Animal Models for Parkinson’s Disease Research: Trends in the 2000s


Abstract
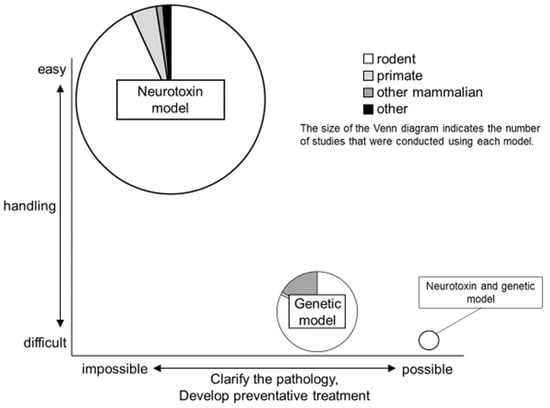
1. Introduction
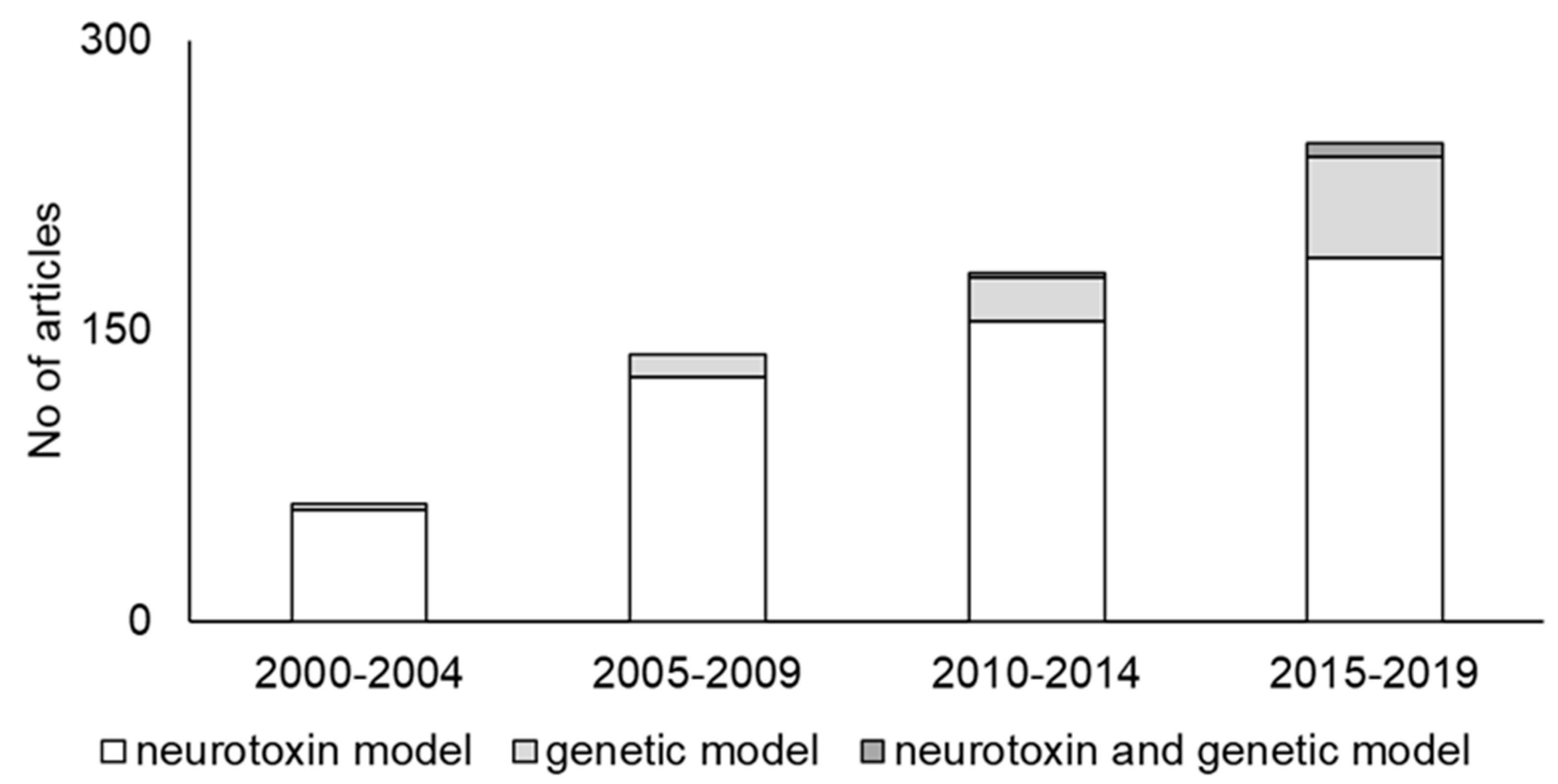
2. Trends in PD Animal Models
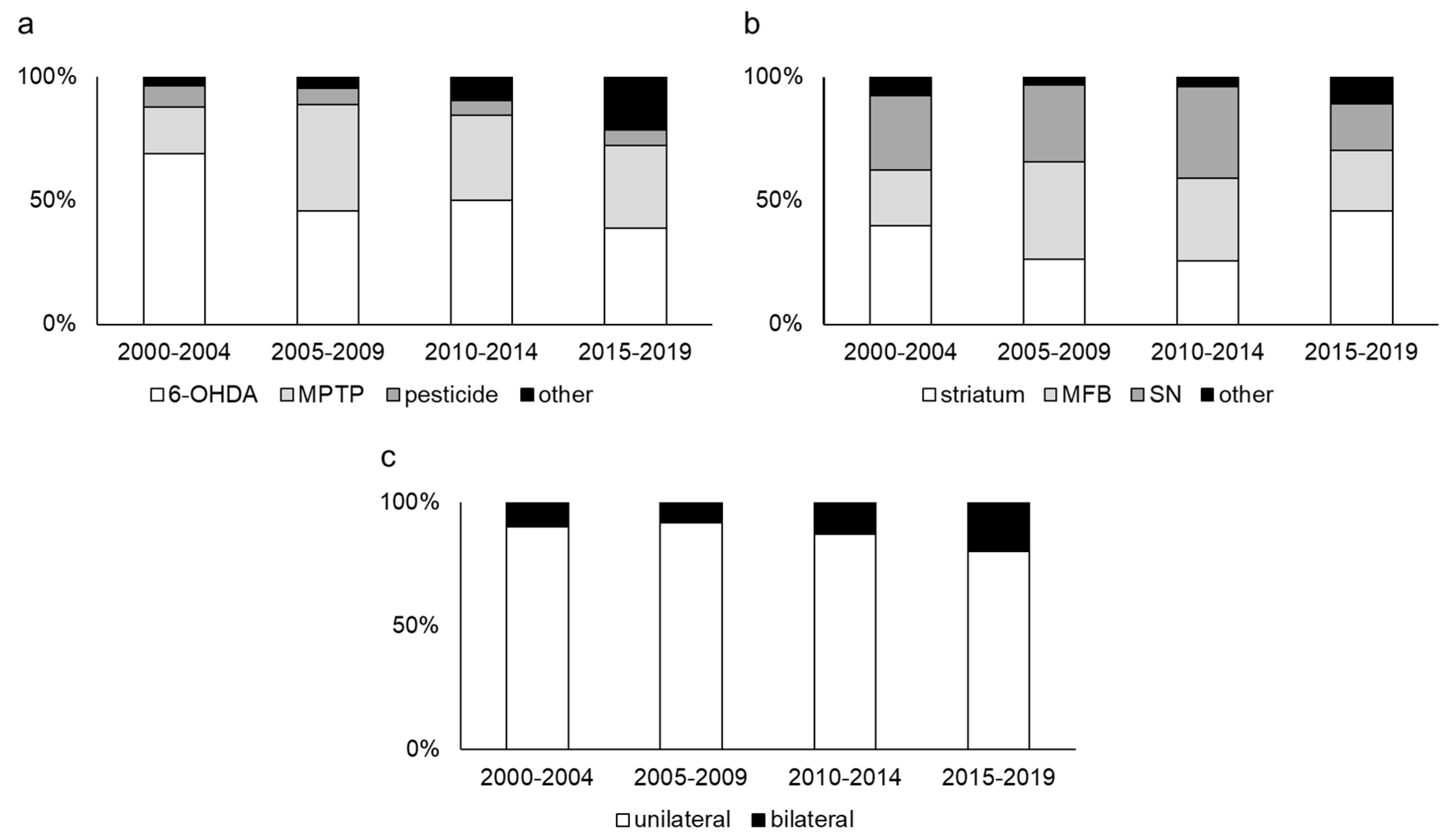
2.1. Neurotoxin Models
2.2. Genetic Models
2.3. Neurotoxin and Genetic Models
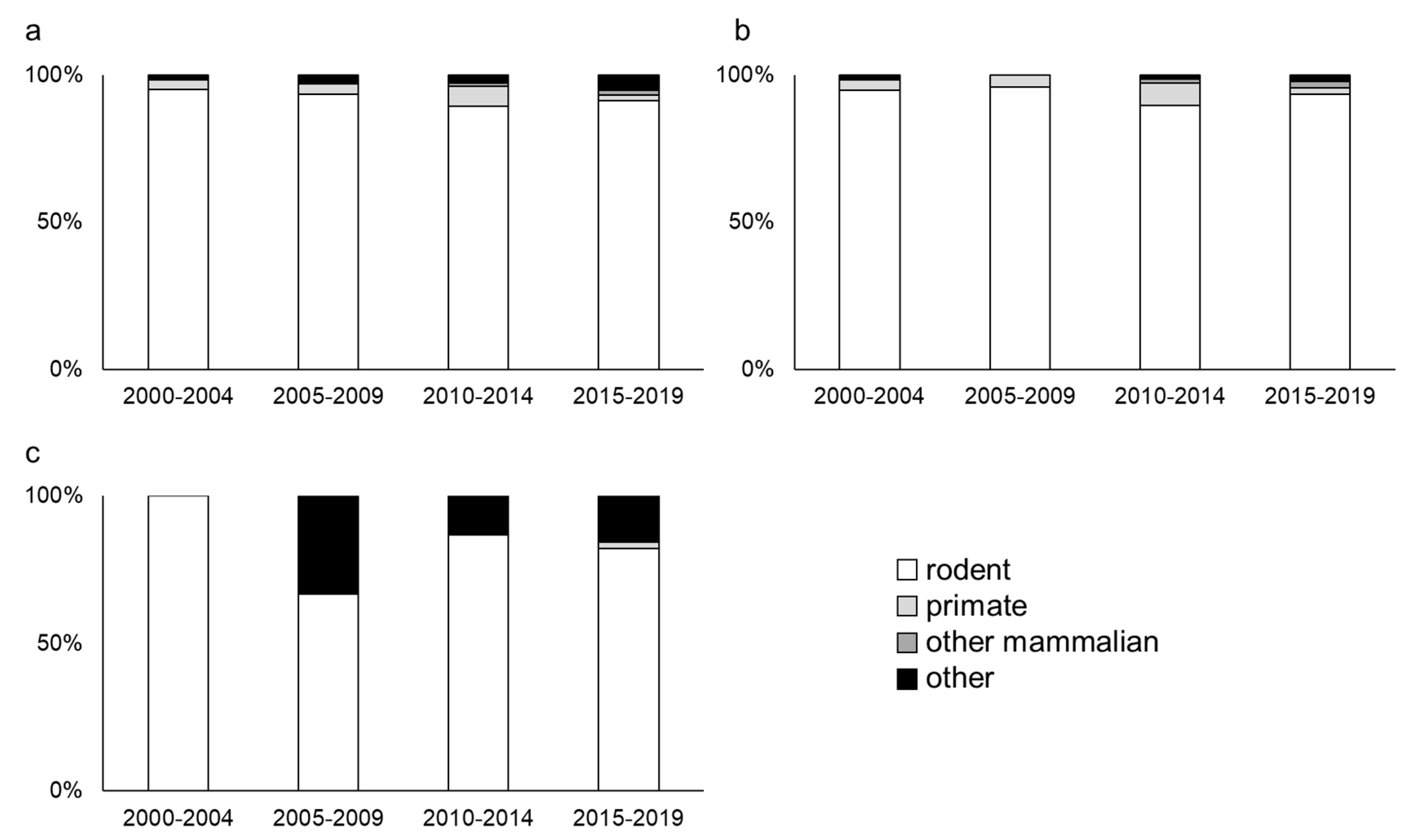
2.4. The Type of Animal
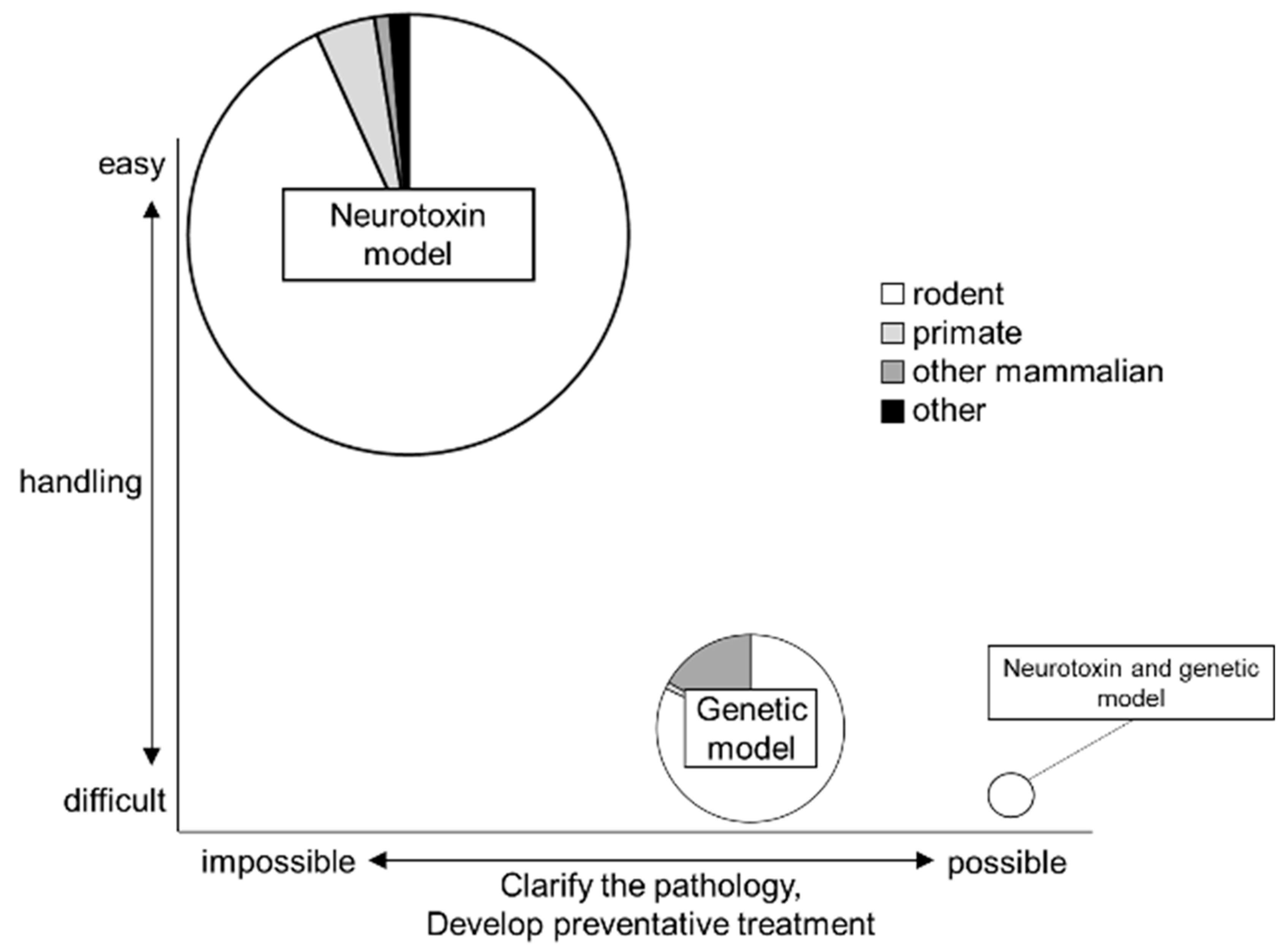
3. Summary and Future Prospective
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| PD | Parkinson’s disease |
| SN | substantia nigra |
| 6-OHDA | 6-hydroxydopamine |
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| MFB | Medial forebrain bundle |
| PINK1 | PTEN-induced putative kinase 1 |
| C. elegans | Caenorhabditis elegans |
References
- Billings, J.L.; Hare, D.J.; Nurjono, M.; Volitakis, I.; Cherny, R.A.; Bush, A.I.; Adlard, P.A.; Finkelstein, D.I. Effects of Neonatal Iron Feeding and Chronic Clioquinol Administration on the Parkinsonian Human A53T Transgenic Mouse. ACS Chem. Neurosci. 2016, 7, 360–366. [Google Scholar] [CrossRef]
- Lang, A.E.; Lozano, A.M. Parkinson’s disease. First of two parts. N. Engl. J. Med. 1998, 339, 1044–1053. [Google Scholar] [CrossRef]
- Lang, A.E.; Lozano, A.M. Parkinson’s disease. Second of two parts. N. Engl. J. Med. 1998, 339, 1130–1143. [Google Scholar] [CrossRef]
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Cacabelos, R. Parkinson’s Disease: From Pathogenesis to Pharmacogenomics. Int. J. Mol. Sci. 2017, 18, 551. [Google Scholar] [CrossRef]
- Damier, P.; Hirsch, E.C.; Agid, Y.; Graybiel, A.M. The substantia nigra of the human brain. II. Patterns of loss of dopamine-containing neurons in Parkinson’s disease. Brain 1999, 122, 1437–1448. [Google Scholar] [CrossRef]
- Obeso, J.A.; Stamelou, M.; Goetz, C.G.; Poewe, W.; Lang, A.E.; Weintraub, D.; Burn, D.; Halliday, G.M.; Bezard, E.; Przedborski, S.; et al. Past, present, and future of Parkinson’s disease: A special essay on the 200th Anniversary of the Shaking Palsy. Mov. Disord. 2017, 32, 1264–1310. [Google Scholar] [CrossRef]
- Carta, A.R.; Carboni, E.; Spiga, S. The MPTP/probenecid model of progressive Parkinson’s disease. Methods Mol. Biol. 2013, 964, 295–308. [Google Scholar] [CrossRef]
