Circulating-Free DNA Analysis in Hepatocellular Carcinoma: A Promising Strategy to Improve Patients’ Management and Therapy Outcomes

 ,
,  ,
,  and
and
Abstract
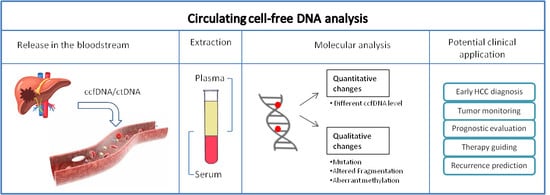
1. Introduction
2. Circulating-Free DNA Investigation in Oncology
3. Liquid Biopsy to Improve Hepatocellular Carcinoma (HCC) Therapeutic Management
3.1. Circulating Cell-Free DNA Level and HCC Clinical Outcome
3.2. Circulating Cell-Free DNA Genetic Profiling and HCC Clinical Outcome
3.3. Circulating Cell-Free DNA Methylation Profiling and HCC Clinical Outcome
3.4. ccfDNA Selection by Length in HCC
4. Potential Issues Related to the Clinical Application of ccfDNA Analysis
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AFP | Alpha-fetoprotein |
| APC | Adenomatous Polyposis Coli |
| BCLC | Barcelona Clinic Liver Cancer |
| CNA | Copy number alteration |
| cfDI | Cell-free DNA integrity |
| ccfDNA | Circulating Cell-free DNA |
| CFRT | Conventionally fractionated RT |
| CNVs | Copy-number variants |
| CTCs | Circulating tumor cells |
| ctDNA | Circulating tumor DNA |
| CTLA4 | Cytotoxic T-lymphocyte antigen 4 |
| CTNNB1 | Catenin beta 1 |
| DCR | Disease control rate |
| DFS | Disease free survival |
| DNMT | DNA methyltransferase |
| EGFR | Epidermal growth factor receptor |
| ESC | Embryonic stem cells |
| GIN | Genome-instability |
| GSTP1 | Glutathione S-transferase P1 |
| HBV | Hepatitis B virus |
| HCC | Hepatocellular carcinoma |
| HCV | Hepatitis C virus |
| hTERT | Human telomerase reverse transcriptase |
| IHFF | Intrahepatic failure-free |
| KRAS | KRAS proto-oncogene GTPase |
| LC | Local control |
| MRD | Minimal residual disease |
| MSRE-PCR | Methylation-sensitive restriction enzyme digestion PCR |
| NGS | Next-generation sequencing |
| OS | Overall survival |
| PCR | Polymerase chain reaction |
| p16 | Protein p16 |
| PD-1 | Programmed cell death protein-1 |
| PD-L1 | Programmed death-ligand 1 |
| PFS | Progression-free survival |
| RASSF1A | Ras association domain family 1 isoform A |
| RT-CT | Radio-chemotherapy |
| SBRT | Stereotactic body radiation therapy; |
| SEPT9 | Septin 9 |
| SFRP1 | Secreted frizzled related protein 1 |
| SNVs | Single-nucleotide variants |
| SPINT2 | Serine peptidase inhibitor, kunitz type 2 |
| TACE | Transarterial chemoembolization |
| TFPI2 | Tissue factor pathway inhibitor 2 |
| TMB | Tumor mutational burden |
| TP53 | Tumor protein p53 |
| TTP | Time to progression |
| VEGFA | Vascular endothelial growth factor A |
| WES | Whole-exome sequencing |
| WGS | Whole-genome sequencing |
References
- Villanueva, A. Hepatocellular Carcinoma. N. Engl. J. Med. 2019, 380, 1450–1462. [Google Scholar] [CrossRef] [PubMed]
- McGlynn, K.A.; Petrick, J.L.; London, W.T. Global epidemiology of hepatocellular carcinoma: An emphasis on demographic and regional variability. Clin. Liver Dis. 2015, 19, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Zucman-Rossi, J.; Pikarsky, E.; Sangro, B.; Schwartz, M.; Sherman, M.; Gores, G. Hepatocellular carcinoma. Nat. Rev. Dis. Prim. 2016, 2, 16018. [Google Scholar] [PubMed]
- Zhu, K.; Dai, Z.; Zhou, J. Biomarkers for hepatocellular carcinoma: Progression in early diagnosis, prognosis, and personalized therapy. Biomark. Res. 2013, 1, 10. [Google Scholar] [CrossRef] [PubMed]
- De Mattia, E.; Cecchin, E.; Guardascione, M.; Foltran, L.; Di Raimo, T.; Angelini, F.; D’Andrea, M.; Toffoli, G. Pharmacogenetics of the systemic treatment in advanced hepatocellular carcinoma. World J. Gastroenterol. 2019, 25, 3870–3896. [Google Scholar] [PubMed]
