Abstract
Tea (Camellia sinensis) is enriched with bioactive secondary metabolites, and is one of the most popular nonalcoholic beverages globally. Two tea reference genomes have been reported; however, the functional analysis of tea genes has lagged, mainly due to tea’s recalcitrance to genetic transformation and the absence of alternative high throughput heterologous expression systems. A full-length cDNA collection with a streamlined cloning system is needed in this economically important woody crop species. RNAs were isolated from nine different vegetative tea tissues, pooled, then used to construct a normalized full-length cDNA library. The titer of unamplified and amplified cDNA library was 6.89 × 106 and 1.8 × 1010 cfu/mL, respectively; the library recombinant rate was 87.2%. Preliminary characterization demonstrated that this collection can complement existing tea reference genomes and facilitate rare gene discovery. In addition, to streamline tea cDNA cloning and functional analysis, a binary vector (pBIG2113SF) was reengineered, seven tea cDNAs isolated from this library were successfully cloned into this vector, then transformed into Arabidopsis. One FL-cDNA, which encodes a putative P1B-type ATPase 5 (CsHMA5), was characterized further as a proof of concept. We demonstrated that overexpression of CsHMA5 in Arabidopsis resulted in copper hyposensitivity. Thus, our data demonstrated that this represents an efficient system for rare gene discovery and functional characterization of tea genes. The integration of a tea FL-cDNA collection with efficient cloning and a heterologous expression system would facilitate functional annotation and characterization of tea genes.
1. Introduction
Full-length cDNA (FL-cDNA) is the DNA complement to an mRNA sequence that covers the region near the 5′ cap structure to the poly(A) tail [1]. It is essential to identify exon–intron boundaries and gene-coding regions within genomic sequences, and for gene-function analysis at the transcriptional and translational levels [2]. In addition, the FL-cDNA sequences can improve the quality of genome annotation [3], and provide accurate transcription start sites (TSSs), which can increase our understanding of the transcriptional regulation and associated network because transcription factor-binding sites are located around TSSs [4]. Furthermore, FL-cDNA makes it possible to manipulate gene expression in heterologous systems, thus aiding the functional analysis of genes by reverse genetics. Due to these advantages, FL-cDNA technology has been widely applied to the genomic researches of different plant species, including Arabidopsis [2], rice [5], soybean [6], corn [7], tomato [8], citrus [3], cotton [9,10], and wheat [11]. However, straight-forward, high-throughput analysis of cDNA libraries is complicated by the differential abundance of various transcripts from any particular cell or tissue; as a result, the identification of rare transcripts from the standard library is often difficult [12]. Thus, a normalized FL-cDNA library is required for rare gene discovery [13]. A normalized cDNA library is that cDNAs derived from abundant mRNAs are greatly reduced, thus originally high- and low-abundance transcripts present in the library at similar frequency.
Tea (Camellia sinensis) is rich in bioactive secondary metabolites, and one of the most popular nonalcoholic beverages globally. In 2017, 6 million tons of tea was produced by 20 top production countries, according to the Food and Agriculture Organization of the United Nations (http://faostat.fao.org/). To this day, tea plants have been cultivated in more than fifty countries, and provide massive job opportunities and social wealth [14]. Due to tea’s economic and health importance, many efforts have been made to characterize its agronomic traits. One popular method in tea research is expressed sequence tags (ESTs). Studies of tea transcriptome have provided new insights of the molecular functions of agronomy important genes [15,16,17,18,19]. So far, tea tree genomes from Camellia sinensis var. assamica (CSA.; Assam type) and Camellia sinensis var. sinensis (CSS.; Chinese type) have been reported, with 36,951 and 33,932 annotated protein-coding genes, respectively [20,21], and provide valuable resources for the functional characterization of tea genes. Recently, Qiao et al. (2018) applied single molecular real-time (SMRT) sequencing to obtain full-length enriched transcripts for the identification of alternative splicing events [22]. However, a normalized FL-cDNA library in tea tree has not been reported.
Molecular biology and functional genetic studies have been limited in tea tree, mainly due to two factors: (1) Tea tree is recalcitrant to genetic transformation. The first transgenic tea was reported more than a decade back [23], since then not much progress has been reported even though extensive efforts have been invested. (2) High throughput heterologous expression systems have not been established in tea. To complement and extend previous works, we combined SMART method with duplex-specific nuclease (DSN) treatment, and successfully constructed a normalized tea FL-cDNA library. In order to accelerate the functional analysis of tea genes, a binary vector was reengineered to streamline gene cloning and heterologous expression in Arabidopsis. The short generation time and efficient transformation frequency of Arabidopsis would accelerate the functional characterization of tea genes. As a proof of the practical application of this FL-cDNA collection, one tea FL-cDNA, which encodes the P1B-type ATPase 5 (CsHMA5), was cloned and expressed in Arabidopsis. The overexpression of CsHMA5 resulted in copper hyposensitivity, and the phenotypes are consistent with a role of CsHMA5 as a bona fide tea orthologs of the Arabidopsis HMA5 gene. The protocols described in this study have thus been proven to be useful for functional characterization of tea genes.
2. Results and Discussion
2.1. Construction of a Normalized Full-Length Enriched cDNA Library of Camellia sinensis
In this study, a new normalized EST collection with enrichment of full-length and rare transcripts of tea was successfully generated by applying an integrated method. Camellia sinensis cv. ‘Fuding Dabaicha’ was selected as material for several considerations: (1) it is a national elite germplasm and is widely cultivated in China for the production of green tea, white tea, oolong tea, and black tea; (2) it has been widely used as a maternal line in multiple tea breeding programs in China and produced dozens of new elite germplasms; (3) it is diploid. To maximize the cDNAs included in this library, total RNA was isolated from nine different tissue types, including buds, tender stems, tender leaves, mature leaves, flower buds, flowers, fruit pericarps, roots, and bark. Equal amounts of total RNAs from these tissues were pooled for cDNA library construction. A λ phage (λTriplEx2) was selected as cloning vector since it is not biased toward short insert size. In total, 580 µL of unamplified cDNA library was obtained with a titer of 6.89 × 106 cfu/mL and recombination rate of 87.2%; the titer reached 1.8 × 1010 cfu/mL after library amplification.
The insert size was evaluated by colony PCR from randomly selected white plaques, which were in the range of 0.8–2.5 kb with medium insertion size of 1.5 kb, as seen in Figure 1.
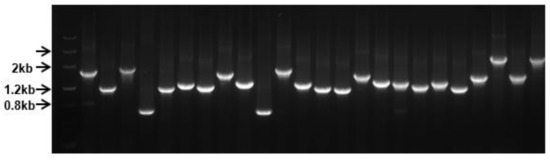
Figure 1.
Library insert size evaluation by clonal PCR. Twenty-four white plaques were randomly picked from plates for PCR amplification.
