Fast Vibrational Modes and Slow Heterogeneous Dynamics in Polymers and Viscous Liquids



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. A model of the Slow Heterogeneous Relaxation and Transport in Terms of Vibrational Dynamics
2.1. Relaxation Time
2.2. Diffusion Coefficient
2.3. Stokes–Einstein Product
3. Transport and Relaxation in Polymeric Melts
4. Correlation between Vibrational Fast Dynamics and Slow Relaxation
4.1. Vibrational Caged Dynamics and Debye–Waller factor
4.2. Debye–Waller Scaling of the Slow Relaxation
5. Signatures of the Heterogeneous Dynamics
5.1. van Hove Function
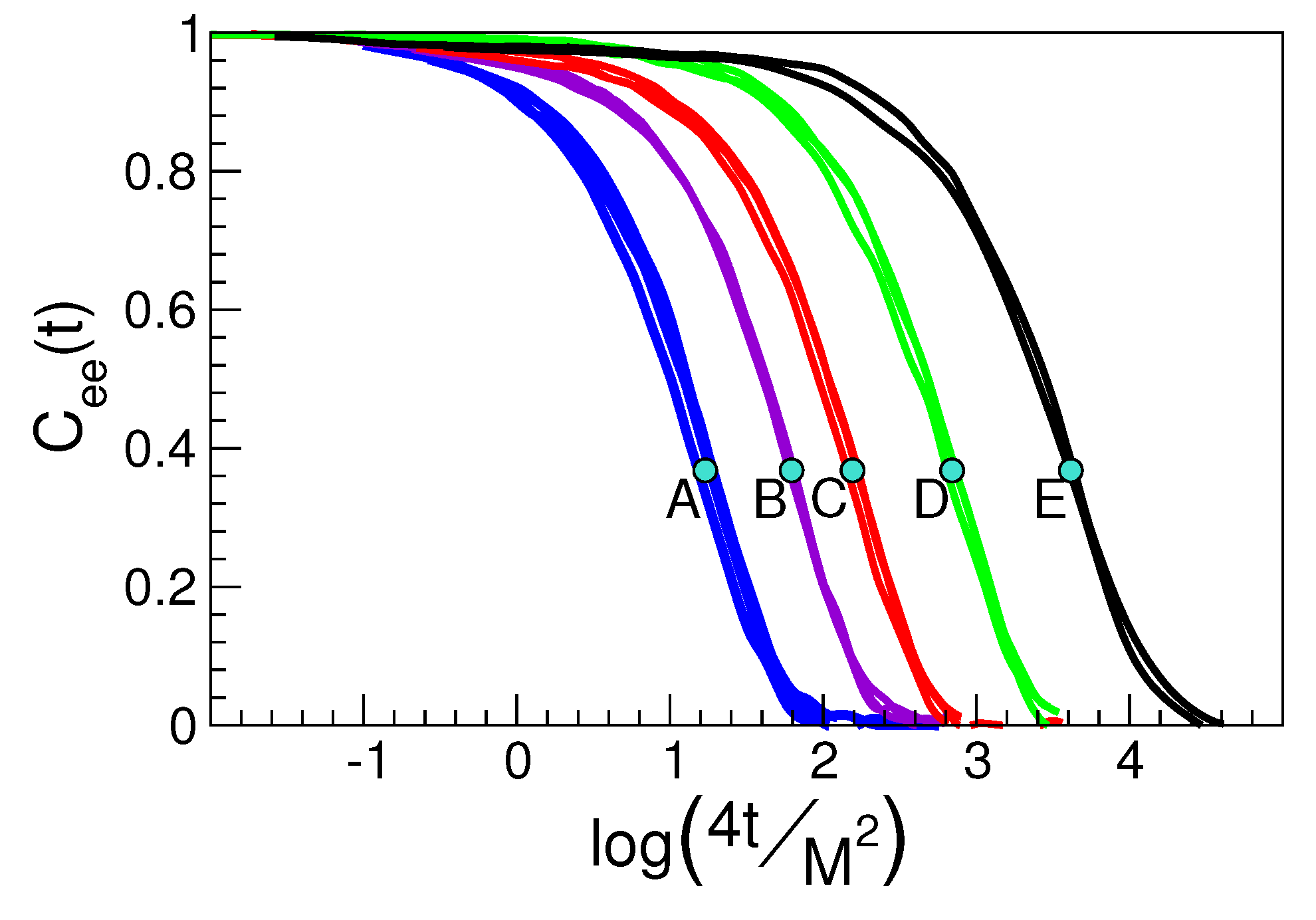
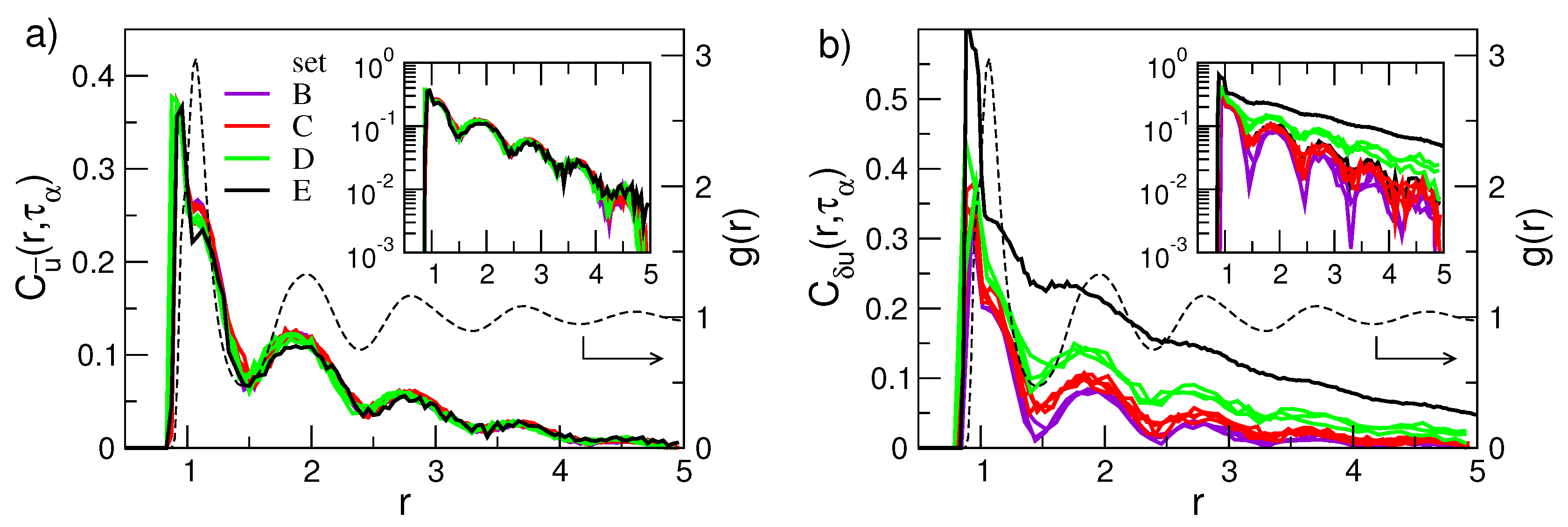
- The self-part of the van Hove function is expressed by suitable correlation functions, see Appendix B. Then, the coincidence of in states with equal DW factor observed in Figure 5a (the sets of states labelled as A, ⋯, E) is in harmony with Equation (3).
- Equation (3) also holds if one inspects the spatial dependence of the correlation function, e.g., the van Hove function, at . In particular, even in the presence of DH.