- De Mattia, E.; Cecchin, E.; Polesel, J.; Bignucolo, A.; Roncato, R.; Lupo, F.; Crovatto, M.; Buonadonna, A.; Tiribelli, C.; Toffoli, G. Genetic biomarkers for hepatocellular cancer risk in a caucasian population. World J. Gastroenterol. 2017, 23, 6674–6684. [Google Scholar] [PubMed]
- De Mattia, E.; Cecchin, E.; Polesel, J.; Lupo, F.; Tiribelli, C.; Crovatto, M.; Buonadonna, A.; Toffoli, G. UGT1A polymorphisms as genetic biomarkers for HCC risk in Caucasian population. Liver Int. 2017, 37, 1345–1353. [Google Scholar] [CrossRef]
- Harding, J.J.; Nandakumar, S.; Armenia, J.; Khalil, D.N.; Albano, M.; Ly, M.; Shia, J.; Hechtman, J.F.; Kundra, R.; El Dika, I.; et al. Prospective Genotyping of Hepatocellular Carcinoma: Clinical Implications of Next Generation Sequencing for Matching Patients to Targeted and Immune Therapies. Clin. Cancer Res. 2018, 25, 2116–2126. [Google Scholar] [CrossRef]
- Ally, A.; Balasundaram, M.; Carlsen, R.; Chuah, E.; Clarke, A.; Dhalla, N.; Holt, R.A.; Jones, S.J.M.; Lee, D.; Ma, Y.; et al. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017, 169, 1327–1341.e23. [Google Scholar] [CrossRef]
- Lu, L.-C.; Hsu, C.-H.; Hsu, C.; Cheng, A.-L. Tumor Heterogeneity in Hepatocellular Carcinoma: Facing the Challenges. Liver Cancer 2016, 5, 128–138. [Google Scholar]
- Labgaa, I.; Villacorta-Martin, C.; D’Avola, D.; Craig, A.J.; von Felden, J.; Martins-Filho, S.N.; Sia, D.; Stueck, A.; Ward, S.C.; Fiel, M.I.; et al. A pilot study of ultra-deep targeted sequencing of plasma DNA identifies driver mutations in hepatocellular carcinoma. Oncogene 2018, 37, 3740–3752. [Google Scholar] [PubMed]
- Ng, C.K.Y.; Di Costanzo, G.G.; Tosti, N.; Paradiso, V.; Coto-Llerena, M.; Roscigno, G.; Perrina, V.; Quintavalle, C.; Boldanova, T.; Wieland, S.; et al. Genetic profiling using plasma-derived cell-free DNA in therapy-naïve hepatocellular carcinoma patients: A pilot study. Ann. Oncol. 2018, 29, 1286–1291. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.-X.; Chen, G.; Zeng, Y.-Y.; Dong, X.-Q.; Lin, M.-J.; Huang, X.-H.; Zhang, D.; Liu, X.-L.; Liu, J.-F. Circulating tumor DNA profiling reveals clonal evolution and real-time disease progression in advanced hepatocellular carcinoma. Int. J. Cancer 2017, 141, 977–985. [Google Scholar] [PubMed]
- Ikeda, S.; Lim, J.S.; Kurzrock, R. Analysis of Tissue and Circulating Tumor DNA by Next-Generation Sequencing of Hepatocellular Carcinoma: Implications for Targeted Therapeutics. Mol. Cancer Ther. 2018, 17, 1114–1122. [Google Scholar] [CrossRef]
- Mandel, P.; Metais, P. Les acides nucleiques du plasma sanguin chez l’homme. C. R. Seances Soc. Biol. Fil. 1948, 142, 241–243. [Google Scholar]
- Leon, S.A.; Shapiro, B.; Sklaroff, D.M.; Yaros, M.J. Free DNA in the serum of cancer patients and the effect of therapy. Cancer Res. 1977, 37, 646–650. [Google Scholar]
- Sorenson, G.D.; Pribish, D.M.; Valone, F.H.; Memoli, V.A.; Bzik, D.J.; Yao, S.L. Soluble normal and mutated DNA sequences from single-copy genes in human blood. Cancer Epidemiol. Biomark. Prev. 1994, 3, 67–71. [Google Scholar]
- Choi, J.-J.; Reich, C.F.; Pisetsky, D.S. The role of macrophages in the in vitro generation of extracellular DNA from apoptotic and necrotic cells. Immunology 2005, 115, 55–62. [Google Scholar]
- Holdenrieder, S.; Stieber, P.; Chan, L.Y.S.; Geiger, S.; Kremer, A.; Nagel, D.; Lo, Y.M.D. Cell-free DNA in serum and plasma: Comparison of ELISA and quantitative PCR. Clin. Chem. 2005, 51, 1544–1546. [Google Scholar]
- Szpechcinski, A.; Chorostowska-Wynimko, J.; Struniawski, R.; Kupis, W.; Rudzinski, P.; Langfort, R.; Puscinska, E.; Bielen, P.; Sliwinski, P.; Orlowski, T. Cell-free DNA levels in plasma of patients with non-small-cell lung cancer and inflammatory lung disease. Br. J. Cancer 2015, 113, 476–483. [Google Scholar] [CrossRef]