- Meredith, G.E.; Sonsalla, P.K.; Chesselet, M.F. Animal models of Parkinson’s disease progression. Acta Neuropathol. 2008, 115, 385–398. [Google Scholar] [CrossRef]
- Olanow, C.W.; Kieburtz, K.; Schapira, A.H. Why have we failed to achieve neuroprotection in Parkinson’s disease? Ann. Neurol. 2008, 64 (Suppl. 2), S101–S110. [Google Scholar] [CrossRef]
- More, S.V.; Kumar, H.; Cho, D.Y.; Yun, Y.S.; Choi, D.K. Toxin-Induced Experimental Models of Learning and Memory Impairment. Int. J. Mol. Sci. 2016, 17, 1447. [Google Scholar] [CrossRef] [PubMed]
- Blesa, J.; Przedborski, S. Parkinson’s disease: Animal models and dopaminergic cell vulnerability. Front. Neuroanat. 2014, 8, 155. [Google Scholar] [CrossRef] [PubMed]
- Creed, R.B.; Goldberg, M.S. New Developments in Genetic rat models of Parkinson’s Disease. Mov. Disord. 2018, 33, 717–729. [Google Scholar] [CrossRef] [PubMed]
- Dauer, W.; Przedborski, S. Parkinson’s disease: Mechanisms and models. Neuron 2003, 39, 889–909. [Google Scholar] [CrossRef]
- Van Kampen, J.M.; Robertson, H.A. The BSSG rat model of Parkinson’s disease: Progressing towards a valid, predictive model of disease. EPMA J. 2017, 8, 261–271. [Google Scholar] [CrossRef]
- Sauer, H.; Oertel, W.H. Progressive degeneration of nigrostriatal dopamine neurons following intrastriatal terminal lesions with 6-hydroxydopamine: A combined retrograde tracing and immunocytochemical study in the rat. Neuroscience 1994, 59, 401–415. [Google Scholar] [CrossRef]
- Przedborski, S.; Levivier, M.; Jiang, H.; Ferreira, M.; Jackson-Lewis, V.; Donaldson, D.; Togasaki, D.M. Dose-dependent lesions of the dopaminergic nigrostriatal pathway induced by intrastriatal injection of 6-hydroxydopamine. Neuroscience 1995, 67, 631–647. [Google Scholar] [CrossRef]
- Kuruvilla, K.P.; Nandhu, M.S.; Paul, J.; Paulose, C.S. Oxidative stress mediated neuronal damage in the corpus striatum of 6-hydroxydopamine lesioned Parkinson’s rats: Neuroprotection by serotonin, GABA and bone marrow cells supplementation. J. Neurol. Sci. 2013, 331, 31–37. [Google Scholar] [CrossRef]
- Blandini, F.; Levandis, G.; Bazzini, E.; Nappi, G.; Armentero, M.T. Time-course of nigrostriatal damage, basal ganglia metabolic changes and behavioural alterations following intrastriatal injection of 6-hydroxydopamine in the rat: New clues from an old model. Eur. J. Neurosci. 2007, 25, 397–405. [Google Scholar] [CrossRef]