- The pattern of the D and E sets of states is not consistent with the Gaussian limit , Equation (20), predicting a progressive decay with r, i.e., the DH dynamics is not Gaussian;
5.2. Non-Gaussian Parameter
6. Breakdown of the Stokes–Einstein (SE) Law in the Presence of Dynamical Heterogeneity
6.1. SE Breakdown in Unentangled Polymers
6.2. Quasi-Universal SE Breakdown of Fragile Glass-Formers
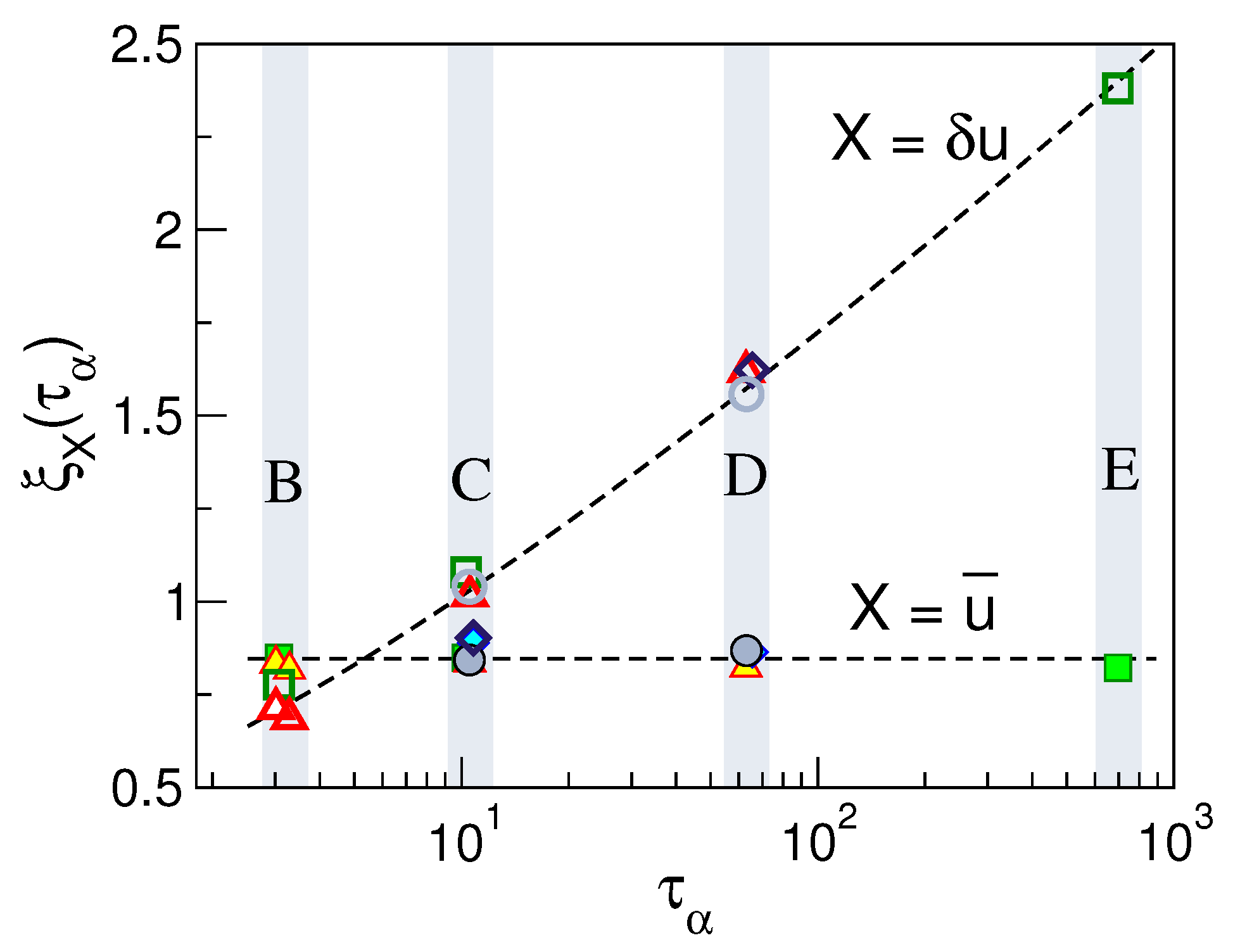
7. Displacement Correlation Length
8. Discussion
9. Methods
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DDC | displacement–displacement correlation |
| DH | dynamical heterogeneity |
| DW | Debye–Waller |
| ISF | Intermediate scattering function |
| MD | Molecular-dynamics |
| MSD | Mean square displacement |
| NGP | non-Gaussian parameter |
| SE | Stokes–Einstein |
Appendix A
Appendix A.1. Structural Relaxation
Appendix A.2. Diffusion Coefficient
Appendix A.3. Stokes–Einstein Product
Appendix B
References
- Angell, C. Relaxation in liquids, polymers and plastic crystals-strong/fragile patterns and problems. J. Non-Cryst. Sol. 1991, 131–133, 13–31. [Google Scholar] [CrossRef]
- Debenedetti, P.G.; Stillinger, F.H. Supercooled liquids and the glass transition. Nature 2001, 410, 259–267. [Google Scholar] [CrossRef]
- Ngai, K.L. Relaxation and Diffusion in Complex Systems; Springer: Berlin, Germany, 2011. [Google Scholar]
- Sillescu, H. Heterogeneity at the glass transition: A review. J. Non-Cryst. Solids 1999, 243, 81–108. [Google Scholar] [CrossRef]
- Ngai, K.L. Why the fast relaxation in the picosecond to nanosecond time range can sense the glass transition. Philos. Mag. 2004, 84, 1341–1353. [Google Scholar] [CrossRef]
- Berthier, L.; Biroli, G. Theoretical perspective on the glass transition and amorphous materials. Rev. Mod. Phys. 2011, 83, 587–645. [Google Scholar] [CrossRef]
- Ashcroft, N.W.; Mermin, N.D. Solid State Physics; Holt, Rinehart and Wiston: New York, NY, USA, 1976. [Google Scholar]
- Simmons, D.S.; Cicerone, M.T.; Zhong, Q.; Tyagic, M.; Douglas, J.F. Generalized localization model of relaxation in glass-forming liquids. Soft Matter 2012, 8, 11455–11461. [Google Scholar] [CrossRef]
- Pazmiño Betancourt, B.A.; Hanakata, P.Z.; Starr, F.W.; Douglas, J.F. Quantitative relations between cooperative motion, emergent elasticity, and free volume in model glass-forming polymer materials. Proc. Natl. Acad. Sci. USA 2015, 112, 2966–2971. [Google Scholar] [CrossRef]
- Douglas, J.F.; Pazmiño Betancourt, B.A.; Tong, X.; Zhang, H. Localization model description of diffusion and structural relaxation in glass-forming Cu–Zr alloys. J. Stat. Mech. Theory Exp. 2016, 2016, 054048. [Google Scholar] [CrossRef]
- Ediger, M.D. Spatially heterogeneous dynamics in supercooled liquids. Annu. Rev. Phys. Chem. 2000, 51, 99–128. [Google Scholar] [CrossRef]