- Murtaza, M.; Dawson, S.-J.; Tsui, D.W.Y.; Gale, D.; Forshew, T.; Piskorz, A.M.; Parkinson, C.; Chin, S.-F.; Kingsbury, Z.; Wong, A.S.C.; et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature 2013, 497, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.D.; Li, L.; Wang, Y.; Thoburn, C.; Afsari, B.; Danilova, L.; Douville, C.; Javed, A.A.; Wong, F.; Mattox, A.; et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 2018, 359, 926–930. [Google Scholar] [PubMed]
- Dawson, S.-J.; Tsui, D.W.Y.; Murtaza, M.; Biggs, H.; Rueda, O.M.; Chin, S.-F.; Dunning, M.J.; Gale, D.; Forshew, T.; Mahler-Araujo, B.; et al. Analysis of Circulating Tumor DNA to Monitor Metastatic Breast Cancer. N. Engl. J. Med. 2013, 368, 1199–1209. [Google Scholar] [CrossRef]
- Jensen, T.J.; Goodman, A.M.; Kato, S.; Ellison, C.K.; Daniels, G.A.; Kim, L.; Nakashe, P.; McCarthy, E.; Mazloom, A.R.; McLennan, G.; et al. Genome-Wide Sequencing of Cell-Free DNA Identifies Copy-Number Alterations That Can Be Used for Monitoring Response to Immunotherapy in Cancer Patients. Mol. Cancer Ther. 2019, 18, 448–458. [Google Scholar] [CrossRef] [PubMed]
- Thress, K.S.; Brant, R.; Carr, T.H.; Dearden, S.; Jenkins, S.; Brown, H.; Hammett, T.; Cantarini, M.; Barrett, J.C. EGFR mutation detection in ctDNA from NSCLC patient plasma: A cross-platform comparison of leading technologies to support the clinical development of AZD9291. Lung Cancer 2015, 90, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Tie, J.; Kinde, I.; Wang, Y.; Wong, H.L.; Roebert, J.; Christie, M.; Tacey, M.; Wong, R.; Singh, M.; Karapetis, C.S.; et al. Circulating tumor DNA as an early marker of therapeutic response in patients with metastatic colorectal cancer. Ann. Oncol. 2015, 26, 1715–1722. [Google Scholar] [PubMed]
- Li, J.; Han, X.; Yu, X.; Xu, Z.; Yang, G.; Liu, B.; Xiu, P. Clinical applications of liquid biopsy as prognostic and predictive biomarkers in hepatocellular carcinoma: Circulating tumor cells and circulating tumor DNA. J. Exp. Clin. Cancer Res. 2018, 37, 213. [Google Scholar] [PubMed]
- Chen, H.; Sun, L.-Y.; Zheng, H.-Q.; Zhang, Q.-F.; Jin, X.-M. Total serum DNA and DNA integrity: Diagnostic value in patients with hepatitis B virus-related hepatocellular carcinoma. Pathology 2012, 44, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.; Zhang, X.; Zhou, S.-L.; Cao, Y.; Huang, X.-W.; Fan, J.; Yang, X.-R.; Zhou, J. Plasma Circulating Cell-free DNA Integrity as a Promising Biomarker for Diagnosis and Surveillance in Patients with Hepatocellular Carcinoma. J. Cancer 2016, 7, 1798–1803. [Google Scholar] [CrossRef]
- Jiang, P.; Chan, C.W.M.; Chan, K.C.A.; Cheng, S.H.; Wong, J.; Wong, V.W.-S.; Wong, G.L.H.; Chan, S.L.; Mok, T.S.K.; Chan, H.L.Y.; et al. Lengthening and shortening of plasma DNA in hepatocellular carcinoma patients. Proc. Natl. Acad. Sci. USA 2015, 112, E1317–E1325. [Google Scholar] [CrossRef]
- Jiang, P.; Sun, K.; Tong, Y.K.; Cheng, S.H.; Cheng, T.H.T.; Heung, M.M.S.; Wong, J.; Wong, V.W.S.; Chan, H.L.Y.; Chan, K.C.A.; et al. Preferred end coordinates and somatic variants as signatures of circulating tumor DNA associated with hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 2018, 115, E10925–E10933. [Google Scholar] [CrossRef] [PubMed]
- Chandrananda, D.; Thorne, N.P.; Bahlo, M. High-resolution characterization of sequence signatures due to non-random cleavage of cell-free DNA. BMC Med. Genom. 2015, 8, 29. [Google Scholar] [CrossRef] [PubMed]