2.2. Comparison with Tea CSS and CSA Genome
Two tea reference genomes from Yunkang 10 (Camellia sinensis var. assamica (CSA.; Assam type)) and Shuchazao (Camellia sinensis var. sinensis (CSS.; Chinese type)) have been published with 36,951 and 33,932 annotated protein-coding genes, respectively [20,21]. The quality of our normalized tea cDNA library was further evaluated by sequencing. Forty-four white plaques were randomly selected and sequenced, after the vector backbone and additional sequences added during cDNA synthesis were trimmed, the obtained sequences of FL-cDNAs were deposited in NCBI database, as seen in Table 1. No redundancy was found, and a total of 44 unigenes were obtained from those 44 clones, including 42 protein-coding RNAs, one long chain noncoding RNA (clone 43, MN027194), and one natural antisense transcript to CSA010175 or TEA005630 (clone 34, MN158199) [24]. Based on gene annotation, these encoded proteins were involved in various cellular processes, including metabolism, transport, protein/RNA modification or transcription factors. Some well-known abundant RNAs, such as chlorophyll a/b binding protein, ribulose-1,5-biphosphate carboxylase, actin, 18S rRNA, alpha-tubulin 10, and ubiquitin [9,25], were not present from this list, suggesting the successful normalization of this cDNA library.

Table 1.
Forty-four unigenes identified from the tea cDNA library.
These 44 FL-cDNA sequences were used to query the CSS and CSA database, respectively. Thirty-three cDNAs were commonly detected in both databases; seven cDNAs (MK795747, MK795755, MK795757, MK795761, MK795762, MK795764, and MK889354) were found from CSS database but not from CSA database; two cDNAs (MN102719 and MK795766) were found from CSA only. The other two cDNAs, one encoding a natural antisense transcript (clone 34), and the other one encoding a putative long chain noncoding RNA (clone number 43), were not found from CSS and CSA databases, as seen in Figure 2. These observations demonstrated that this normalized tea FL-cDNA library can complement the existing tea genome databases, and identify rare protein coding cDNAs, noncoding RNAs, or natural antisense transcripts. In addition, once this library was extensively sequenced, these sequences’ information can be used to improve the prediction of tea genetic structures.
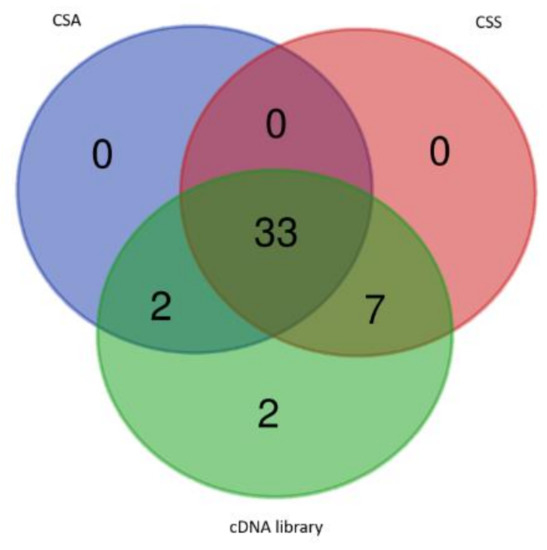
Figure 2.
The 44 FL-cDNAs identified in this study showed partial overlap with the Camellia sinensis var. assamica (CSA.; Assam type) and Camellia sinensis var. sinensis (CSS.; Chinese type) databases.
2.3. 5′-UTR and 3′-UTR Analysis
At the whole-genome level, UTRs provide functional specificity to genes in a length-dependent manner [26]. For the calculation of the average length of 5′-UTR and 3′-UTR of tea, three cDNAs were excluded, including clone 29 (incomplete 5′- and 3′-UTR), clone 34 (natural antisense transcript), and clone 43 (noncoding RNA), as seen in Table 1. For the other 41 cDNAs, clone 7 has the shortest (4 bp) 5′-UTR, while clone 40 has the longest (601 bp); clone 42 shows the shortest (39 bp) 3′-UTR, while clone 18 shows and the longest (627 bp). The 5′-UTR lengths for MN027184, MK795746, and MK795755 were only 4, 8, and 11 nt, respectively, as seen in Table 1, raising the question of whether they were truncated during reverse transcription. Thus, they were further verified by RT-PCR. Two flanking forward primers were designed for each cDNA: the first forward primer (F1) ends before the first nucleotide of our identified 5′-UTR sequence, and the second forward primer (F2) starts from the first nucleotide of our identified 5′-UTR sequence; the reverse primers (R) were designed from the respective coding region, as seen in Table S1 A hexamer was used for reverse transcription reaction. If the identified 5′-UTRs were truncated during the library construction, or represent a short version of alternative splicing variant, both F1/R and F2/R primer sets can amplify a predicted fragment; if the identified 5′-UTRs were indeed complete, one would expect that the F2/R primer sets to amplify a predicted fragment, whereas the F1/R primer sets cannot, as seen in Figure S1.
For MN027184, the annotated CDS from the tea genome database (CSA015215 and TEA028910) has an additional 900 nt before our identified 5′-UTR sequence, as seen in Figure S2A. Both F1/R and F2/R primer sets amplified the predicted size of band, as seen in Figure S2B, suggesting that MN027184 could represent a short isoform of alternative splicing variant. MK795746 showed the same start codon as the annotated CDS from the tea genome database (CSA022245), as seen in Figure S3A, both F1/R and F2/R primer sets amplified a predicted size of band, as seen in Figure S3B, suggesting that 5′-UTR of MK795746 could be truncated. The start codon usage of MK795755 is 13 nt shorter than that of CSS024713, as seen in Figure S4A, both F1/R and F2/R primer sets amplified a predicted size of band, as seen in Figure S4B, suggesting that MK795755 could represent a shorter alternative splicing variant. Global dissection of alternative spicing in tea identified 28,980 AS events [22,26].
Based on the validation results above, these three clones were excluded from the calculation of the average length of 5′-UTR, but they are included for the calculation of the average length of 3′-UTR. Our data indicated that the average lengths for the 5′-UTR and 3′-UTR are 128 and 296 bp, respectively. In rice, the average lengths of 5′- and 3′-UTRs are 259 and 469 bp, respectively, but only 155 and 242 bp in Arabidopsis [27]. Thus, UTRs in tea tree share higher similarity with Arabidopsis than rice. This is unexpected, since the tea genome (3.02 Gb) is 24-fold larger than that of Arabidopsis (129 Mb); In addition, as a perennial species tea tree may require more complicated gene regulations compared to annual plant species such as Arabidopsis or rice. Thus, it is anticipated that the average length of 5′-UTR and 3′-UTR of tea should be longer than that of Arabidopsis or rice. Arabidopsis tea tree is a dicot, which may suggest that the 5′-UTR and 3′-UTR from dicot and monocot have been independently evolved after their evolutionary divergence. Since only 41 randomly selected tea FL-cDNAs were used here for UTR length calculation, this tentative estimation requires further validation. The extensive sequencing of this cDNA library in the future would provide more conclusive evidence.