- Richert, R. Heterogeneous dynamics in liquids: Fluctuations in space and time. J. Phys. Condens. Matter 2002, 14, R703–R738. [Google Scholar] [CrossRef]
- Tarjus, G.; Kivelson, D. Breakdown of the Stokes–Einstein relation in supercooled liquids. J. Chem. Phys. 1995, 103, 3071–3073. [Google Scholar] [CrossRef]
- Tracht, U.; Wilhelm, M.; Heuer, A.; Feng, H.; Schmidt-Rohr, K.; Spiess, H.W. Length Scale of Dynamic Heterogeneities at the Glass Transition Determined by Multidimensional Nuclear Magnetic Resonance. Phys. Rev. Lett. 1998, 81, 2727–2730. [Google Scholar] [CrossRef]
- Douglas, J.; Leporini, D. Obstruction model of the fractional Stokes–Einstein relation in glass-forming liquids. J. Non-Cryst. Solids 1998, 235–237, 137–141. [Google Scholar] [CrossRef]
- Jung, Y.; Garrahan, J.P.; Chandler, D. Excitation lines and the breakdown of Stokes–Einstein relations in supercooled liquids. Phys. Rev. E 2004, 69, 061205. [Google Scholar] [CrossRef]
- Adam, G.; Gibbs, J.H. On the Temperature Dependence of Cooperative Relaxation Properties in Glass-Forming Liquids. J. Chem. Phys. 1965, 43, 139–146. [Google Scholar] [CrossRef]
- Starr, F.W.; Douglas, J.F.; Sastry, S. The relationship of dynamical heterogeneity to the Adam-Gibbs and random first-order transition theories of glass formation. J. Chem. Phys. 2013, 138, 12A541. [Google Scholar] [CrossRef]
- Karmakar, S.; Dasgupta, C.; Sastry, S. Length scales in glass-forming liquids and related systems: A review. Rep. Prog. Phys. 2015, 79, 016601. [Google Scholar] [CrossRef]
- Albert, S.; Bauer, T.; Michl, M.; Biroli, G.; Bouchaud, J.P.; Loidl, A.; Lunkenheimer, P.; Tourbot, R.; Wiertel-Gasquet, C.; Ladieu, F. Fifth-order susceptibility unveils growth of thermodynamic amorphous order in glass-formers. Science 2016, 352, 1308–1311. [Google Scholar] [CrossRef]
- Biroli, G.; Karmakar, S.; Procaccia, I. Comparison of Static Length Scales Characterizing the Glass Transition. Phys. Rev. Lett. 2013, 111, 165701. [Google Scholar] [CrossRef]
- Wyart, M.; Cates, M.E. Does a Growing Static Length Scale Control the Glass Transition? Phys. Rev. Lett. 2017, 119, 195501. [Google Scholar] [CrossRef]
- Andreozzi, L.; Schino, A.D.; Giordano, M.; Leporini, D. Evidence of a fractional DebyeStokesEinstein law in supercooled oterphenyl. Europhys. Lett. 1997, 38, 669–674. [Google Scholar] [CrossRef]
- De Michele, C.; Leporini, D. Viscous flow and jump dynamics in molecular supercooled liquids. II. Rotations. Phys. Rev. E 2001, 63, 036702. [Google Scholar] [CrossRef] [PubMed]
- Tyrrell, H.J.V.; Harris, K.R. Diffusion in Liquids; Butterworths: London, UK, 1984. [Google Scholar]
- Hansen, J.P.; McDonald, I.R. Theory of Simple Liquids, 3rd ed.; Academic Press: New York, NY, USA, 2006. [Google Scholar]
- De Michele, C.; Leporini, D. Viscous flow and jump dynamics in molecular supercooled liquids. I. Translations. Phys. Rev. E 2001, 63, 036701. [Google Scholar] [CrossRef] [PubMed]