- Ren, N.; Ye, Q.-H.; Qin, L.-X.; Zhang, B.-H.; Liu, Y.-K.; Tang, Z.-Y. Circulating DNA level is negatively associated with the long-term survival of hepatocellular carcinoma patients. World J. Gastroenterol. 2006, 12, 3911–3914. [Google Scholar] [CrossRef]
- Huang, Z.-H.; Hu, Y.; Hua, D.; Wu, Y.-Y.; Song, M.-X.; Cheng, Z.-H. Quantitative analysis of multiple methylated genes in plasma for the diagnosis and prognosis of hepatocellular carcinoma. Exp. Mol. Pathol. 2011, 91, 702–707. [Google Scholar] [CrossRef]
- Tokuhisa, Y.; Iizuka, N.; Moribe, T.; Fujita, N.; Miura, T.; Tamatsukuri, S.; Ishitsuka, H.; Uchida, K.; Terai, S.; Sakamoto, K.; et al. Circulating cell-free DNA as a predictive marker for distant metastasis of hepatitis C virus-related hepatocellular carcinoma. Br. J. Cancer 2007, 97, 1399–1403. [Google Scholar] [CrossRef]
- Piciocchi, M.; Cardin, R.; Vitale, A.; Vanin, V.; Giacomin, A.; Pozzan, C.; Maddalo, G.; Cillo, U.; Guido, M.; Farinati, F. Circulating free DNA in the progression of liver damage to hepatocellular carcinoma. Hepatol. Int. 2013, 7, 1050–1057. [Google Scholar] [CrossRef]
- Oh, C.R.; Kong, S.-Y.; Im, H.-S.; Kim, H.J.; Kim, M.K.; Yoon, K.-A.; Cho, E.-H.; Jang, J.-H.; Lee, J.; Kang, J.; et al. Genome-wide copy number alteration and VEGFA amplification of circulating cell-free DNA as a biomarker in advanced hepatocellular carcinoma patients treated with Sorafenib. BMC Cancer 2019, 19, 292. [Google Scholar] [CrossRef]
- Park, S.; Lee, E.J.; Rim, C.H.; Seong, J. Plasma Cell-Free DNA as a Predictive Marker after Radiotherapy for Hepatocellular Carcinoma. Yonsei Med. J. 2018, 59, 470–479. [Google Scholar] [CrossRef]
- Lee, T.H.; Montalvo, L.; Chrebtow, V.; Busch, M.P. Quantitation of genomic DNA in plasma and serum samples: Higher concentrations of genomic DNA found in serum than in plasma. Transfusion 2001, 41, 276–282. [Google Scholar] [CrossRef]
- Swarup, V.; Moganty, R. Circulating (cell-free) nucleic acids–A promising, non-invasive tool for early detection of several human diseases. FEBS Lett. 2007, 581, 795–799. [Google Scholar] [CrossRef]
- Lou, X.; Hou, Y.; Liang, D.; Peng, L.; Chen, H.; Ma, S.; Zhang, L. A novel Alu-based real-time PCR method for the quantitative detection of plasma circulating cell-free DNA: Sensitivity and specificity for the diagnosis of myocardial infarction. Int. J. Mol. Med. 2015, 35, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Nault, J.-C.; Galle, P.R.; Marquardt, J.U. The role of molecular enrichment on future therapies in hepatocellular carcinoma. J. Hepatol. 2018, 69, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-J.; Wu, H.-C.; Shen, J.; Ahsan, H.; Tsai, W.Y.; Yang, H.-I.; Wang, L.-Y.; Chen, S.-Y.; Chen, C.-J.; Santella, R.M. Predicting hepatocellular carcinoma by detection of aberrant promoter methylation in serum DNA. Clin. Cancer Res. 2007, 13, 2378–2384. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.; Yang, H.; Xu, H.; Wang, Y.; Ge, P.; Ren, J.; Xu, W.; Lu, X.; Sang, X.; Zhong, S.; et al. Noninvasive detection of tumor-associated mutations from circulating cell-free DNA in hepatocellular carcinoma patients by targeted deep sequencing. Oncotarget 2016, 7, 40481–40490. [Google Scholar] [CrossRef]
- Cai, Z.; Chen, G.; Zeng, Y.; Dong, X.; Li, Z.; Huang, Y.; Xin, F.; Qiu, L.; Xu, H.; Zhang, W.; et al. Comprehensive Liquid Profiling of Circulating Tumor DNA and Protein Biomarkers in Long-Term Follow-Up Patients with Hepatocellular Carcinoma. Clin. Cancer Res. 2019, 25, 5284–5294. [Google Scholar] [CrossRef]
- Robertson, K.D. DNA methylation and human disease. Nat. Rev. Genet. 2005, 6, 597–610. [Google Scholar] [CrossRef]