2.4. Ectopic Expression of Tea Genes in Arabidopsis
Arabidopsis has several advantages for functional analysis of heterologous genes, such as its small size, short generation time, and its high transformation efficiency [28]. Thus, it was used for heterologous expression of tea FL-cDNA. To streamline tea FL-cDNA cloning into the binary vector, the binary vector pBIG2113SF was reengineered to make Sfi I cloning sites identical to those of the λ phage vector [29], thus tea FL-cDNAs recovered from this cDNA library can be directly cloned into the pBIG2113SF-M vector with correct orientation for gene expression analysis.
To validate this cloning and expression system, seven tea FL-cDNAs were selected (clone numbers 4, 8, 9, 12, 21, 27, and 28), as seen in Table 1, and their FL-cDNAs were recovered from their respective plasmids by Sfi I digestion, ligated into pBIG2113SF–M vector, and transformed into Arabidopsis. RT-PCR analysis of transgenic plants demonstrated that all seven tea genes were successfully transformed and expressed in Arabidopsis, as seen in Figure 3.
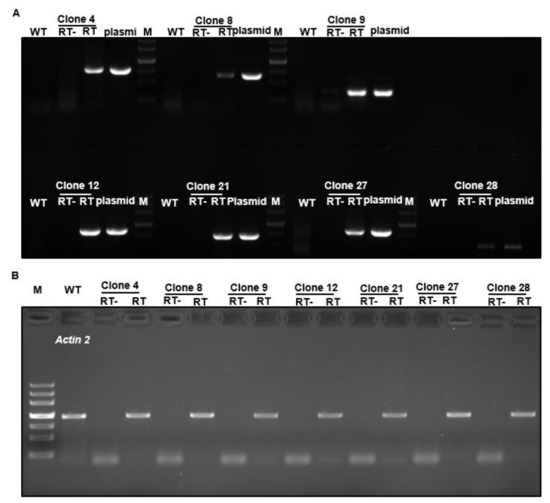
Figure 3.
RT-PCR confirmation of transgene expression in Arabidopsis. Camellia sinensis FL-cDNAs from clones 4, 8, 9, 12, 21, 27, and 28 were transformed into Arabidopsis. (A) Total RNA was isolated from respective transgenic plants, tea transgene-specific primer pairs were used for RT-PCR. The respective plasmids were used as positive controls for the PCR. (B) Actin 2 was amplified as the loading control. RT-: no reverse transcriptase added into RT reaction mixture; RT: with reverse transcriptase added into RT reaction mixture.
2.5. Overexpression of a Tea Gene Encoding P1B-Type ATPase 5 in Arabidopsis Resulted in Hyposensitivity to Copper
Clone 8 corresponds to a 1183 nt transcript with a 209-nt 5′-UTR and 248-nt 3′-UTR, and encodes a deduced 25.6 kDa protein of 241 amino acids. Its amino acid sequence showed high similarity to Arabidopsis HMA5 protein, as seen in Figure 4, indicating that clone 8 encodes a tea ortholog of AtHMA5. Thus, we renamed the clone 8 cDNA as CsHMA5. CsHMA5 was transformed into Arabidopsis. Several hygromycin-resistant homozygous lines were identified, and two overexpression lines (OX-5-1 and OX-6-2) were characterized further. When germinated on standard 0.5× MS solid medium, transgenic plants showed shorter primary and lateral roots than that of WT control, as seen in Figure 5A. RT-qPCR results demonstrated that CsHMA5 was highly and similarly expressed from both overexpressors, but was undetected in the WT control, as seen in Figure 5B. HMA5 mainly catalyzes copper export from the root cells in Arabidopsis. The standard 1× MS medium contains 0.1 µM copper. To test if the short root phenotype of the overexpressors was a result of their copper hypersensitivity, WT and two overexpressors were germinated on synthetic 1× MS medium without copper or low copper (0.01 µM). Under both growth conditions, the root lengths of OX-5-1 and OX-6-2 were still shorter than that of control, as seen in Figure 5C,D. These data suggested that overexpression of CsHMA5 in Arabidopsis did not result in copper hypersensitivity. To test if CsHMA5 overexpressors could show altered response to higher concentrations of copper, seeds of WT and two CsHMA5 overexpressors were germinated on standard 0.5× MS medium supplemented with 12.5–50 µM copper. The exogenously supplementation of copper into standard MS medium would increase its free copper concentration accordingly. After plant growth for 10 days, WT root length from control plate was almost two-fold longer as that of CsHMA5 overexpressors. With the increasing concentration of copper, WT root length became gradually shorter, and this growth inhibition became more evident when exogenously supplied copper reached above 12.5 µM.; in contrast, the root growth from CsHMA5 overexpressors was not affected by as high as 37.5 µM copper, as seen in Figure 5D. These observations demonstrated that overexpression of CsHMA5 in Arabidopsis led to copper hyposensitivity.

Figure 4.
Sequence comparison of CsHMA5 and Arabidopsis orthologs. (A) Protein sequence alignment of CsHMA5 with Arabidopsis orthologs HMA1 (At4g37270), HMA2 (At4g30110), HMA3 (At4g30120), HMA4 (At2g19110), HMA5 (At1g63440), RAN1 (At5g44790), and PAA1 (At4g33520). (B) Phylogenetic tree of the amino acid sequence of CsHMA5 and the seven Arabidopsis HMA protein sequences.
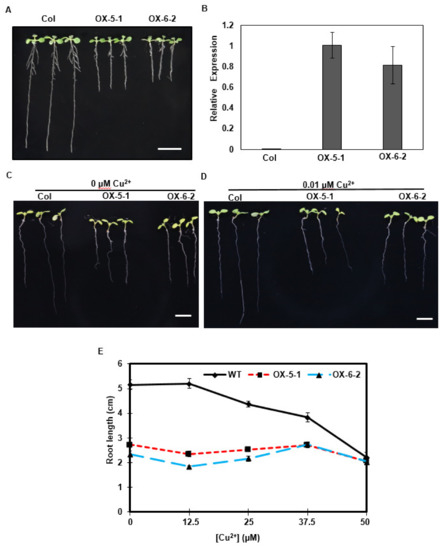
Figure 5.
