyr77, Gln80, Ile81, Lys84 |
| Site 2 | ||
| DIMBOA | Asp89 | Trp78, Lys79, Leu82, Tyr85, Gln90, Tyr91 |
| Nicotine | No H-bond formation | Trp78, Lys79, Ala83, Tyr85, Asp89, Tyr91 |
| Boost Parameters (kcal mol−1) | Folding Simulation of SlitCSP3, Iamd = 3 | SlitCSP3-Site1 with DIMBOA, Iamd = 1 | SlitCSP3-Site 2 with DIMBOA, iamd = 1 |
|---|---|---|---|
| EPtot | −48643 | −62150 | −62056 |
| Edihed | 1325 | 1408 | 1400 |
| a1, a2 | 4.0 | NA | NA |
| b1, b2 | 0.16 | 0.10 | 0.10 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, S.; Tyagi, C.; Rather, I.A.; Sabir, J.S.M.; Hassan, M.I.; Singh, A.; Singh, I.K. Molecular Modeling of Chemosensory Protein 3 from Spodoptera litura and Its Binding Property with Plant Defensive Metabolites. Int. J. Mol. Sci. 2020, 21, 4073. https://doi.org/10.3390/ijms21114073
Singh S, Tyagi C, Rather IA, Sabir JSM, Hassan MI, Singh A, Singh IK. Molecular Modeling of Chemosensory Protein 3 from Spodoptera litura and Its Binding Property with Plant Defensive Metabolites. International Journal of Molecular Sciences. 2020; 21(11):4073. https://doi.org/10.3390/ijms21114073
Chicago/Turabian StyleSingh, Sujata, Chetna Tyagi, Irfan A. Rather, Jamal S.M. Sabir, Md. Imtaiyaz Hassan, Archana Singh, and Indrakant Kumar Singh. 2020. "Molecular Modeling of Chemosensory Protein 3 from Spodoptera litura and Its Binding Property with Plant Defensive Metabolites" International Journal of Molecular Sciences 21, no. 11: 4073. https://doi.org/10.3390/ijms21114073
APA StyleSingh, S., Tyagi, C., Rather, I. A., Sabir, J. S. M., Hassan, M. I., Singh, A., & Singh, I. K. (2020). Molecular Modeling of Chemosensory Protein 3 from Spodoptera litura and Its Binding Property with Plant Defensive Metabolites. International Journal of Molecular Sciences, 21(11), 4073. https://doi.org/10.3390/ijms21114073