- Guerriero, P.; Moshiri, F.; Lupini, L.; Sabbioni, S.; Negrini, M.; Callegari, E. Circulating tumor DNAs and non-coding RNAs as potential biomarkers for hepatocellular carcinoma diagnosis, prognosis and response to therapy. Hepatoma Res. 2019. [Google Scholar] [CrossRef]
- Esteller, M. Epigenetics in cancer. N. Engl. J. Med. 2008, 358, 1148–1159. [Google Scholar] [CrossRef]
- Xu, R.-H.; Wei, W.; Krawczyk, M.; Wang, W.; Luo, H.; Flagg, K.; Yi, S.; Shi, W.; Quan, Q.; Li, K.; et al. Circulating tumour DNA methylation markers for diagnosis and prognosis of hepatocellular carcinoma. Nat. Mater. 2017, 16, 1155–1161. [Google Scholar] [CrossRef]
- Shames, D.S.; Minna, J.D.; Gazdar, A.F. DNA methylation in health, disease, and cancer. Curr. Mol. Med. 2007, 7, 85–102. [Google Scholar] [CrossRef]
- Warren, J.D.; Xiong, W.; Bunker, A.M.; Vaughn, C.P.; Furtado, L.V.; Roberts, W.L.; Fang, J.C.; Samowitz, W.S.; Heichman, K.A. Septin 9 methylated DNA is a sensitive and specific blood test for colorectal cancer. BMC Med. 2011, 9, 133. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Qin, Y.; Li, B.; Sun, Z.; Yang, B. Detection of aberrant promoter methylation of GSTP1 in the tumor and serum of Chinese human primary hepatocellular carcinoma patients. Clin. Biochem. 2006, 39, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Wong, I.H.; Lo, Y.M.; Zhang, J.; Liew, C.T.; Ng, M.H.; Wong, N.; Lai, P.B.; Lau, W.Y.; Hjelm, N.M.; Johnson, P.J. Detection of aberrant p16 methylation in the plasma and serum of liver cancer patients. Cancer Res. 1999, 59, 71–73. [Google Scholar] [PubMed]
- Yeo, W.; Wong, N.; Wong, W.-L.; Lai, P.B.S.; Zhong, S.; Johnson, P.J. High frequency of promoter hypermethylation of RASSF1A in tumor and plasma of patients with hepatocellular carcinoma. Liver Int. 2005, 25, 266–272. [Google Scholar] [CrossRef]
- Chan, K.C.A.; Lai, P.B.S.; Mok, T.S.K.; Chan, H.L.Y.; Ding, C.; Yeung, S.W.; Lo, Y.M.D. Quantitative analysis of circulating methylated DNA as a biomarker for hepatocellular carcinoma. Clin. Chem. 2008, 54, 1528–1536. [Google Scholar] [CrossRef]
- Thierry, A.R.; El Messaoudi, S.; Gahan, P.B.; Anker, P.; Stroun, M. Origins, structures, and functions of circulating DNA in oncology. Cancer Metast. Rev. 2016, 35, 347–376. [Google Scholar] [CrossRef]
- Jahr, S.; Hentze, H.; Englisch, S.; Hardt, D.; Fackelmayer, F.O.; Hesch, R.D.; Knippers, R. DNA fragments in the blood plasma of cancer patients: Quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 2001, 61, 1659–1665. [Google Scholar]
- Wang, B.G.; Huang, H.-Y.; Chen, Y.-C.; Bristow, R.E.; Kassauei, K.; Cheng, C.-C.; Roden, R.; Sokoll, L.J.; Chan, D.W.; Shih, I.-M. Increased plasma DNA integrity in cancer patients. Cancer Res. 2003, 63, 3966–3968. [Google Scholar]
- Jiang, W.-W.; Zahurak, M.; Goldenberg, D.; Milman, Y.; Park, H.; Westra, W.; Koch, W.; Sidransky, D.; Califano, J. Increased plasma DNA integrity index in head and neck cancer patients. J. Int. Cancer 2006, 119, 2673–2676. [Google Scholar] [CrossRef]
- Arko-Boham, B.; Aryee, N.A.; Blay, R.M.; Owusu, E.D.A.; Tagoe, E.A.; Doris Shackie, E.-S.; Debrah, A.B.; Adu-Aryee, N.A. Circulating cell-free DNA integrity as a diagnostic and prognostic marker for breast and prostate cancers. Cancer Genet. 2019, 235, 65–71. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Study Population | Therapy | Analyte | Measure Methods | Serum/ Plasma | Clinical Endpoint | Main Finding | Ref |
|---|---|---|---|---|---|---|---|