Overexpression of CsHMA5 in Arabidopsis reduced copper sensitivity. (A) CsHMA5 overexpressors showed short roots on standard 0.5× MS medium. (B) RT-qPCR analysis of the expression levels of control and two CsHMA5 overexpressors. (C,D) Col and two CsHMA5 overexpressors (OX-5-1 and OX-6-2) were germinated on synthetic 1× MS medium without copper or low copper concentration (0.01 µM). (E) Col and two CsHMA5 overexpressors (OX-5-1 and OX-6-2) were germinated on standard 0.5× MS medium supplemented with 12.5–50 µM copper. The seeds were germinated vertically under continuous light, the root lengths were measured from 10-day-old seedlings. Data are expressed as mean ± SE (n = 28). Scale Bar = 1 cm.
In Arabidopsis, HMA5 is primarily expressed in roots, and strongly and specifically induced by copper in whole plants [30]. To further investigate the mechanisms of how CsHMA5 overexpression rendered copper sensitivity of transgenic plants, 16 genes from copper signaling pathways were selected, and their expression levels in the root tissues were compared between WT and CsHMA5 overexpressors. We found that the expression levels of the endogenous AtHMA5 from CsHMA5 overexpressors were not affected, as seen in Figure S5A–F. However, the expression levels of COPT3 and COX17-1 in transgenic lines were significantly higher than that of WT, while COPT5, RAN1, and PAA2 showed significantly lower expression levels than that of WT, as seen in Figure S5B. The expression levels for the rest 10 genes, including HMA1, PPA1, CPOT1, CPOT2, CPOT4, COX17-2, ZIP2, ZIP4, ATX1, and CCS1, were not significantly affected, as seen in Figure S5G–P. COX17-1 is a soluble protein located in the mitochondrial intermembrane space and participates in the transfer of copper for cytochrome oxidase assembly [31], the expression of which is induced by high copper concentrations [32]. COPT5 is a tonoplast copper exporter responsible for the transport of copper ions from root to reproductive organs [33]. RAN1 plays an essential role in the biogenesis of the ethylene receptors and copper homeostasis [34]. PAA2 is a chloroplast copper transporter of the thylakoid membrane [35]. These observations suggested that these changes in gene expression might affect copper transport and interorgan copper reallocation. Genetic analysis demonstrated that knockout HMA5 in Arabidopsis results in hypersensitivity to copper. In contrast, expression of SvHMA5II in Arabidopsis increases copper tolerance [36]. In this study, we demonstrated that overexpression of CsHMA5 in Arabidopsis resulted in hyposensitivity to copper, as seen in Figure 5. These data demonstrated that Arabidopsis can be used as a heterologous host to study the functions of tea genes.
3. Materials and Methods
3.1. Plant Materials
Tea plants (Camellia sinensis (L.) O. Kuntze cv. ‘Fuding Dabaicha’) were grown in the tea garden of Fujian Agriculture and Forestry University. Nine different tissues, including roots, tender stems, tender leaves, mature leaves, flower buds, flowers, fruit pericarps, and bark, were collected on September 2015.
3.2. Total RNA Extraction
Total RNA was isolated by using modified CTAB method [37]. A total of 1.5 g tissue was ground into fine powder by mortar and pestle in the presence of liquid nitrogen and a small amount of polyvinyl pyrrolidone (PVP, A610436, sangon, shanghai, China); the powder was transferred into 50 mL tube containing 9 mL of CTAB solution and 450 µL of β-mercaptoethanol(80076928, Sinopharm chemical reagent, shanghai, China), heated at 65 °C water bath for 20 min, then centrifuged at 12,000× g for 10 min at 4 °C. The supernatant was transferred into a fresh tube containing equal volume of prechilled phenol:chloroform:isopropanol (25:24:1, v/v/v) ( P1012, Solabio, Beijing, China), mixed well, incubated on ice water bath for 10 min, then centrifuged at 12,000× g for 10 min at 4 °C. The above extraction steps were repeated, and the supernatant was then transferred into fresh tube containing equal volume of prechilled chloroform:isopropanol (24:1, v/v) ( P1014, Solabio, Beijing, China), mixed well, and centrifuged at 12,000× g for 15 min at 4 °C. The supernatant was once again transferred into a fresh tube containing half volume of prechilled 8 M LiCl (A100416, sangon, Shanghai, China) and 1% β-mercaptoethanol, and then stored at −20 °C overnight. The next day, the mixture was spun at 12,000× g for 30 min at 4 °C, the supernatant was discarded, and the pellet was rinsed with prechilled 75% ethanol, air dried for 10 min, and dissolved in 200–500 µL DEPC-treated water, as seen in Figure S6.
3.3. mRNA Isolation
A 500 µg RNA aliquot from each tissue type was pooled together, then used to isolate mRNA by applying PolyATract mRNA Isolation System (Promega, Madison, WI, USA). The isolated mRNA was eluted into 1 mL of RNase-free water, as seen in Figure S7.
3.4. cDNA Synthesis
The first cDNA strand was synthesized from 1 µg of purified mRNA by using SMART cDNA library construction kit (Clontech, Mountain view, CA, USA), SMART IV Oligonucleotide and CDS-4M adapter were used for amplification, as seen in Figure S8. dsDNA was synthesized by using Advantage 2 PCR kit (Clontech, Mountain view, CA, USA), PCR Primer M1 and CDS-4M adapter were used as primers, as seen in Figure S9. The amplified ds cDNA was purified by QIAquick PCR Purification Kit (Qiagen, Hilden, Germany).
3.5. cDNA Denaturation, Hybridization, and DSN Treatment for Normalization
DSN (Evrogen, Moscow, Russia) isolated from Kamchatka crab was used for normalization. A total of 0.6–1.2 µg purified ds cDNA, 4 µL of 4× hybridization buffer, and an appropriate volume of sterile RNase-free water was combined to make total volume of 16 µL, mixed well, and spun briefly. A 4 µL aliquot of the reaction mixture was taken into each of the four appropriately labeled sterile PCR tubes, the reaction mixture was overlaid with a drop of mineral oil, and the tubes were centrifuged for 2 min at maximum speed in a microcentrifuge. The tubes were incubated in a thermal cycler at 98 °C for 2 min, followed by incubation at 68 °C for 5 h. Shortly before the end of the hybridization procedure, two DSN dilutions were prepared in two sterile tubes to final DSN concentrations of 0.5 and 0.25 U/µL, then placed on ice. The DSN master buffer was preheated at 68 °C for 3–5 min, 5 µL of the hot DSN master buffer was added to each tube containing hybridized cDNA, and the tube was briefly spun in a microcentrifuge and returned quickly to the thermal cycler, and incubated at 68 °C for 10 min. Then 0, 0.25, 0.5, and 1 U DSN were added into each tube, then immediately returned to the thermal cycler, incubated at 68 °C for 25 min. A total of 5 µL of DSN stop solution was mixed in the tubes and spun briefly, incubated in the thermal cycler at 68 °C for 5 min, and then the tubes were placed on ice. A total of 25 µL of sterile RNase-free water was mixed in each tube and spun briefly, then placed on ice, as seen in Figure S10.
