Structural Requirements of N-alpha-Mercaptoacetyl Dipeptide (NAMdP) Inhibitors of Pseudomonas Aeruginosa Virulence Factor LasB: 3D-QSAR, Molecular Docking, and Interaction Fingerprint Studies

 ,
,
Abstract
1. Introduction
2. Results and Discussion
2.1. Results of the QSAR Models
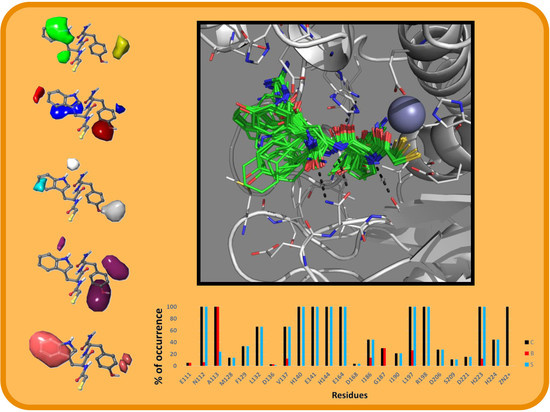
2.2. Molecular Docking Results
3. Materials and Methods
3.1. Dataset Collection and Pre-Processing
3.2. QSAR Methodology
3.3. Molecular Docking
3.4. IFP Calculations
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 3D | three-dimensional |
| AA | amino acid |
| HB | Hydrogen bonding |
| IFPs | interaction fingerprints analysis |
| LasB | Pseudomona elastase |
| NAMdP | N-alpha-mercaptoacetyl dipeptide |
| PDB | Protein Data Bank |
| RMSD | root mean square deviation |
| SAR | structure–activity relationship |
References
- O’Neill, J. Tackling Drug–resistant Infections Globally: Final Report and Recommendations; IICA: San José, Costa Rica, 2016. [Google Scholar]
- Fischbach, M.A.; Walsh, C.T. Antibiotics for emerging pathogens. Science 2009, 325, 1089–1093. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.D.; Wright, G.D. Antibacterial drug discovery in the resistance era. Nature 2016, 529, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Silver, L.L. Challenges of antibacterial discovery. Clin. Microbiol. Rev. 2011, 24, 71–109. [Google Scholar] [CrossRef] [PubMed]
- Payne, D.J.; Gwynn, M.N.; Holmes, D.J.; Pompliano, D.L. Drugs for bad bugs: Confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discov. 2007, 6, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Walsh, C.T.; Wencewicz, T.A. Prospects for new antibiotics: A molecule-centered perspective. J. Antibiot. 2014, 67, 7–22. [Google Scholar] [CrossRef] [PubMed]
- Tommasi, R.; Iyer, R.; Miller, A.A. Antibacterial drug discovery: Some assembly required. Acs Infect. Dis. 2018, 4, 686–695. [Google Scholar] [CrossRef]
- Crofts, T.S.; Gasparrini, A.J.; Dantas, G. Next-generation approaches to understand and combat the antibiotic resistome. Nat. Rev. Microbiol. 2017, 15, 422–434. [Google Scholar] [CrossRef]
- Clatworthy, A.E.; Pierson, E.; Hung, D.T. Targeting virulence: A new paradigm for antimicrobial therapy. Nat. Chem. Biol. 2007, 3, 541–548. [Google Scholar] [CrossRef]
- Lyczak, J.B.; Cannon, C.L.; Pier, G.B. Establishment of Pseudomonas aeruginosa infection: Lessons from a versatile opportunist. Microbes Infect. 2000, 2, 1051–1060. [Google Scholar] [CrossRef]
- Burrows, L.L. The Therapeutic pipeline for Pseudomonas aeruginosa infections. Acs Infect. Dis. 2018, 4, 1041–1047. [Google Scholar] [CrossRef]