| HCC patients (n = 79) Cirrhotic patients (n = 20) Healthy volunteers (n = 20) (Chinese) | Surgery | ccfDNA level | Ultraviolet transilluminator system | Plasma | 3 years DFS, OS, tumor feature | Compared with the healthy volunteers (17.6 ± 9.5 ng/mL), a significant higher ccfDNA level was found in the patients with HCC (47.1 ± 43.7 ng/mL, p = 0.000) or with liver cirrhosis (30.0 ± 13.3 ng/mL, p = 0.002). ccfDNA was closely associated with tumor size (p = 0.008) and TNM stage (p = 0.040), negatively associated with the 3-DFS (p = 0.017) and OS (p = 0.001). | [33] |
| HCC patients (n = 72) Cirrhotic/chronic hepatitis patients (n = 37) Healthy volunteers (n = 41) (Chinese) | Surgery | ccfDNA level | Quantitative RT-PCR | Plasma | OS, tumor feature | Plasma DNA concentrations were significantly higher in HCC patients compared with those in healthy controls or in benign controls (median 173 ng/mL, 9 ng/mL; 46 ng/mL, Mann–Whitney U test, p < 0.01). ccfDNA levels were positively associated with tumor size (p = 0.012), and were significantly elevated in HCC patients with intrahepatic spreading or vascular invasion (p = 0.035). Patients with ccfDNA level higher than the cut off value (173 ng/mL) (n = 29) showed a no-significant shorter OS respect those with low ccfDNA level (p = 0.017). | [34] |
| HCV-related HCC patients (n = 87) HCV carriers (n = 100) (Japanese) | Surgery | GSTP1 | Quantitative RT-PCR | Serum | OS, DFS, tumor feature | Serum ccfDNA levels were significantly higher in HCC patients than in HCV carriers without HCC. ccfDNA levels were not associated with any clinic-pathologic factors. Patients with ccfDNA level higher than the cut off value (117.8 ng/mL) (n = 29) showed a significantly shorter OS compared to those with low ccfDNA level (n = 58) (p = 0.017) Serum ccfDNA levels were not associated with DFS. | [35] |
| HCC patients (n = 55) (Korean) | CFRT (n = 34) −45 Gy/25 fractions (n = 6) −45 Gy/25 fractions + chemotherapy (5-FU, cisplatin) (n = 28). SBRT (n = 21) (60 Gy/4 fractions) | ccfDNA level | Ultraviolet-visible spectrophotometry (Nanodrop2000) | Plasma | Tumor feature, response, OS, PF, IHFF, LC. | Pre-RT and post-RT ccfDNA level were measured. Patients were divided in high DNA (HDNA) and low DNA (LDNA) level group, both for pre-RT and post-RT using cut-off value of 33.65 ng/mL and 37.25 ng/mL respectively. Pre-RT HDNA group tended to have larger tumors (p = 0.017). Mean pre-RT ccfDNA values were similar for both groups (responders vs. non responders: 39.5 vs. 39.6 ng/mL, p = 0.988), but were significantly different post-RT (responders vs. non responders: 35.9 vs. 56.1 ng/mL, p = 0.002). Treatment response was significantly better in the post-RT LDNA group than the post-RT HDNA group (81.8% vs. 47.8%, p = 0.017). OS and PF rates were not significantly associated with different post-RT ccfDNA level. Tumor response, IHFF and LC rates were significantly better in the post-RT LDNA group r compared to the HDNA group (p = 0.017, p = 0.035, and p = 0.006, respectively). | [38] |
| Advance/metastatic HCC patients (n = 151) Healthy volunteers (n = 14) (Korean) | Systemic therapy (sorafenib 400 mg twice daily) | ccfDNA level | Plasma | DCR TTP OS | ccfDNA concentration in HCC patients was significantly higher than in healthy volunteers (0.71 ng/μL vs 0.34 ng/μL, p < 0.0001). Regarding HCC patients, DCR was significantly lower in ccfDNA-high group than in ccfDNA-low group using a cut off value of 0.82 ng/μL (p = 0.003). Moreover, the ccfDNA-high group had worse TTP (2.2 vs. 4.1 months; HR = 1.71; p = 0.002) and OS (4.1 vs. 14.8 months; HR = 3.50; p < 0.0001) than the ccfDNA-low group. In the multivariable analyses, the ccfDNA remained an independent prognostic factor for OS (p < 0.0001). | [37] | |