3.6. Amplification of Normalized cDNA
Well-normalized cDNA was amplified by Advantage 2 PCR Kit (Clontech, CA, USA). PCR Primer M1 was used for amplification, as seen in Figure S11. The amplified ds cDNA was purified by using QIAquick PCR Purification Kit (Qiagen, Hilden, Germany), and eluted with 79 µL of sterile RNase-free water.
3.7. Sfi I Digestion and cDNA Size Fractionation
Purified cDNA was digested by Sfi I (R0123V, NEB, Ipswich, MA, USA) at 50 °C for 2 h, then size fractioned by using CHROMA SPIN TM-400 column. The first four fractions (lanes 5–8) were collected and pooled into a clean tube, as seen in Figure S12, precipitated with 95% ethanol at −20 °C overnight, air dried, and the pellet was resuspended in 7 μL of deionized H2O. The Sfi I-digested cDNA is then ready to be ligated into the Sfi I-digested, dephosphorylated λTriplEx2 vector (Clontech, CA, USA).
3.8. cDNA Ligation into λTriplEx2 Vector
To ligate Sfi I digested cDNA to λTriplEx2 vector (500 ng), three different ligations with varying insert to vector ratio were performed, as seen in Figure S13. The ligation was conducted at 16 °C overnight, then heated in a 65 °C water bath for 5 min to deactivate the T4 DNA ligase.
3.9. Package Ligation Mixture
To package into the λTriplEx2 vector, 25 µL of MaxPlax Lambda Packaging Extracts (Epicentre, Madison, WI, USA) was added into above ligation mixture, the detailed packaging procedure followed manufacturer’s instructions.
3.10. Titering the Unamplified Library and Determination of the Rate of Recombination
To titer the unamplified library, the library was diluted to 10−2–10−3 with phage dilution buffer, then a 100 µL aliquot was mixed with 100 µL E. coli XL1-blue cell, incubated at 37 °C for 15 min, and then plated onto LB/MgSO4 medium. To determine the recombinant rate, before plating onto LB/MgSO4 medium, 3.0 mL of melted LB/MgSO4 top agar (45 °C) containing 75 µL IPTG Fand 75 µL X-gal was added and mixed well. The plates were incubated at 37 °C for 16–18 h, then plaque numbers were counted. The recombinant rate was expressed as the ratio of white (recombinant) to blue (nonrecombinant) plaques.
3.11. Library Amplification and Titering
The library was amplified and titered according to product manual.
3.12. Converting λTriplEx2 to pTriplEx2 Plasmid
A well-isolated positive plaque from secondary or tertiary screening plate was picked, coincubated with 200 µL of E. coli BM25.8 at 31 °C for 30 min without shaking, and 1–10 µL of infected cell suspension was spread onto LB/carbenicillin plate to obtain isolated colonies.
3.13. cDNA Sequencing
Plasmid DNAs were prepared from randomly selected 44 white clones and sequenced by using standard ds-DNA sequencing protocols. The forward and reverse sequencing primers were: 5′-TAATACGACTCACTATAGGGC-3′; and 5′-CTCGGGAAGCGCGCCATTGTG-3′, respectively. The 5′-UTR and 3′-tail sequences were determined from individual cDNA. The CDS sequence was used to query the CSA and CSS genomes using BLAST [38].
3.14. Binary Vector pBIG2113SF-M Construction
Binary vector pBIG2113SF was selected to express tea FL-cDNAs in planta [29]. The Sfi IA and Sfi IB cutting sites of pBIG2113SF vector are different from λTriplEx2 vector and were re-engineered to make them same as λTriplEx2 vector. As a first step, the Sfi IA and Sfi IB cutting sites from vector U12445 were first modified by using Fast Site-Directed Mutagenesis Kit (TIANGEN, Beijing, China). The primer sequences for Sfi IA mutagenesis were: 5′-ATCTGGAATTCGGCCATTACGGCCAGAAGGAGATA-3′; and 5′-TATCTCCTTCTGGCCGTAATGGCCGAATTCCAGAT-3′. The primer sequences for Sfi IB mutagenesis were: 5′-AAAGTGCCTAAGGCCGCCTCGGCCGTCGACTAGAA-3′; and 5′-TTCTAGTCGACGGCCCGGAGGGCCTTAGGCACTTT-3′. The new U12445 vector with altered Sfi IA and Sfi IB recognition sequence was renamed as U12445M.
The DNA fragment spanning Sfi IA and Sfi IB from U12445M vector was amplified by using following pair of primers: 5′-TACAACTACATCTAGAGTTATCTGGAATTCGGCCATTA-3′; and 5′-CCGGGGATCCTCTAGAAATTCTAGTCGACGGCCGAG-3′. The PCR products were separated by agarose gel electrophoresis and purified by using E.Z.N.A.® Gel Extraction Kit (Omega Bio-tek, Norcross, GA, USA), then cloned into pBIG2113SF vector by gene fusion method. The engineered pBIG2113SF vector with new Sfi IA and Sfi IB sites was renamed as pBIG2113SF-M.
3.15. Cloning Tea FL-cDNAs into pBIG2113SF-M Vector
Individual clones from the tea cDNA library were converted into pTriplEx2 plasmid, the tea FL-cDNA insert was recovered from the plasmid by Sfi I digestion at 50 °C for 3 h, purified by gel extraction kit (Omega Bio-tek, Norcross, GA, USA), and then ligated into Sfi I sites of pBIG2113SF-M vector by T4 DNA ligase (New England Bio Labs, Beverly, MA, USA) at 16 °C overnight. In this report, seven tea FL-cDNAs were cloned into the pBIG2113SF-M vector.
3.16. Transforming Tea FL-cDNA into Arabidopsis
Individual recombinant pBIG2113SF-M vectors containing tea FL-cDNA insert were introduced into Agrobacterium tumefaciens GV3101 and transformed into Arabidopsis thaliana Columbia by the floral dip method. The T1 seeds were harvested and screened in 0.5× MS medium containing 50 μg mL−1 hygromycin. Resistant seedlings were transferred into soil pots, and T2 seeds were harvested from individual plants. T2 seeds were screened in 0.5× MS medium containing 50 μg mL−1 hygromycin. Six resistant seedlings per line were transferred into soil pot, and T3 seeds were harvested individually. Homozygote transgenic T3 seeds were identified and used for further characterization. Tea gene-specific primers were used to confirm transgene expression by RT-PCR, as seen in Table S1.