- Morihara, K.; Tsuzuki, H.; Oda, K. Protease and elastase of Pseudomonas aeruginosa: Inactivation of human plasma alpha 1-proteinase inhibitor. Infect. Immun. 1979, 24, 188–193. [Google Scholar] [PubMed]
- Kessler, E.; Israel, M.; Landshman, N.; Chechick, A.; Blumberg, S. In vitro inhibition of Pseudomonas aeruginosa elastase by metal-chelating peptide derivatives. Infect. Immun. 1982, 38, 716–723. [Google Scholar] [PubMed]
- Jacobsen, J.A.; Fullagar, J.L.; Miller, M.T.; Cohen, S.M. Identifying chelators for metalloprotein inhibitors using a fragment-based approach. J. Med. Chem. 2011, 54, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Fullagar, J.L.; Garner, A.L.; Struss, A.K.; Day, J.A.; Martin, D.P.; Yu, J.; Cai, X.; Janda, K.D.; Cohen, S.M. Antagonism of a zinc metalloprotease using a unique metal-chelating scaffold: Tropolones as inhibitors of P. aeruginosa elastase. Chem. Commun. 2013, 49, 3197. [Google Scholar] [CrossRef]
- Thayer, M.M.; Flaherty, K.M.; Mckay, D.B. Three-dimensional structure of the elastase of Pseudomona aeruginosa at 1.5 angstroms resolution. J. Biol. Chem. 1991, 266, 2864–2871. [Google Scholar]
- Cathcart, G.R.A.; Quinn, D.; Greer, B.; Harriott, P.; Lynas, J.F.; Gilmore, B.F.; Walker, B. Novel inhibitors of the Pseudomonas aeruginosa virulence factor LasB: A potential therapeutic approach for the attenuation of virulence mechanisms in Pseudomonal infection. Antimicrob. Agents Chemother. 2011, 55, 2670–2678. [Google Scholar] [CrossRef]
- Zhu, J.; Cai, X.; Harris, T.L.; Gooyit, M.; Wood, M.; Lardy, M.; Janda, K.D. Disarming Pseudomonas aeruginosa virulence factor LasB by leveraging a caenorhabditis elegans infection model. Chem. Biol. 2015, 22, 483–491. [Google Scholar] [CrossRef]
- Kany, A.M.; Sikandar, A.; Haupenthal, J.; Yahiaoui, S.; Maurer, C.K.; Proschak, E.; Köhnke, J.; Hartmann, R.W. Binding mode characterization and early in vivo evaluation of fragment-like thiols as inhibitors of the virulence factor LasB from Pseudomonas aeruginosa. ACS Infect. Dis. 2018, 4, 988–997. [Google Scholar] [CrossRef]
- Cathcart, G.R.; Gilmore, B.F.; Greer, B.; Harriott, P.; Walker, B. Inhibitor profiling of the Pseudomonas aeruginosa virulence factor LasB using N-alpha mercaptoamide template-based inhibitors. Bioorganic Med. Chem. Lett. 2009, 19, 6230–6232. [Google Scholar] [CrossRef]
- Garner, A.L.; Struss, A.K.; Fullagar, J.L.; Agrawal, A.; Moreno, A.Y.; Cohen, S.M.; Janda, K.D. 3-Hydroxy-1-alkyl-2-methylpyridine-4(1 H)-thiones: Inhibition of the Pseudomonas aeruginosa Virulence Factor LasB. Acs Med. Chem. Lett. 2012, 3, 668–672. [Google Scholar] [CrossRef]
- Golbraikh, A.; Tropsha, A. Beware of q2! J. Mol. Graph. Model. 2002, 20, 269–276. [Google Scholar] [CrossRef]