- Perese, D.A.; Ulman, J.; Viola, J.; Ewing, S.E.; Bankiewicz, K.S. A 6-hydroxydopamine-induced selective parkinsonian rat model. Brain Res. 1989, 494, 285–293. [Google Scholar] [CrossRef]
- Larramendy, C.; Taravini, I.R.; Saborido, M.D.; Ferrario, J.E.; Murer, M.G.; Gershanik, O.S. Cabergoline and pramipexole fail to modify already established dyskinesias in an animal model of parkinsonism. Behav. Brain Res. 2008, 194, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Liu, K.; Agari, T.; Yasuhara, T.; Morimoto, J.; Okazaki, M.; Takeuchi, H.; Toyoshima, A.; Sasada, S.; Shinko, A.; et al. Anti-high mobility group box 1 antibody exerts neuroprotection in a rat model of Parkinson’s disease. Exp. Neurol. 2016, 275, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Shinko, A.; Agari, T.; Kameda, M.; Yasuhara, T.; Kondo, A.; Tayra, J.T.; Sato, K.; Sasaki, T.; Sasada, S.; Takeuchi, H.; et al. Spinal cord stimulation exerts neuroprotective effects against experimental Parkinson’s disease. PLoS ONE 2014, 9, e101468. [Google Scholar] [CrossRef] [PubMed]
- Thomas Tayra, J.; Kameda, M.; Yasuhara, T.; Agari, T.; Kadota, T.; Wang, F.; Kikuchi, Y.; Liang, H.; Shinko, A.; Wakamori, T.; et al. The neuroprotective and neurorescue effects of carbamylated erythropoietin Fc fusion protein (CEPO-Fc) in a rat model of Parkinson’s disease. Brain Res. 2013, 1502, 55–70. [Google Scholar] [CrossRef]
- Tajiri, N.; Yasuhara, T.; Shingo, T.; Kondo, A.; Yuan, W.; Kadota, T.; Wang, F.; Baba, T.; Tayra, J.T.; Morimoto, T.; et al. Exercise exerts neuroprotective effects on Parkinson’s disease model of rats. Brain Res. 2010, 1310, 200–207. [Google Scholar] [CrossRef]
- Kostrzewa, J.P.; Kostrzewa, R.A.; Kostrzewa, R.M.; Brus, R.; Nowak, P. Perinatal 6-Hydroxydopamine to Produce a Lifelong Model of Severe Parkinson’s Disease. Curr. Top. Behav. Neurosci. 2016, 29, 313–332. [Google Scholar] [CrossRef]
- Dovero, S.; Gross, C.; Bezard, E. Unexpected toxicity of very low dose MPTP in mice: A clue to the etiology of Parkinson’s disease? Synapse 2016, 70, 49–51. [Google Scholar] [CrossRef]
- Langston, J.W.; Ballard, P. Parkinsonism induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP): Implications for treatment and the pathogenesis of Parkinson’s disease. Can. J. Neurol. Sci. 1984, 11, 160–165. [Google Scholar] [CrossRef]