- Lad, K.N.; Jakse, N.; Pasturel, A. Signatures of fragile-to-strong transition in a binary metallic glass-forming liquid. J. Chem. Phys. 2012, 136, 104509. [Google Scholar] [CrossRef] [PubMed]
- Puosi, F.; Leporini, D. Communication: Fast and local predictors of the violation of the Stokes- Einstein law in polymers and supercooled liquids. J. Chem. Phys. 2012, 136, 211101. [Google Scholar] [CrossRef] [PubMed]
- Puosi, F.; Leporini, D. Communication: Fast dynamics perspective on the breakdown of the Stokes–Einstein law in fragile glass-formers. J. Chem. Phys. 2018, 148, 131102. [Google Scholar] [CrossRef]
- Chang, I.; Fujara, F.; Geil, B.; Heuberger, G.; Mangel, T.; Sillescu, H. Translational and rotational molecular motion in supercooled liquids studied by NMR and forced Rayleigh scattering. J. Non-Cryst. Solids 1994, 172–175, 248–255. [Google Scholar] [CrossRef]
- Puosi, F.; Leporini, D. Communication: Correlation of the instantaneous and the intermediate-time elasticity with the structural relaxation in glass-forming systems. J. Chem. Phys. 2012, 136, 041104. [Google Scholar] [CrossRef]
- Tobolsky, A.; Powell, R.E.; Eyring, H. Elastic-viscous properties of matter. In Frontiers in Chemistry; Burk, R.E., Grummit, O., Eds.; Interscience: New York, NY, USA, 1943; Volume 1, pp. 125–190. [Google Scholar]
- Hall, R.W.; Wolynes, P.G. The aperiodic crystal picture and free energy barriers in glasses. J. Chem. Phys. 1987, 86, 2943–2948. [Google Scholar] [CrossRef]
- Angell, C.A. Formation of glasses from liquids and biopolymers. Science 1995, 267, 1924–1935. [Google Scholar] [CrossRef]
- Dyre, J.C.; Olsen, N.B.; Christensen, T. Local elastic expansion model for viscous-flow activation energies of glass-forming molecular liquids. Phys. Rev. B 1996, 53, 2171–2174. [Google Scholar] [CrossRef] [PubMed]
- Martinez, L.M.; Angell, C.A. A thermodynamic connection to the fragility of glass-forming liquids. Nature 2001, 410, 663–667. [Google Scholar] [CrossRef] [PubMed]
- Sastry, S. The relationship between fragility, configurational entropy and the potential energy landscape of glass-forming liquids. Nature 2001, 409, 164–167. [Google Scholar] [CrossRef] [PubMed]
- Ngai, K.L. Dynamic and thermodynamic properties of glass-forming substances. J. Non-Cryst. Solids 2000, 275, 7–51. [Google Scholar] [CrossRef]
- Xia, X.; Wolynes, P.G. Fragilities of liquids predicted from the random first order transition theory of glasses. Proc. Natl. Acad. Sci. USA 2000, 97, 2990–2994. [Google Scholar] [CrossRef] [PubMed]
- Buchenau, U.; Zorn, R. A relation between fast and slow motions in glassy and liquid selenium. Europhys. Lett. 1992, 18, 523–528. [Google Scholar] [CrossRef]
- Starr, F.; Sastry, S.; Douglas, J.F.; Glotzer, S. What do we learn from the local geometry of glass-forming liquids? Phys. Rev. Lett. 2002, 89, 125501. [Google Scholar] [CrossRef]