| Viral-related (i.e., HBV or HCV) advanced chronic hepatitis or cirrhotic HCC patients (n = 66) Cirrhotic patients (n = 35) Advanced HCV-related chronic hepatitis patients (n = 41) (Italian) | Not available | h-TERT | Quantitative RT-PCR | Plasma | OS | HCC patients ccfDNA concentration was higher than in the other groups, but not statistically significant (p = 0.02, one-way analysis of variance (ANOVA)). Patients with ccfDNA level below the cut off value (2ng/μL) showed an improvement in OS compared with patients with ccfDNA level above the cut off value (37 months vs 24 months, p = 0.03). | [36] |
| Study Population | Therapy | Analyte | Measure Methods | Serum/Plasma | Clinical Endpoint | Main Finding | Ref |
|---|---|---|---|---|---|---|---|
| Early-stage HCC patients (n = 41) Healthy volunteers (n = 6) (Chinese) | Surgery | TERT, CTNNB1, TP53 | MiSeq sequencing | Plasma | RFS | Eight of the 40 patients successfully analyzed presented tumor-associated mutations. Patients with mutations in ctDNA were more likely to relapse (89 days for patients with somatic mutation vs. 365 days for patients without somatic mutation, p < 0.001). | [44] |
| Long-term follow-up patients with HCC (n = 34) (Chinese) | Surgery plus other adjuvant therapies (e.g., TACE radiofrequency ablation, target therapy) during follow-up. | Tumor somatic SNVs and CNVs | Target sequencing and low-coverage WGS | Plasma | MRD RFS OS | All plasma samples before surgery showed somatic genetic variations profile resembling corresponding primary matched tumor tissues. Patient groups with high SNV/CNV fractions evaluated in preoperative plasma samples have significantly poorer RFS (SNV, p = 0.0019; CNV, p = 0.001) and OS (SNV, p = 0.003; CNV, p = 0.0067) when compared with low SNV/CNV fractions. Moreover, increasing SNV fraction and CNV fraction were related to increasing tumor size, presence of microvascular invasion, and more severe tumor differentiation. During follow-up, SNVs and CNVs dynamically changed correlating to patients′ tumor burden. A model based on acquired SNV information was developed and was shown to accurately assess patients′ tumor burden with high consistence compared with imaging results. This model could discover tumor occurrence in advance of imaging for an average of 4.6 months, and showed superior performance than serum biomarkers (i.e., AFP, AFP-L3%, DCP). The model could also precisely detect MRD in advance and predict patients′ RFS (p = 0.001) and OS (p = 0.001). Furthermore combining ctDNA with DCP could increase the sensitivity for MRD detection, providing better prognostic value for both RFS (log-rank, p < 0.0001) and OS (log-rank, p < 0.0001) than ctDNA or DCP alone | [45] |
| Advance/metastatic HCC patients (n = 151) (Korean) | Systemic therapy (sorafenib 400 mg twice daily) | CNA, EIF2C1 (VEGFA-to-EIF2C1 ratio) | NextSeq 500 illumina low depth whole-genome sequencing | Plasma | DCR, TTP, OS | DCR and TTP did not significantly differ between the VEGFA-high and VEGFA-low group (p = 0.309 and p = 0.781). OS was reported shorter in VEGFA-high group than in VEGFA-low group even if it was not statistically significant (7.5 and 12.8 months respectively, p = 0.180). An high i-score, used as a CNA variation alternative, was correlated with worse DCR, TTP and OS (p = 0.0003, p < 0.0001 and p < 0.0001). | [37] |
| Study Population | Therapy | Analyte | Measure Methods | Serum/plasma | Clinical Endpoint | Main Finding | Ref |
|---|---|---|---|---|---|---|---|