- Bezard, E.; Dovero, S.; Prunier, C.; Ravenscroft, P.; Chalon, S.; Guilloteau, D.; Crossman, A.R.; Bioulac, B.; Brotchie, J.M.; Gross, C.E. Relationship between the appearance of symptoms and the level of nigrostriatal degeneration in a progressive 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-lesioned macaque model of Parkinson’s disease. J. Neurosci. 2001, 21, 6853–6861. [Google Scholar] [CrossRef]
- Cui, M.; Aras, R.; Christian, W.V.; Rappold, P.M.; Hatwar, M.; Panza, J.; Jackson-Lewis, V.; Javitch, J.A.; Ballatori, N.; Przedborski, S.; et al. The organic cation transporter-3 is a pivotal modulator of neurodegeneration in the nigrostriatal dopaminergic pathway. Proc. Natl. Acad. Sci. USA 2009, 106, 8043–8048. [Google Scholar] [CrossRef]
- Javitch, J.A.; D’Amato, R.J.; Strittmatter, S.M.; Snyder, S.H. Parkinsonism-inducing neurotoxin, N-methyl-4-phenyl-1,2,3,6 -tetrahydropyridine: Uptake of the metabolite N-methyl-4-phenylpyridine by dopamine neurons explains selective toxicity. Proc. Natl. Acad. Sci. USA 1985, 82, 2173–2177. [Google Scholar] [CrossRef] [PubMed]
- Bezard, E.; Gross, C.E.; Fournier, M.C.; Dovero, S.; Bloch, B.; Jaber, M. Absence of MPTP-induced neuronal death in mice lacking the dopamine transporter. Exp. Neurol. 1999, 155, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Guillot, T.S.; Miller, G.W. Protective actions of the vesicular monoamine transporter 2 (VMAT2) in monoaminergic neurons. Mol. Neurobiol. 2009, 39, 149–170. [Google Scholar] [CrossRef] [PubMed]
- Meredith, G.E.; Rademacher, D.J. MPTP mouse models of Parkinson’s disease: An update. J. Parkinson’s Dis. 2011, 1, 19–33. [Google Scholar] [CrossRef]
- Barrientos, A.; Moraes, C.T. Titrating the effects of mitochondrial complex I impairment in the cell physiology. J. Biol. Chem. 1999, 274, 16188–16197. [Google Scholar] [CrossRef]
- Palmeira, C.M.; Moreno, A.J.; Madeira, V.M. Mitochondrial bioenergetics is affected by the herbicide paraquat. Biochim. Biophys. Acta 1995, 1229, 187–192. [Google Scholar] [CrossRef]
- Domico, L.M.; Zeevalk, G.D.; Bernard, L.P.; Cooper, K.R. Acute neurotoxic effects of mancozeb and maneb in mesencephalic neuronal cultures are associated with mitochondrial dysfunction. Neurotoxicology 2006, 27, 816–825. [Google Scholar] [CrossRef]