- Roy, P.P.; Roy, K. On Some aspects of variable selection for partial least squares regression models. Qsar Comb. Sci. 2008, 27, 302–313. [Google Scholar] [CrossRef]
- Aptula, A.O.; Jeliazkova, N.G.; Schultz, T.W.; Cronin, M.T.D. The better predictive model: High q2 for the training set or low root mean square error of prediction for the test set? Qsar Comb. Sci. 2005, 24, 385–396. [Google Scholar] [CrossRef]
- Doweyko, A.M. 3D-QSAR illusions. J. Comput. Aided Mol. Des. 2004, 18, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Velázquez-Libera, J.L.; Navarro-Retamal, C.; Caballero, J. Insights into the structural requirements of 2(S)-amino-6-boronohexanoic acid derivatives as arginase I inhibitors: 3D-QSAR, docking, and interaction fingerprint studies. Int. J. Mol. Sci. 2018, 19, 2956. [Google Scholar] [CrossRef]
- Ramírez, D.; Caballero, J. Is It Reliable to take the molecular docking top scoring position as the best solution without considering available structural data? Molecules 2018, 23, 1038. [Google Scholar] [CrossRef]
- Gohlke, H.; Hendlich, M.; Klebe, G. Knowledge-based scoring function to predict protein-ligand interactions. J. Mol. Biol. 2000, 295, 337–356. [Google Scholar] [CrossRef]
- Caballero, J.; Morales-Bayuelo, A.; Navarro-Retamal, C. Mycobacterium tuberculosis serine/threonine protein kinases: Structural information for the design of their specific ATP-competitive inhibitors. J. Comput. Aided Mol. Des. 2018, 32, 1315–1336. [Google Scholar] [CrossRef]
- Velázquez-Libera, J.L.; Rossino, G.; Navarro-Retamal, C.; Collina, S.; Caballero, J. Docking, interaction fingerprint, and three-dimensional quantitative structure-activity relationship (3D-QSAR) of sigma1 receptor ligands, analogs of the neuroprotective agent RC-33. Front. Chem. 2019, 7, 496. [Google Scholar]
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A software program for pK a prediction and protonation state generation for drug-like molecules. J. Comput. -Aided Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef]
- Greenwood, J.R.; Calkins, D.; Sullivan, A.P.; Shelley, J.C. Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aqueous solution. J. Comput. -Aided Mol. Des. 2010, 24, 591–604. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Tirado-Rives, J. The OPLS [optimized potentials for liquid simulations] potential functions for proteins, energy minimizations for crystals of cyclic peptides and crambin. J. Am. Chem. Soc. 1988, 110, 1657–1666. [Google Scholar] [CrossRef] [PubMed]