3.17. Root Length Measurement and Gene Expression Analysis
The standard 0.5× MS medium was prepared from Murashige & Skoog basal medium vitamins (Phyto Technology Laboratories, Shawnee Mission, KS, USA). A total of 2.2 g of MS basal medium and 0.5 g of MES were dissolved in 1 L of ddH2O, the pH adjusted to 5.7, and 8 g of agar powder (Solarbio, Beijing, China) was added, then copper sulfate was supplemented to specified concentrations. The composition of the synthetic copper deficiency 1× MS medium was (per liter of medium): 1650 mg NH4NO3, 1900 mg KNO3, 440 mg CaCl2.2H2O, 370 mg MgSO4.7H2O, 170 mg KH2PO4, 6.2 mg H3BO3, 16.9 mg MnSO4.H2O, 8.6 mg ZnSO4.H2O, 0.83 mg KI, 0.25 mg NaMoO4.2H2O, 0.025 mg CoCl2.6H2O, 27.8 mg FeSO4.7H2O, 37.3 mg Na2EDTA.2H2O, 103 mg B5 vitamin mix, 15,000 mg sucrose, 8000 mg agar. All the macrosalts and sucrose used in this study were analytical grade. Homozygote transgenic T3 seeds were germinated vertically on growth medium under continuous light (100 µmol photon m−2 s−1). After plant growth for 10 days, root lengths were measured by using Image J software (version 1.48, NIH, Washington, DC, USA). Data were expressed as mean ± SE (n = 28). After root length measurement, root tissues were harvested to isolate total RNA. One microgram of total RNA was reverse transcribed using PrimeScript™ RT Reagent Kit (Perfect Real Time) (TaKaRa, Japan) and a mixture of oligo (dT)12–18 primer and random hexamers were added. The RT products were diluted three-fold before use as qPCR templates. The primer pairs for copper signaling genes are listed in Table S2. qPCR was performed using a SYBR Premix Ex Taq™ II (TliRNase H Plus) kit (TaKaRa, Dalian, China) and CFX Connect™ Optics Module (Bio-Rad, Pleasanton, CA, USA) according to the manufactures’ instructions. Arabidopsis ADAPTOR PROTEIN-2 MU-ADAPTIN (AP2M, At5g46630) was used as loading control. Each reaction was performed in triplicate along with an internal control reaction. Relative gene expression levels were calculated according to the 2−ΔΔCt comparative CT method [39].
3.18. Bioinformatic Analysis
Protein sequence comparison and the phylogenetic tree construction of Arobidopsis P1B-type ATPases and CsHMA5 were done by using DNAMAN software (version: 7.0.2.176, Lynnon Corporation, San Ramon, CA, USA).
4. Conclusions
Tea is an obligate out-breeding plant species, which makes its genome highly heterozygous. Its seeds contain genome from two different tea germplasms. To avoid RNA contamination from other tea germplasms, in this study, only vegetative tissues from a single cultivar ‘Fuding Dabaicha’ were used to make the RNA pool. As a result, our cDNA library also excludes those genes that are specifically expressed in tea seeds.
There are several procedures to make a FL-cDNA library, mostly based on the mRNA cap structure [40,41]. These methods require high quantities of starting material and complicated multistep manipulations of the cap structure of mRNA and cDNA intermediates, which often cause mRNA degradation [42]. In this study, we applied the SMART™ method, which has been demonstrated to yield longer average ORF length [42,43]. To decrease the prevalence of clones from abundant transcripts, DSN from the Kamchatka crab was applied to specifically cleave ds-DNA in both DNA-DNA and DNA-RNA duplexes, which allowed us to separate and then PCR amplified the normalized ss- fraction. We also used λ phage (λTriplEx2) as vector for this library construction. Compared with plasmid vectors, the λ phage can accommodate cDNAs in broad ranges of sizes and shows high efficiency for long cDNA fragments. In contrast, plasmid vectors are often biased toward short cDNA fragments [44]. Furthermore, λ phage can be easily converted into plasmid for downstream applications.
Here we performed a small-scale trial and demonstrated that this FL-cDNA library is a valuable resource for new tea gene discovery and functional characterization. A more depth sequencing of this library would be simply a matter of cost. The establishment of easy tea gene cloning in combination with highly efficient Arabidopsis transformation will obviate the technical hurdle of direct tea transformation, thus facilitating functional analysis of tea genes. This system has the potential to be scaled up by using pooled inserts for cloning and transformation, and a large transgenic population can be easily generated for phenotypic observation. Once phenotypes were scored and corresponding tea transgenes identified, the functions of corresponding tea genes can be elucidated. Such strategies have been applied to study gene functions of model plant species, such as Arabidopsis and rice [45,46], and should be more applicable to nonmodel plant species such as tea tree.
Supplementary Materials
The following are available online at https://www.mdpi.com/1422-0067/20/23/5929/s1.
Author Contributions
M.C. and W.S. conceived and designed this research. L.L., W.C., Z.D., W.Z., and Q.X. performed the experiment and analyzed the data. M.C. wrote the manuscript. All authors participated in manuscript editing and approved its final version for submission.
Funding
This work was supported by the Department of Science and Technology of Fujian Province (2017NZ0002-1) and the National Science Foundation of China (31870803).