- Gash, D.M.; Rutland, K.; Hudson, N.L.; Sullivan, P.G.; Bing, G.; Cass, W.A.; Pandya, J.D.; Liu, M.; Choi, D.Y.; Hunter, R.L.; et al. Trichloroethylene: Parkinsonism and complex 1 mitochondrial neurotoxicity. Ann. Neurol. 2008, 63, 184–192. [Google Scholar] [CrossRef]
- Betarbet, R.; Sherer, T.B.; MacKenzie, G.; Garcia-Osuna, M.; Panov, A.V.; Greenamyre, J.T. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat. Neurosci. 2000, 3, 1301–1306. [Google Scholar] [CrossRef]
- Hoglinger, G.U.; Feger, J.; Prigent, A.; Michel, P.P.; Parain, K.; Champy, P.; Ruberg, M.; Oertel, W.H.; Hirsch, E.C. Chronic systemic complex I inhibition induces a hypokinetic multisystem degeneration in rats. J. Neurochem. 2003, 84, 491–502. [Google Scholar] [CrossRef]
- Cicchetti, F.; Drouin-Ouellet, J.; Gross, R.E. Environmental toxins and Parkinson’s disease: What have we learned from pesticide-induced animal models? Trends Pharmacol. Sci. 2009, 30, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Brooks, A.I.; Chadwick, C.A.; Gelbard, H.A.; Cory-Slechta, D.A.; Federoff, H.J. Paraquat elicited neurobehavioral syndrome caused by dopaminergic neuron loss. Brain Res. 1999, 823, 1–10. [Google Scholar] [CrossRef]
- McCormack, A.L.; Thiruchelvam, M.; Manning-Bog, A.B.; Thiffault, C.; Langston, J.W.; Cory-Slechta, D.A.; Di Monte, D.A. Environmental risk factors and Parkinson’s disease: Selective degeneration of nigral dopaminergic neurons caused by the herbicide paraquat. Neurobiol. Dis. 2002, 10, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Fernagut, P.O.; Hutson, C.B.; Fleming, S.M.; Tetreaut, N.A.; Salcedo, J.; Masliah, E.; Chesselet, M.F. Behavioral and histopathological consequences of paraquat intoxication in mice: Effects of alpha-synuclein over-expression. Synapse 2007, 61, 991–1001. [Google Scholar] [CrossRef] [PubMed]
- Manning-Bog, A.B.; McCormack, A.L.; Li, J.; Uversky, V.N.; Fink, A.L.; Di Monte, D.A. The herbicide paraquat causes up-regulation and aggregation of alpha-synuclein in mice: Paraquat and alpha-synuclein. J. Biol. Chem. 2002, 277, 1641–1644. [Google Scholar] [CrossRef] [PubMed]
- LoPachin, R.M.; Gavin, T. Response to Paraquat: The red herring of Parkinson’s disease research. Toxicol. Sci. 2008, 103, 219–221. [Google Scholar] [CrossRef] [PubMed]