- Shivakumar, D.; Williams, J.; Wu, Y.; Damm, W.; Shelley, J.; Sherman, W. Prediction of absolute solvation free energies using molecular dynamics free energy perturbation and the OPLS force field. J. Chem. Theory Comput. 2010, 6, 1509–1519. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein−ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef]
- Quesada-Romero, L.; Mena-Ulecia, K.; Tiznado, W.; Caballero, J. Insights into the interactions between maleimide derivates and GSK3β combining molecular docking and QSAR. PLoS ONE 2014, 9, e102212. [Google Scholar] [CrossRef]
- Quesada-Romero, L.; Caballero, J. Docking and quantitative structure–activity relationship of oxadiazole derivates as inhibitors of GSK3beta. Mol. Divers. 2014, 18, 149–159. [Google Scholar] [CrossRef]
- Mena-Ulecia, K.; Tiznado, W.; Caballero, J. Study of the differential activity of thrombin inhibitors using docking, QSAR, molecular dynamics, and MM-GBSA. PLoS ONE 2015, 10, e0142774. [Google Scholar] [CrossRef]
- Muñoz-Gutierrez, C.; Adasme-Carreño, F.; Fuentes, E.; Palomo, I.; Caballero, J. Computational study of the binding orientation and affinity of PPARγ agonists: Inclusion of ligand-induced fit by cross-docking. Rsc. Adv. 2016, 6, 64756–64768. [Google Scholar] [CrossRef]
- Deng, Z.; Chuaqui, C.; Singh, J. Structural interaction fingerprint (SIFt): A novel method for analyzing three-dimensional protein-ligand binding interactions. J. Med. Chem. 2004, 47, 337–344. [Google Scholar] [CrossRef]
- Singh, J.; Deng, Z.; Narale, G.; Chuaqui, C. Structural interaction fingerprints: A new approach to organizing, mining, analyzing, and designing protein-small molecule complexes. Chem. Biol. Drug Des. 2006, 67, 5–12. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NAMdP | Experimental pKi | Predicted pKi | QSAR Set | Glide Score (kcal/mol) | RMSD (Å) | %RefMatch | %MolMatch | |
|---|---|---|---|---|---|---|---|---|
| 1 | HSCH2CO–Ala–Arg–NH2 | −2.0607 | −2.3215 | Training | −7.652 | 0.95 | 38 | 67 |
| 2 | HSCH2CO–Ala–Asp–NH2 | −2.4997 | −2.1689 | Training | −5.239 | 0.95 | 38 | 78 |
| 3 | HSCH2CO–Ala–Leu–NH2 | −1.3222 | −2.1287 | Training | −5.067 | 0.96 | 38 | 78 |
| 4 | HSCH2CO–Ala–Lys–NH2 | −2.1847 | −2.3482 | Training | −5.975 | 0.94 | 38 | 74 |
| 5 | HSCH2CO–Ala–Trp–NH2 | −2.5966 | −2.3854 | Training | −5.161 | 3.09 | 62 | 96 |
| 6 | HSCH2CO–Ala–Val–NH2 | −1.7076 | −2.1121 | Test | −4.774 | 0.95 | 38 | 82 |
| 7 | HSCH2CO–Arg–Asp–NH2 | −2.8129 | −2.3112 | Test | −5.663 | 0.94 | 43 | 67 |
| 8 | HSCH2CO–Arg–Lys–NH2 | −2.1303 | −2.4909 | Training | −7.267 | 0.91 | 43 | 64 |
| 9 | HSCH2CO–Arg–Phe–NH2 | −2.3502 | −2.2621 | Training | −6.318 | 1.05 | 46 | 63 |
| 10 | HSCH2CO–Arg–Trp–NH2 | −2.0969 | −2.5236 | Training | −6.212 | 3.01 | 68 | 83 |
| 11 | HSCH2CO–Asn–Arg–NH2 | −2.4472 | −2.4689 | Training | −8.582 | 0.98 | 46 | 71 |