Acknowledgments
The authors thank Liuying Ma from Fujian Agriculture and Forestry University providing pBIG2113SF vector and R. Shyama Prasad Rao from Yenepoya University and Zijian Chen from Engineering School of University of Missouri for helping with manuscript editing.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Suzuki, Y.; Yoshitomo-Nakagawa, K.; Maruyama, K.; Suyama, A.; Sugano, S. Construction and characterization of a full length-enriched and a 5′-end-enriched cDNA library. Gene 1997, 200, 149–156. [Google Scholar] [CrossRef]
- Seki, M.; Narusaka, M.; Kamiya, A.; Ishida, J.; Satou, M.; Sakurai, T.; Nakajima, M.; Enju, A.; Akiyama, K.; Oono, Y.; et al. Functional annotation of a full-length Arabidopsis cDNA collection. Science 2002, 296, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Marques, M.C.; Alonso-Cantabrana, H.; Forment, J.; Arribas, R.; Alamar, S.; Conejero, V.; Perez-Amador, M.A. A new set of ESTs and cDNA clones from full-length and normalized libraries for gene discovery and functional characterization in citrus. BMC Genom. 2009, 10, 428. [Google Scholar] [CrossRef] [PubMed]
- Makita, Y.; Shimada, S.; Kawashima, M.; Kondou-Kuriyama, T.; Toyoda, T.; Matsui, M. MOROKOSHI: Transcriptome database in sorghum bicolor. Plant Cell Physiol. 2015, 56, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, S.; Satoh, K.; Nagata, T.; Kawagashira, N.; Doi, K.; Kishimoto, N.; Yazaki, J.; Ishikawa, M.; Yamada, H.; Ooka, H.; et al. Collection, mapping, and annotation of over 28,000 cDNA clones from japonica rice. Science 2003, 301, 376–379. [Google Scholar] [CrossRef]
- Umezawa, T.; Sakurai, T.; Totoki, Y.; Toyoda, A.; Seki, M.; Ishiwata, A.; Akiyama, K.; Kurotani, A.; Yoshida, T.; Mochida, K.; et al. Sequencing and analysis of approximately 40,000 soybean cDNA clones from a full length-enriched cDNA library. DNA Res. 2008, 15, 333–346. [Google Scholar] [CrossRef] [PubMed]
- Soderlund, C.; Descour, A.; Kudrna, D.; Bomhoff, M.; Boyd, L.; Currie, J.; Angelova, A.; Collura, K.; Wissotski, M.; Ashley, E.; et al. Sequencing, mapping, and analysis of 27,455 Maize full-length cDNAs. PLoS Genet. 2009, 5, e1000740. [Google Scholar] [CrossRef]
- Aoki, K.; Yano, K.; Suzuki, A.; Kawamura, S.; Sakurai, N.; Suda, K.; Kurabayashi, A.; Suzuki, T.; Tsugane, T.; Watanabe, M.; et al. Large-scale analysis of full-length cDNAs from the tomato (Solanum lycopersicum) cultivar Micro-Tom, a reference system for the Solanaceae genomics. BMC Genom. 2010, 11, 210. [Google Scholar] [CrossRef]
- Lin, M.; Lai, D.; Pang, C.; Fan, S.; Song, M.; Yu, S. Generation and analysis of a large-scale expressed sequence tag database from a full-length enriched cDNA library of developing leaves of Gossypium hirsutum L. PLoS ONE 2013, 8, e76443. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, H.; Qi, F.; Jian, G. Generation of transcriptome profiling and gene functional analysis in Gossypium hirsutum upon Verticillium dahliae infection. Biochem. Biophys. Res. Commun. 2016, 473, 879–885. [Google Scholar] [CrossRef]
- Ogihara, Y.; Mochida, K.; Kawaura, K.; Murai, K.; Seki, M.; Kamiya, A.; Shinozaki, K.; Carninci, P.; Hayashizaki, H.; Shin-I, T.; et al. Construction of a full-length cDNA library from young spikelets of hexaploid wheat and its characterization by large-scale sequencing of expressed sequence tags. Genes Genet. Syst. 2004, 79, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Carninci, P.; Shibata, Y.; Hayatsu, N.; Sugahara, Y.; Shibata, K.; Itoh, M.; Konno, H.; Okazaki, Y.; Muramatsu, M.; Hayashizaki, Y. Normalization and subtraction of CAP-trapper-selected cDNAs to prepare full-length cDNA libraries for rapid discovery of new genes. Genome Res. 2000, 10, 1617–1630. [Google Scholar] [CrossRef] [PubMed]
- Zhulidov, P.A.; Bogdanova, E.A.; Shcheglov, A.S.; Shagina, I.A.; Vagner, L.L.; Khazpekov, G.L.; Kozhemiako, V.V.; Luk’ianov, S.A.; Shagin, D.A. A method for the preparation of normalized cDNA libraries enriched with full-length sequences. Russ. J. Bioorg. Chem. 2005, 31, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Apoaroliswa, Z.; Chen, Z.M. Global Tea Breeding: Achievement, Challenges and Perspectives; Zhejiang University Press: Zhejiang, China, 2012. [Google Scholar]
- Di, T.; Zhao, L.; Chen, H.; Qian, W.; Wang, P.; Zhang, X.; Xia, T. Transcriptomic and metabolic insights into the distinctive effects of exogenous melatonin and gibberellin on terpenoid synthesis and plant hormone signal transduction pathway in Camellia sinensis. J. Agric. Food Chem. 2019, 67, 4689–4699. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Zhu, C.; Zhao, S.; Zhang, S.; Wang, W.; Fu, H.; Li, X.; Zhou, C.; Chen, L.; Lin, Y.; et al. De novo transcriptome and phytochemical analyses reveal differentially expressed genes and characteristic secondary metabolites in the original oolong tea (Camellia sinensis) cultivar ‘Tieguanyin’ compared with cultivar ‘Benshan’. BMC Genom. 2019, 20, 265. [Google Scholar] [CrossRef]
- Gao, Y.; Zhao, M.; Wu, X.; Li, D.; Borthakur, D.; Ye, J.; Zheng, X.; Lu, J. Analysis of differentially expressed genes in tissues of Camellia sinensis during dedifferentiation and root redifferentiation. Sci. Rep. 2019, 9, 2935. [Google Scholar] [CrossRef]
- Xin, Z.; Ge, L.; Chen, S.; Sun, X. Enhanced transcriptome responses in herbivore-infested tea plants by the green leaf volatile (Z)-3-hexenol. J. Plant Res. 2019, 132, 285–293. [Google Scholar] [CrossRef]
- Wu, L.; Fang, Z.; Lin, J.; Sun, Y.; Du, Z.; Guo, Y.; Liu, J.; Liang, Y.; Ye, J. Complementary iTRAQ proteomic and transcriptomic analyses of leaves in tea plant (Camellia sinensis L.) with different maturity and regulatory network of flavonoid biosynthesis. J. Proteome Res. 2019, 18, 252–264. [Google Scholar]
- Xia, E.; Zhang, H.; Sheng, J.; Li, K.; Zhang, Q.; Kim, C.; Zhang, Y.; Liu, Y.; Zhu, T.; Li, W.; et al. The tea tree genome provides insights into tea flavor and independent evolution of caffeine biosynthesis. Mol. Plant 2017, 10, 866–877. [Google Scholar] [CrossRef]
- Wei, C.; Yang, H.; Wang, S.; Zhao, J.; Liu, C.; Gao, L.; Xia, E.; Lu, Y.; Tai, Y.; She, G.; et al. Draft genome sequence of Camellia sinensis var. sinensis provides insights into the evolution of the tea genome and tea quality. Proc. Natl. Acad. Sci. USA 2018, 115, E4145–E4158. [Google Scholar] [CrossRef]