- Thiruchelvam, M.; Brockel, B.J.; Richfield, E.K.; Baggs, R.B.; Cory-Slechta, D.A. Potentiated and preferential effects of combined paraquat and maneb on nigrostriatal dopamine systems: Environmental risk factors for Parkinson’s disease? Brain Res. 2000, 873, 225–234. [Google Scholar] [CrossRef]
- Grandi, L.C.; Di Giovanni, G.; Galati, S. Animal models of early-stage Parkinson’s disease and acute dopamine deficiency to study compensatory neurodegenerative mechanisms. J. Neurosci. Methods 2018, 308, 205–218. [Google Scholar] [CrossRef]
- Smith, G.A.; Isacson, O.; Dunnett, S.B. The search for genetic mouse models of prodromal Parkinson’s disease. Exp. Neurol. 2012, 237, 267–273. [Google Scholar] [CrossRef]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Bonifati, V.; Rizzu, P.; van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Dekker, M.C.; Squitieri, F.; Ibanez, P.; Joosse, M.; et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 2003, 299, 256–259. [Google Scholar] [CrossRef] [PubMed]
- Moore, D.J.; Dawson, T.M. Value of genetic models in understanding the cause and mechanisms of Parkinson’s disease. Curr. Neurol. Neurosci. Rep. 2008, 8, 288–296. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Dawson, V.L.; Dawson, T.M. Animal models of Parkinson’s disease: Vertebrate genetics. Cold Spring Harb. Perspect. Med. 2012, 2. [Google Scholar] [CrossRef]
- Potashkin, J.A.; Blume, S.R.; Runkle, N.K. Limitations of animal models of Parkinson’s disease. Parkinson’s Dis. 2010, 2011, 658083. [Google Scholar] [CrossRef]
- Lim, K.L.; Ng, C.H. Genetic models of Parkinson disease. Biochim. Biophys. Acta 2009, 1792, 604–615. [Google Scholar] [CrossRef]
- Murphy, D.D.; Rueter, S.M.; Trojanowski, J.Q.; Lee, V.M. Synucleins are developmentally expressed, and alpha-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J. Neurosci. 2000, 20, 3214–3220. [Google Scholar] [CrossRef]
- Sharon, R.; Goldberg, M.S.; Bar-Josef, I.; Betensky, R.A.; Shen, J.; Selkoe, D.J. alpha-Synuclein occurs in lipid-rich high molecular weight complexes, binds fatty acids, and shows homology to the fatty acid-binding proteins. Proc. Natl. Acad. Sci. USA 2001, 98, 9110–9115. [Google Scholar] [CrossRef]
- Goedert, M.; Spillantini, M.G.; Del Tredici, K.; Braak, H. 100 years of Lewy pathology. Nat. Rev. Neurol. 2013, 9, 13–24. [Google Scholar] [CrossRef]