| 12 | HSCH2CO–Asn–Leu–NH2 | −2.7059 | −2.2637 | Training | −5.526 | 0.97 | 46 | 81 |
| 13 | HSCH2CO–Asn–Lys–NH2 | −2.4609 | −2.4835 | Test | −6.439 | 0.97 | 46 | 77 |
| 14 | HSCH2CO–Asn–Phe–NH2 | −1.5682 | −2.2525 | Training | −6.015 | 1.00 | 49 | 75 |
| 15 | HSCH2CO–Asn–Trp–NH2 | −1.8451 | −2.5190 | Training | −5.518 | 2.87 | 70 | 96 |
| 16 | HSCH2CO–Asn–Val–NH2 | −2.2553 | −2.2475 | Training | −5.682 | 0.99 | 46 | 85 |
| 17 | HSCH2CO–Asp–Arg–NH2 | −2.7412 | −2.5774 | Training | −8.351 | 0.98 | 46 | 71 |
| 18 | HSCH2CO–Asp–Leu–NH2 | −2.0719 | −2.3721 | Test | −5.413 | 0.95 | 46 | 81 |
| 19 | HSCH2CO–Asp–Lys–NH2 | −2.6911 | −2.5914 | Training | −5.397 | 0.96 | 46 | 77 |
| 20 | HSCH2CO–Asp–Trp–NH2 | −2.3160 | −2.6275 | Training | −4.564 | 2.86 | 70 | 96 |
| 21 | HSCH2CO–Cys–Arg–NH2 | −2.8102 | −2.4454 | Training | −5.857 | 1.18 | 41 | 68 |
| 22 | HSCH2CO–Cys–Lys–NH2 | −2.4378 | −2.4601 | Training | −3.691 | 1.18 | 41 | 75 |
| 23 | HSCH2CO–Cys–Phe–NH2 | −2.1173 | −2.2302 | Test | −3.155 | 0.97 | 43 | 73 |
| 24 | HSCH2CO–Cys–Trp–NH2 | −2.0334 | −2.4966 | Test | −3.091 | 3.05 | 65 | 96 |
| 25 | HSCH2CO–Cys–Val–NH2 | −2.2068 | −2.2235 | Training | −2.802 | 0.95 | 41 | 83 |
| 26 | HSCH2CO–Gln–Arg–NH2 | −2.3365 | −2.4675 | Test | −8.794 | 0.97 | 43 | 64 |
| 27 | HSCH2CO–Gln–Leu–NH2 | −2.7324 | −2.2627 | Test | −6.350 | 0.95 | 43 | 73 |
| 28 | HSCH2CO–Gln–Lys–NH2 | −2.5798 | −2.4815 | Test | −6.717 | 0.94 | 43 | 70 |
| 29 | HSCH2CO–Gln–Trp–NH2 | −1.9590 | −2.5192 | Training | −6.093 | 2.93 | 68 | 89 |
| 30 | HSCH2CO–Gln–Val–NH2 | −2.9717 | −2.2451 | Training | −6.254 | 0.93 | 43 | 76 |
| 31 | HSCH2CO–Glu–Lys–NH2 | −2.7427 | −2.6041 | Training | −5.895 | 0.93 | 43 | 70 |
| 32 | HSCH2CO–Glu–Phe–NH2 | −2.1644 | −2.3753 | Training | −4.813 | 1.15 | 46 | 68 |
| 33 | HSCH2CO–Glu–Trp–NH2 | −2.8476 | −2.6418 | Training | −5.828 | 2.97 | 68 | 89 |
| 34 | HSCH2CO–Glu–Val–NH2 | −2.8633 | −2.3677 | Training | −6.019 | 0.96 | 43 | 76 |
| 35 | HSCH2CO–Gly–Arg–NH2 | −2.8069 | −2.5394 | Training | −8.393 | 0.94 | 35 | 65 |
| 36 | HSCH2CO–Gly–Leu–NH2 | −2.1399 | −2.3336 | Training | −5.225 | 0.94 | 35 | 76 |
| 37 | HSCH2CO–Gly–Lys–NH2 | −2.6542 | −2.5543 | Training | −6.274 | 0.94 | 35 | 72 |
| 38 | HSCH2CO–Gly–Phe–NH2 | −1.7076 | −2.3247 | Training | −5.604 | 0.97 | 38 | 70 |
| 39 | HSCH2CO–Gly–Trp–NH2 | −2.0864 | −2.5911 | Training | −5.584 | 3.16 | 59 | 96 |
| 40 | HSCH2CO–Gly–Val–NH2 | −2.6599 | −2.3163 | Training | −5.022 | 0.94 | 35 | 81 |
| 41 | HSCH2CO–His–Ala–NH2 | −0.5587 | −1.6658 | Training | −3.155 | 0.93 | 38 | 70 |
| 42 | HSCH2CO–His–Leu–NH2 | −2.4857 | −1.68 | Training | −3.252 | 0.92 | 38 | 61 |