- Qiao, D.; Yang, C.; Chen, J.; Guo, Y.; Li, Y.; Niu, S.; Cao, K.; Che, Z. Comprehensive identification of the full-length transcripts and alternative splicing related to the secondary metabolism pathways in the tea plant (Camellia sinensis). Sci. Rep. 2019, 9, 2709. [Google Scholar] [CrossRef] [PubMed]
- Mondal, T.K.; Bhattacharya, A.; Ahuja, P.S.; Chand, P.K. Factor effecting Agrobacterium tumefaciens mediated transformation of tea (Camellia sinensis (L). O. Kuntze. Plant Cell Rep. 2001, 20, 712–720. [Google Scholar] [CrossRef]
- Osato, N.; Yamada, H.; Satoh, K.; Ooka, H.; Yamamoto, M.; Suzuki, K.; Kawai, J.; Carninci, P.; Ohtomo, Y.; Murakami, K.; et al. Antisense transcripts with rice full-length cDNAs. Genome Biol. 2003, 5, R5. [Google Scholar] [CrossRef] [PubMed]
- Brandle, J.E.; Richman, A.; Swanson, A.K.; Chapman, B.P. Leaf ESTs from Stevia rebaudiana: A resource for gene discovery in diterpene synthesis. Plant Mol. Biol. 2002, 50, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.Y.; Wang, X.W.; Xu, Q.S.; Zhao, S.Q.; Tai, Y.L.; Wei, C.L. Global dissection of alternative splicing uncovers transcriptional diversity in tissues and associates with the flavonoid pathway in tea plant (Camellia sinensis). BMC Plant Biol. 2018, 18, 266. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.K.; Lu, Y.; Zinta, G.; Lang, Z.; Zhu, J.K. UTR-Dependent Control of Gene Expressionin Plants. Trends Plant Sci. 2018, 23, 248–259. [Google Scholar] [CrossRef]
- Pang, P.P.; Meyerowitz, E.M. Arabidopsis thaliana: A model system for plant molecular biology. Nat. Biotechnol. 1987, 5, 1177–1181. [Google Scholar] [CrossRef]
- Taji, T.; Ohsumi, C.; Iuchi, S.; Seki, M.; Kasuga, M.; Kobayashi, M.; Yamaguchi-Shinozaki, K.; Shinozaki, K. Important roles of drought- and cold-inducible genes for galactinol synthase in stress tolerance in Arabidopsis thaliana. Plant J. 2002, 29, 417–426. [Google Scholar] [CrossRef]
- Andrés-Colás, N.; Sancenón, V.; Rodríguez-Navarro, S.; Mayo, S.; Thiele, D.J.; Ecker, J.R.; Puig, S.; Peń arrubia, L. The Arabidopsis heavy metal P-type ATPase HMA5 interacts with metallochaperones and functions in copper detoxification of roots. Plant. J. 2006, 45, 225–236. [Google Scholar] [CrossRef]
- Lucila, G.; Elina, W.; Uta, G.; Agustín, L.A.; Iris, S.; Daniel, H.G. The cytochrome c oxidase biogenesis factor AtCOX17 modulates stress responses in Arabidopsis. Plant Cell Environ. 2016, 39, 628–644. [Google Scholar]
- Attallah, C.V.; Welchen, E.; Gonzalez, D.H. The promoters of Arabidopsis thaliana genes AtCOX17-1 and -2, encoding a copper chaperone involved in cytochrome c oxidase biogenesis, are preferentially active in roots and anthers and induced by biotic and abiotic stress. Physiol. Plant. 2007, 129, 123–134. [Google Scholar] [CrossRef]
- Klaumann, S.; Nickolaus, S.D.; Fürst, S.H.; Starck, S.; Schneider, S.; Ekkehard Neuhaus, H.; Trentmann, O. The tonoplast copper transporter COPT5 acts as an exporter and is required for interorgan allocation of copper in Arabidopsis thaliana. New Phytol. 2011, 192, 393–404. [Google Scholar] [CrossRef] [PubMed]
- Binder, B.M.; Rodríguez, F.I.; Bleecker, A.B. The Copper Transporter RAN1 Is Essential for Biogenesis of Ethylene Receptors in Arabidopsis. J. Biol. Chem. 2010, 285, 37263–37270. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Ghany, S.E.; Müller-Moulé, P.; Niyogi, K.K.; Pilon, M.; Shikanai, T. Two P-Type ATPases Are Required for Copper Delivery inArabidopsis thaliana Chloroplasts. Plant Cell 2005, 17, 1233–1251. [Google Scholar] [CrossRef]
- Li, Y.; Iqbal, M.; Zhang, Q.; Spelt, C.; Bliek, M.; Hakvoort, H.W.J.; Quattrocchio, F.M.; Koes, R.; Schat, H. Two Silene vulgaris copper transporters residing in different cellular compartments confer copper hypertolerance by distinct mechanisms when expressed in Arabidopsis thaliana. New Phytol. 2017, 215, 1102–1114. [Google Scholar] [CrossRef]
- Porebski, S.; Bailey, L.G.; Baum, B.R. Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Mol. Biol. Rep. 1997, 15, 8–15. [Google Scholar] [CrossRef]
- Xia, E.; Li, F.; Tong, W.; Li, P.; Wu, Q.; Zhao, H.; Ge, R.; Li, R.; Li, Y.; Zhang, Z.; et al. Tea plant information archive (TPIA): A comprehensive genomics and bioinformatics platform for tea plant. Plant Biotechnol. J. 2019. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Carninci, P.; Kvam, C.; Kitamura, A.; Ohsumi, T.; Okazaki, Y.; Itoh, M.; Kamiya, M.; Shibata, K.; Sasaki, N.; Izawa, M.; et al. High-efficiency full-length cDNA cloning by biotinylated CAP trapper. Genomics 1996, 37, 327–336. [Google Scholar] [CrossRef]
- Edery, I.; Chu, L.; Sonenberg, N.; Pelletier, J. An efficient strategy to isolate full-length cDNAs based on an mRNA cap retention procedure (CAPture). Mol. Cell Biol. 1995, 15, 3363–3371. [Google Scholar] [CrossRef]
- Zhu, Y.Y.; Machleder, E.M.; Chenchik, A.; Li, R.; Siebert, P.D. Reverse transcriptase template switching: A SMART (TM) approach for full-length cDNA library construction. Biotechniques 2001, 30, 892–897. [Google Scholar] [CrossRef] [PubMed]
- Belyavsky, A.; Vinogradova, T.V.; Rajewsky, K. PCR-based cDNA library construction: General cDNA libraries at the level of a few cells. Nucl. Acids Res. 1989, 17, 2919–2932. [Google Scholar] [CrossRef] [PubMed]
- Carninci, P.; Shibata, Y.; Hayatsu, N.; Itoh, M.; Shiraki, T.; Hirozane, T.; Watahiki, A.; Shibata, K.; Konno, H.; Muramatsu, M.; et al. Balanced-size and long-size cloning of full-length, cap-trapped cDNAs into vectors of the novel lambda-FLC family allows enhanced gene discovery rate and functional analysis. Genomics 2001, 77, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, T.; Nakazawa, M.; Kawashima, M.; Iizumi, H.; Kuroda, H.; Kondou, Y.; Tsuhara, Y.; Suzuki, K.; Ishikawa, A.; Seki, M.; et al. The FOX hunting system: An alternative gain-of-function gene hunting technique. Plant. J. 2006, 48, 974–985. [Google Scholar] [CrossRef] [PubMed]
- Kondou, Y.; Higuchi, M.; Takahashi, S.; Sakurai, T.; Ichikawa, T.; Kuroda, H.; Yoshizumi, T.; Tsumoto, Y.; Horii, Y.; Kawashima, M.; et al. Systematic approaches to using the FOX hunting system to identify useful rice genes. Plant. J. 2009, 57, 883–894. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).