- Bordat, P.; Affouard, F.; Descamps, M.; Ngai, K.L. Does the interaction potential determine both the fragility of a liquid and the vibrational properties of its glassy state? Phys. Rev. Lett. 2004, 93, 105502. [Google Scholar] [CrossRef]
- Widmer-Cooper, A.; Harrowell, P. Predicting the Long-Time Dynamic Heterogeneity in a Supercooled Liquid on the Basis of Short-Time Heterogeneities. Phys. Rev. Lett. 2006, 96, 185701. [Google Scholar] [CrossRef]
- Zhang, H.; Srolovitz, D.J.; Douglas, J.F.; Warren, J.A. Grain boundaries exhibit the dynamics of glass-forming liquids. Proc. Natl. Acad. Sci. USA 2009, 106, 7735–7740. [Google Scholar] [CrossRef]
- Widmer-Cooper, A.; Perry, H.; Harrowell, P.; Reichman, D.R. Irreversible reorganization in a supercooled liquid originates from localized soft modes. Nat. Phys. 2008, 4, 711–715. [Google Scholar] [CrossRef]
- Larini, L.; Ottochian, A.; De Michele, C.; Leporini, D. Universal scaling between structural relaxation and vibrational dynamics in glass-forming liquids and polymers. Nat. Phys. 2008, 4, 42–45. [Google Scholar] [CrossRef]
- Ottochian, A.; De Michele, C.; Leporini, D. Universal divergenceless scaling between structural relaxation and caged dynamics in glass-forming systems. J. Chem. Phys. 2009, 131, 224517. [Google Scholar] [CrossRef] [PubMed]
- Puosi, F.; Leporini, D. Scaling between Relaxation, Transport, and Caged Dynamics in Polymers: From Cage Restructuring to Diffusion. J. Phys. Chem. B 2011, 115, 14046–14051. [Google Scholar] [CrossRef]
- Ottochian, A.; Leporini, D. Scaling between structural relaxation and caged dynamics in Ca0.4K0.6(NO3)1.4 and glycerol: Free volume, time scales and implications for the pressure-energy correlations. Philos. Mag. 2011, 91, 1786–1795. [Google Scholar] [CrossRef][Green Version]
- Ottochian, A.; Leporini, D. Universal scaling between structural relaxation and caged dynamics in glass-forming systems: Free volume and time scales. J. Non-Cryst. Solids 2011, 357, 298–301. [Google Scholar] [CrossRef]
- De Michele, C.; Del Gado, E.; Leporini, D. Scaling between Structural Relaxation and Particle Caging in a Model Colloidal Gel. Soft Matter 2011, 7, 4025–4031. [Google Scholar] [CrossRef]
- Puosi, F.; Leporini, D. Spatial displacement correlations in polymeric systems. J. Chem. Phys. 2012, 136, 164901. [Google Scholar] [CrossRef]
- Puosi, F.; Leporini, D. Erratum: “Spatial displacement correlations in polymeric systems” [J. Chem. Phys. 136, 164901 (2012)]. J. Chem. Phys. 2013, 139, 029901. [Google Scholar] [CrossRef]
- Puosi, F.; Michele, C.D.; Leporini, D. Scaling between relaxation, transport and caged dynamics in a binary mixture on a per-component basis. J. Chem. Phys. 2013, 138, 12A532. [Google Scholar] [CrossRef]
- Ottochian, A.; Puosi, F.; Michele, C.D.; Leporini, D. Comment on “Generalized localization model of relaxation in glass-forming liquids”. Soft Matter 2013, 9, 7890–7891. [Google Scholar] [CrossRef]