| 43 | HSCH2CO–His–Lys–NH2 | −2.5211 | −1.8976 | Training | −3.563 | 0.96 | 38 | 58 |
| 44 | HSCH2CO–His–Phe–NH2 | −1.3222 | −1.6688 | Training | −3.128 | 0.95 | 41 | 58 |
| 45 | HSCH2CO–His–Trp–NH2 | −1.2553 | −1.9353 | Training | −3.052 | 3.10 | 62 | 79 |
| 46 | HSCH2CO–His–Val–NH2 | −1.6721 | −1.6632 | Training | −2.932 | 0.94 | 38 | 64 |
| 47 | HSCH2CO–Ile–Asp–NH2 | −2.1523 | −1.5871 | Training | −4.167 | 1.00 | 43 | 76 |
| 48 | HSCH2CO–Ile–Gln–NH2 | −0.5717 | −0.5951 | Training | −6.036 | 0.92 | 43 | 73 |
| 49 | HSCH2CO–Ile–Leu–NH2 | −0.1004 | −1.5472 | Training | −4.684 | 0.94 | 43 | 76 |
| 50 | HSCH2CO–Ile–Lys–NH2 | −2.2788 | −1.7662 | Training | −5.633 | 0.95 | 43 | 73 |
| 51 | HSCH2CO–Ile–Thr–NH2 | −0.5328 | −1.3756 | Test | −6.040 | 0.93 | 43 | 80 |
| 52 | HSCH2CO–Ile–Trp–NH2 | −2.5635 | −1.8041 | Training | −5.822 | 2.92 | 68 | 93 |
| 53 | HSCH2CO–Ile–Tyr–NH2 | −0.3139 | 0.226 | Training | −5.897 | 0.94 | 46 | 68 |
| 54 | HSCH2CO–Ile–Val–NH2 | −0.2625 | −1.5298 | Training | −4.851 | 0.93 | 43 | 80 |
| 55 | HSCH2CO–Leu–Arg–NH2 | −2.7945 | −2.013 | Test | −7.750 | 1.16 | 46 | 71 |
| 56 | HSCH2CO–Leu–Asp–NH2 | −2.7686 | −1.8479 | Training | −4.075 | 1.10 | 46 | 81 |
| 57 | HSCH2CO–Leu–Leu–NH2 | −1.7243 | −1.8061 | Training | −5.417 | 1.23 | 46 | 81 |
| 58 | HSCH2CO–Leu–Lys–NH2 | −1.1461 | −2.0253 | Test | −5.291 | 1.10 | 46 | 77 |
| 59 | HSCH2CO–Leu–Phe–NH2 | −2.0531 | −1.7966 | Training | −3.977 | 1.26 | 49 | 75 |
| 60 | HSCH2CO–Leu–Trp–NH2 | −2.4771 | −2.0634 | Training | −4.790 | 2.98 | 70 | 96 |
| 61 | HSCH2CO–Lys–Asp–NH2 | −2.9872 | −2.4763 | Test | −6.095 | 0.97 | 43 | 73 |
| 62 | HSCH2CO–Lys–Leu–NH2 | −2.0899 | −2.4284 | Training | −6.349 | 0.96 | 43 | 73 |
| 63 | HSCH2CO–Lys–Lys–NH2 | −2.6365 | −2.6554 | Training | −7.149 | 0.96 | 43 | 70 |
| 64 | HSCH2CO–Lys–Phe–NH2 | −2.1004 | −2.4267 | Training | −6.502 | 0.97 | 46 | 68 |
| 65 | HSCH2CO–Lys–Val–NH2 | −2.7443 | −2.4166 | Training | −6.072 | 0.96 | 43 | 76 |
| 66 | HSCH2CO–Met–Arg–NH2 | −0.8195 | −1.8487 | Training | −7.798 | 1.00 | 43 | 67 |
| 67 | HSCH2CO–Met–Asp–NH2 | −0.8451 | −1.6836 | Training | −4.891 | 0.99 | 43 | 76 |
| 68 | HSCH2CO–Met–Lys–NH2 | −0.5866 | −1.8627 | Training | −6.684 | 0.97 | 43 | 73 |
| 69 | HSCH2CO–Met–Phe–NH2 | −2.9380 | −1.6339 | Training | −5.564 | 1.19 | 46 | 71 |
| 70 | HSCH2CO–Met–Trp–NH2 | −2.3096 | −1.9004 | Training | −4.916 | 2.89 | 68 | 93 |
| 71 | HSCH2CO–Met–Tyr–NH2 | −0.5623 | 0.1316 | Training | −5.955 | 1.26 | 46 | 68 |
| 72 | HSCH2CO–Met–Val–NH2 | −1.9912 | −1.6213 | Training | −5.815 | 0.97 | 43 | 80 |
| 73 | HSCH2CO–Phe–Gln–NH2 | 0.1226 | −0.2028 | Training | −6.245 | 0.96 | 38 | 56 |
| 74 | HSCH2CO–Phe–Ile–NH2 | −0.6503 | −1.1362 | Training | −5.120 | 0.93 | 38 | 58 |