- Kruger, R.; Kuhn, W.; Muller, T.; Woitalla, D.; Graeber, M.; Kosel, S.; Przuntek, H.; Epplen, J.T.; Schols, L.; Riess, O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat. Genet. 1998, 18, 106–108. [Google Scholar] [CrossRef]
- Zarranz, J.J.; Alegre, J.; Gomez-Esteban, J.C.; Lezcano, E.; Ros, R.; Ampuero, I.; Vidal, L.; Hoenicka, J.; Rodriguez, O.; Atares, B.; et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 2004, 55, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. alpha-Synuclein locus triplication causes Parkinson’s disease. Science 2003, 302, 841. [Google Scholar] [CrossRef] [PubMed]
- Vekrellis, K.; Xilouri, M.; Emmanouilidou, E.; Rideout, H.J.; Stefanis, L. Pathological roles of alpha-synuclein in neurological disorders. Lancet Neurol. 2011, 10, 1015–1025. [Google Scholar] [CrossRef]
- Schapira, A.H.; Olanow, C.W.; Greenamyre, J.T.; Bezard, E. Slowing of neurodegeneration in Parkinson’s disease and Huntington’s disease: Future therapeutic perspectives. Lancet 2014, 384, 545–555. [Google Scholar] [CrossRef]
- Kara, E.; Kiely, A.P.; Proukakis, C.; Giffin, N.; Love, S.; Hehir, J.; Rantell, K.; Pandraud, A.; Hernandez, D.G.; Nacheva, E.; et al. A 6.4 Mb duplication of the alpha-synuclein locus causing frontotemporal dementia and Parkinsonism: Phenotype-genotype correlations. JAMA Neurol. 2014, 71, 1162–1171. [Google Scholar] [CrossRef]
- Lucking, C.B.; Durr, A.; Bonifati, V.; Vaughan, J.; De Michele, G.; Gasser, T.; Harhangi, B.S.; Meco, G.; Denefle, P.; Wood, N.W.; et al. Association between early-onset Parkinson’s disease and mutations in the parkin gene. N. Engl. J. Med. 2000, 342, 1560–1567. [Google Scholar] [CrossRef]
- Klein, C.; Lohmann, K. Parkinson disease(s): Is “Parkin disease” a distinct clinical entity? Neurology 2009, 72, 106–107. [Google Scholar] [CrossRef]
- Klein, C.; Schlossmacher, M.G. Parkinson disease, 10 years after its genetic revolution: Multiple clues to a complex disorder. Neurology 2007, 69, 2093–2104. [Google Scholar] [CrossRef]
- Pilcher, H. Parkin implicated in sporadic Parkinson’s disease. Lancet Neurol. 2005, 4, 798. [Google Scholar] [CrossRef]
- Schulte, C.; Gasser, T. Genetic basis of Parkinson’s disease: Inheritance, penetrance, and expression. Appl. Clin. Genet. 2011, 4, 67–80. [Google Scholar] [CrossRef]