- Novikov, V.N.; Sokolov, A.P. Role of Quantum Effects in the Glass Transition. Phys. Rev. Lett. 2013, 110, 065701. [Google Scholar] [CrossRef] [PubMed]
- Puosi, F.; Chulkin, O.; Bernini, S.; Capaccioli, S.; Leporini, D. Thermodynamic scaling of vibrational dynamics and relaxation. J. Chem. Phys. 2016, 145, 234904. [Google Scholar] [CrossRef] [PubMed]
- Guillaud, E.; Joly, L.; de Ligny, D.; Merabia, S. Assessment of elastic models in supercooled water: A molecular dynamics study with the TIP4P/2005f force field. J. Chem. Phys. 2017, 147, 014504. [Google Scholar] [CrossRef]
- Horstmann, R.; Vogel, M. Common behaviours associated with the glass transitions of water-like models. J. Chem. Phys. 2017, 147, 034505. [Google Scholar] [CrossRef]
- Becchi, M.; Giuntoli, A.; Leporini, D. Molecular layers in thin supported films exhibit the same scaling as the bulk between slow relaxation and vibrational dynamics. Soft Matter 2018, 14, 8814–8820. [Google Scholar] [CrossRef]
- Bernini, S.; Puosi, F.; Leporini, D. Thermodynamic scaling of relaxation: Insights from anharmonic elasticity. J. Phys. Condens. Matter 2017, 29, 135101. [Google Scholar] [CrossRef]
- Puosi, F.; Leporini, D. The kinetic fragility of liquids as manifestation of the elastic softening. Eur. Phys. J. E 2015, 38, 87. [Google Scholar] [CrossRef]
- Bernini, S.; Leporini, D. Cage effect in supercooled molecular liquids: Local anisotropies and collective solid-like response. J. Chem. Phys. 2016, 144, 144505. [Google Scholar] [CrossRef]
- Allen, M.P.; Tildesley, D.J. Computer Simulations of Liquids; Oxford University Press: New York, NY, USA, 1987. [Google Scholar]
- Doi, M.; Edwards, S.F. The Theory of Polymer Dynamics; Clarendon Press: Oxford, UK, 1988. [Google Scholar]
- Boon, J.P.; Yip, S. Molecular Hydrodynamics; Dover Publications: New York, NY, USA, 1980. [Google Scholar]
- Gedde, U.W. Polymer Physics; Chapman and Hall: London, UK, 1995. [Google Scholar]
- Kröger, M. Simple models for complex nonequilibrium fluids. Phys. Rep. 2004, 390, 453–551. [Google Scholar] [CrossRef]
- Kob, W.; Donati, C.; Plimpton, S.J.; Poole, P.H.; Glotzer, S.C. Dynamical Heterogeneities in a Supercooled Lennard–Jones Liquid. Phys. Rev. Lett. 1997, 79, 2827–2830. [Google Scholar] [CrossRef]
- Baschnagel, J.; Varnik, F. Computer simulations of supercooled polymer melts in the bulk and in confined geometry. J. Phys. Condens. Matter 2005, 17, R851–R953. [Google Scholar] [CrossRef]
- Bennemann, C.; Donati, C.; Baschnagel, J.; Glotzer, S.C. Growing range of correlated motion in a polymer melt on cooling towards the glass transition. Nature 1999, 399, 246–249. [Google Scholar] [CrossRef]
- Ottochian, A.; De Michele, C.; Leporini, D. Non-Gaussian effects in the cage dynamics of polymers. Philos. Mag. 2008, 88, 4057–4062. [Google Scholar] [CrossRef][Green Version]
- Tölle, A. Neutron scattering studies of the model glass former ortho -terphenyl. Rep. Prog. Phys. 2001, 64, 1473. [Google Scholar] [CrossRef]