| 75 | HSCH2CO–Phe–Leu–NH2 | −2.8096 | −1.146 | Training | −4.450 | 0.95 | 38 | 58 |
| 76 | HSCH2CO–Phe–Lys–NH2 | −1.8808 | −1.3642 | Test | −5.413 | 0.95 | 38 | 56 |
| 77 | HSCH2CO–Phe–Met–NH2 | −0.4502 | −0.7559 | Training | −6.914 | 0.91 | 38 | 58 |
| 78 | HSCH2CO–Phe–Phe–NH2 | −2.1644 | −1.1354 | Training | −5.606 | 1.23 | 41 | 56 |
| 79 | HSCH2CO–Phe–Trp–NH2 | −2.3139 | −1.4029 | Training | −7.568 | 1.76 | 62 | 77 |
| 80 | HSCH2CO–Phe–Tyr–NH2 | 1.3872 | 0.6276 | Training | −6.918 | 0.95 | 41 | 54 |
| 81 | HSCH2CO–Phe–Val–NH2 | −1.0414 | −1.1315 | Training | −4.537 | 0.96 | 38 | 61 |
| 82 | HSCH2CO–Pro–Arg–NH2 | −1.7482 | −2.2055 | Training | −7.338 | 1.53 | 38 | 73 |
| 83 | HSCH2CO–Pro–Leu–NH2 | −2.3909 | −2.0021 | Training | −7.612 | 1.40 | 38 | 85 |
| 84 | HSCH2CO–Pro–Lys–NH2 | −2.8842 | −2.2260 | Training | −8.277 | 1.75 | 38 | 81 |
| 85 | HSCH2CO–Pro–Trp–NH2 | −2.7497 | −2.2623 | Training | −5.630 | 2.96 | 62 | 96 |
| 86 | HSCH2CO–Pro–Val–NH2 | −2.1959 | −1.9837 | Test | −7.628 | 1.48 | 38 | 92 |
| 87 | HSCH2CO–Ser–Arg–NH2 | −2.6474 | −2.4949 | Test | −8.952 | 0.94 | 41 | 68 |
| 88 | HSCH2CO–Ser–Leu–NH2 | −2.7076 | −2.2896 | Training | −6.262 | 1.14 | 41 | 79 |
| 89 | HSCH2CO–Ser–Phe–NH2 | −1.8751 | −2.2801 | Training | −6.316 | 1.15 | 43 | 73 |
| 90 | HSCH2CO–Ser–Val–NH2 | −2.3598 | −2.2737 | Training | −5.362 | 1.12 | 41 | 83 |
| 91 | HSCH2CO–Thr–Arg–NH2 | −2.7896 | −2.2227 | Training | −9.096 | 1.07 | 41 | 65 |
| 92 | HSCH2CO–Thr–Phe–NH2 | −2.3522 | −2.0078 | Test | −5.473 | 1.13 | 43 | 70 |
| 93 | HSCH2CO–Thr–Trp–NH2 | −1.8129 | −2.2743 | Training | −6.166 | 2.99 | 65 | 92 |
| 94 | HSCH2CO–Thr–Val–NH2 | −2.5763 | −2.0035 | Training | −5.808 | 1.06 | 41 | 79 |
| 95 | HSCH2CO–Trp–Arg–NH2 | −1.3979 | −1.1114 | Test | −8.417 | 0.94 | 38 | 47 |
| 96 | HSCH2CO–Trp–Asp–NH2 | −1.5798 | −0.9463 | Training | −6.044 | 0.96 | 38 | 52 |
| 97 | HSCH2CO–Trp–Glu–NH2 | −1.9590 | −0.9930 | Training | −6.483 | 0.99 | 38 | 50 |
| 98 | HSCH2CO–Trp–Ile–NH2 | −0.0128 | −0.8916 | Training | −7.636 | 0.51 | 38 | 52 |
| 99 | HSCH2CO–Trp–Leu–NH2 | −0.5635 | −0.9039 | Training | −5.190 | 0.95 | 38 | 52 |
| 100 | HSCH2CO–Trp–Lys–NH2 | −1.0000 | −1.1254 | Training | −8.613 | 0.66 | 38 | 50 |
| 101 | HSCH2CO–Trp–Phe–NH2 | −0.0414 | −0.8966 | Test | −6.127 | 1.04 | 41 | 50 |
| 102 | HSCH2CO–Trp–Trp–NH2 | −1.6902 | −1.1641 | Training | −5.961 | 3.04 | 62 | 70 |
| 103 | HSCH2CO–Trp–Tyr–NH2 | 1.3925 | 0.8661 | Training | −5.518 | 0.98 | 41 | 48 |
| 104 | HSCH2CO–Trp–Val–NH2 | −0.6096 | −0.8870 | Training | −5.893 | 0.98 | 38 | 54 |
| 105 | HSCH2CO–Tyr–Arg–NH2 | −0.4698 | −1.0968 | Training | −10.083 | 0.59 | 38 | 50 |