| HCC patients (n = 25) Hepatitis/cirrhotic patients (n = 35) Healthy volunteers (n = 20) (Chinese) | Surgery | p15, p16 | MSP, southern blot | Serum/plasma | Recurrence | Methylation of p15 and p16 were found in 92% of tumor sample and in 74% plasma/serum sample. During a median follow-up time of 14 months post-surgery, 75% (9 of 12) of HCC patients with concurrent p15 and p16 methylation in tumors, 3 of 12 with only p16 methylation and 1 of 12 with only p15 methylation developed liver recurrence or lung metastasis. No p15 or p16 methylation were found in healthy or in hepatitis/cirrhotic non-HCC patients. | [53] |
| HCC patients (n = 63) HBV patients (n = 63) Healthy volunteers (n = 50) (Chinese) | Surgery | RASSF1A | MSP | Serum | DFS | Hypermethylated RASSF1A was detected in 93% of HCC patients, 58% of HBV carriers, and 8% of the healthy volunteers. The median RASSF1A concentrations for the HCC patients and HBV carriers were 7.70 × 105 copies/L and 1.18 × 105 copies/L, respectively. Patients with higher RASSF1A concentrations at diagnosis or 1 year after tumor resection showed poorer DFS (p < 0.01). | [55] |
| Training data set: HCC patients (n = 680) Validation data set: HCC patients (n = 369) (Chinese) | Heterogeneous treatment | 401 genes (training data set) 8 genes (validation data set) | Target bisulfite sequencing-illumina sequencing | Plasma | OS | A prognostic prediction model was constructed with an independent 8-genes panel and a combined prognosis score system was generated (cp-score). Patients were divided in high- and low-risk groups, based on cp-score. OS was longer in the low risk group than in high risk group (cut off value −0.24). | [49] |
| HCC patients (n = 72) Cirrhotic patients (n = 25) Chronic inactive hepatitis (n = 12) Healthy volunteers (n = 41) (Chinese) | Not available | APC, GSTP1, RASSF1A, SFRP1 | MSRE-qPCR | Plasma | OS | Elevated plasma methylation levels of APC or RASSF1A was associated with significantly poorer OS (Log-rank test, p < 0.05), while no significant association was found between plasma GSTP1 or SFRP1 methylation and OS (Log-rank test, p > 0.05). Cox multivariate analysis demonstrated that the methylation level of RASSF1A in plasma was an independent prognostic factor for OS (HR = 3.262, 95% CI:1.476–7.209. p = 0.003). | [34] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mezzalira, S.; De Mattia, E.; Guardascione, M.; Dalle Fratte, C.; Cecchin, E.; Toffoli, G. Circulating-Free DNA Analysis in Hepatocellular Carcinoma: A Promising Strategy to Improve Patients’ Management and Therapy Outcomes. Int. J. Mol. Sci. 2019, 20, 5498. https://doi.org/10.3390/ijms20215498
Mezzalira S, De Mattia E, Guardascione M, Dalle Fratte C, Cecchin E, Toffoli G. Circulating-Free DNA Analysis in Hepatocellular Carcinoma: A Promising Strategy to Improve Patients’ Management and Therapy Outcomes. International Journal of Molecular Sciences. 2019; 20(21):5498. https://doi.org/10.3390/ijms20215498
Chicago/Turabian StyleMezzalira, Silvia, Elena De Mattia, Michela Guardascione, Chiara Dalle Fratte, Erika Cecchin, and Giuseppe Toffoli. 2019. "Circulating-Free DNA Analysis in Hepatocellular Carcinoma: A Promising Strategy to Improve Patients’ Management and Therapy Outcomes" International Journal of Molecular Sciences 20, no. 21: 5498. https://doi.org/10.3390/ijms20215498
APA StyleMezzalira, S., De Mattia, E., Guardascione, M., Dalle Fratte, C., Cecchin, E., & Toffoli, G. (2019). Circulating-Free DNA Analysis in Hepatocellular Carcinoma: A Promising Strategy to Improve Patients’ Management and Therapy Outcomes. International Journal of Molecular Sciences, 20(21), 5498. https://doi.org/10.3390/ijms20215498