- Chang, I.; Sillescu, H. Heterogeneity at the Glass Transition: Translational and Rotational Self-Diffusion. J. Phys. Chem. B 1997, 101, 8794–8801. [Google Scholar] [CrossRef]
- Mallamace, F.; Branca, C.; Corsaro, C.; Leone, N.; Spooren, J.; Chen, S.H.; Stanley, H.E. Transport properties of glass-forming liquids suggest that dynamic crossover temperature is as important as the glass transition temperature. Proc. Natl. Acad. Sci. USA 2010, 107, 22457–22462. [Google Scholar] [CrossRef]
- Donati, C.; Glotzer, S.C.; Poole, P.H. Growing Spatial Correlations of Particle Displacements in a Simulated Liquid on Cooling toward the Glass Transition. Phys. Rev. Lett. 1999, 82, 5064–5067. [Google Scholar] [CrossRef]
- Doliwa, B.; Heuer, A. Cooperativity and spatial correlations near the glass transition: Computer simulation results for hard spheres and disks. Phys. Rev. E 2000, 61, 6898–6908. [Google Scholar] [CrossRef]
- Weeks, E.R.; Crocker, J.C.; Weitz, D.A. Short and long-range correlated motion observed in colloidal glasses and liquids. J. Phys. Condens. Matter 2007, 19, 205131. [Google Scholar] [CrossRef]
- Narumi, T.; Tokuyama, M. Simulation study of spatial‚Äìtemporal correlation functions in supercooled liquids. Philos. Mag. 2008, 88, 4169–4175. [Google Scholar] [CrossRef]
- Kremer, K.; Grest, G.S. Dynamics of entangled linear polymer melts: A molecular-dynamics simulation. J. Chem. Phys. 1990, 92, 5057–5086. [Google Scholar] [CrossRef]
- Paul, W.; Smith, G.D. Structure and dynamics of amorphous polymers: Computer simulations compared to experiment and theory. Rep. Prog. Phys. 2004, 67, 1117–1185. [Google Scholar] [CrossRef]
- Luo, C.; Sommer, J.U. Coding coarse grained polymer model for LAMMPS and its application to polymer crystallization. Comput. Phys. Commun. 2009, 180, 1382–1391. [Google Scholar] [CrossRef]










© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Puosi, F.; Tripodo, A.; Leporini, D. Fast Vibrational Modes and Slow Heterogeneous Dynamics in Polymers and Viscous Liquids. Int. J. Mol. Sci. 2019, 20, 5708. https://doi.org/10.3390/ijms20225708
Puosi F, Tripodo A, Leporini D. Fast Vibrational Modes and Slow Heterogeneous Dynamics in Polymers and Viscous Liquids. International Journal of Molecular Sciences. 2019; 20(22):5708. https://doi.org/10.3390/ijms20225708
Chicago/Turabian StylePuosi, Francesco, Antonio Tripodo, and Dino Leporini. 2019. "Fast Vibrational Modes and Slow Heterogeneous Dynamics in Polymers and Viscous Liquids" International Journal of Molecular Sciences 20, no. 22: 5708. https://doi.org/10.3390/ijms20225708
APA StylePuosi, F., Tripodo, A., & Leporini, D. (2019). Fast Vibrational Modes and Slow Heterogeneous Dynamics in Polymers and Viscous Liquids. International Journal of Molecular Sciences, 20(22), 5708. https://doi.org/10.3390/ijms20225708