| 106 | HSCH2CO–Tyr–Asp–NH2 | −0.7404 | −0.9317 | Training | −6.700 | 0.43 | 38 | 56 |
| 107 | HSCH2CO–Tyr–Glu–NH2 | −1.4314 | −0.9771 | Training | −7.712 | 0.47 | 38 | 54 |
| 108 | HSCH2CO–Tyr–Leu–NH2 | −1.5185 | −0.8919 | Training | −7.353 | 0.43 | 38 | 56 |
| 109 | HSCH2CO–Tyr–Lys–NH2 | −0.9294 | −1.1111 | Training | −8.097 | 0.52 | 38 | 54 |
| 110 | HSCH2CO–Tyr–Phe–NH2 | −0.8129 | −0.8823 | Test | −7.380 | 0.50 | 41 | 54 |
| 111 | HSCH2CO–Tyr–Trp–NH2 | −1.1461 | −1.1498 | Training | −7.392 | 2.81 | 62 | 74 |
| 112 | HSCH2CO–Tyr–Tyr–NH2 | −0.3181 | 0.8790 | Test | −7.686 | 0.87 | 41 | 52 |
| 113 | HSCH2CO–Tyr–Val–NH2 | 0.1146 | −0.8746 | Training | −7.274 | 0.46 | 38 | 58 |
| 114 | HSCH2CO–Val–Arg–NH2 | −1.8388 | −1.8918 | Training | −8.183 | 1.00 | 41 | 65 |
| 115 | HSCH2CO–Val–Leu–NH2 | −1.8388 | −1.6874 | Test | −5.767 | 0.94 | 41 | 75 |
| 116 | HSCH2CO–Val–Lys–NH2 | −1.3424 | −1.9057 | Training | −5.501 | 0.95 | 41 | 71 |
| 117 | HSCH2CO–Val–Phe–NH2 | −1.8573 | −1.6755 | Training | −5.001 | 0.95 | 43 | 70 |
| 118 | HSCH2CO–Val–Val–NH2 | −1.0000 | −1.6676 | Test | −5.225 | 0.95 | 41 | 79 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Velázquez-Libera, J.L.; Murillo-López, J.A.; F. de la Torre, A.; Caballero, J. Structural Requirements of N-alpha-Mercaptoacetyl Dipeptide (NAMdP) Inhibitors of Pseudomonas Aeruginosa Virulence Factor LasB: 3D-QSAR, Molecular Docking, and Interaction Fingerprint Studies. Int. J. Mol. Sci. 2019, 20, 6133. https://doi.org/10.3390/ijms20246133
Velázquez-Libera JL, Murillo-López JA, F. de la Torre A, Caballero J. Structural Requirements of N-alpha-Mercaptoacetyl Dipeptide (NAMdP) Inhibitors of Pseudomonas Aeruginosa Virulence Factor LasB: 3D-QSAR, Molecular Docking, and Interaction Fingerprint Studies. International Journal of Molecular Sciences. 2019; 20(24):6133. https://doi.org/10.3390/ijms20246133
Chicago/Turabian StyleVelázquez-Libera, José Luis, Juliana Andrea Murillo-López, Alexander F. de la Torre, and Julio Caballero. 2019. "Structural Requirements of N-alpha-Mercaptoacetyl Dipeptide (NAMdP) Inhibitors of Pseudomonas Aeruginosa Virulence Factor LasB: 3D-QSAR, Molecular Docking, and Interaction Fingerprint Studies" International Journal of Molecular Sciences 20, no. 24: 6133. https://doi.org/10.3390/ijms20246133
APA StyleVelázquez-Libera, J. L., Murillo-López, J. A., F. de la Torre, A., & Caballero, J. (2019). Structural Requirements of N-alpha-Mercaptoacetyl Dipeptide (NAMdP) Inhibitors of Pseudomonas Aeruginosa Virulence Factor LasB: 3D-QSAR, Molecular Docking, and Interaction Fingerprint Studies. International Journal of Molecular Sciences, 20(24), 6133. https://doi.org/10.3390/ijms20246133