- Klein, C.; Westenberger, A. Genetics of Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a008888. [Google Scholar] [CrossRef] [PubMed]
- Dawson, T.M.; Dawson, V.L. The role of parkin in familial and sporadic Parkinson’s disease. Mov. Disord. 2010, 25 (Suppl. 1), S32–S39. [Google Scholar] [CrossRef] [PubMed]
- Kazlauskaite, A.; Muqit, M.M. PINK1 and Parkin—Mitochondrial interplay between phosphorylation and ubiquitylation in Parkinson’s disease. FEBS J 2015, 282, 215–223. [Google Scholar] [CrossRef]
- Vercammen, L.; Van der Perren, A.; Vaudano, E.; Gijsbers, R.; Debyser, Z.; Van den Haute, C.; Baekelandt, V. Parkin protects against neurotoxicity in the 6-hydroxydopamine rat model for Parkinson’s disease. Mol. Ther. 2006, 14, 716–723. [Google Scholar] [CrossRef] [PubMed]
- Yamada, M.; Mizuno, Y.; Mochizuki, H. Parkin gene therapy for alpha-synucleinopathy: A rat model of Parkinson’s disease. Hum. Gene Ther. 2005, 16, 262–270. [Google Scholar] [CrossRef]
- Lo Bianco, C.; Schneider, B.L.; Bauer, M.; Sajadi, A.; Brice, A.; Iwatsubo, T.; Aebischer, P. Lentiviral vector delivery of parkin prevents dopaminergic degeneration in an alpha-synuclein rat model of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 17510–17515. [Google Scholar] [CrossRef]
- Manfredsson, F.P.; Burger, C.; Sullivan, L.F.; Muzyczka, N.; Lewin, A.S.; Mandel, R.J. rAAV-mediated nigral human parkin over-expression partially ameliorates motor deficits via enhanced dopamine neurotransmission in a rat model of Parkinson’s disease. Exp. Neurol. 2007, 207, 289–301. [Google Scholar] [CrossRef]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.S.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef]
- Puschmann, A.; Fiesel, F.C.; Caulfield, T.R.; Hudec, R.; Ando, M.; Truban, D.; Hou, X.; Ogaki, K.; Heckman, M.G.; James, E.D.; et al. Heterozygous PINK1 p.G411S increases risk of Parkinson’s disease via a dominant-negative mechanism. Brain 2017, 140, 98–117. [Google Scholar] [CrossRef]
- Valente, E.M.; Salvi, S.; Ialongo, T.; Marongiu, R.; Elia, A.E.; Caputo, V.; Romito, L.; Albanese, A.; Dallapiccola, B.; Bentivoglio, A.R. PINK1 mutations are associated with sporadic early-onset parkinsonism. Ann. Neurol. 2004, 56, 336–341. [Google Scholar] [CrossRef] [PubMed]
- Poulopoulos, M.; Levy, O.A.; Alcalay, R.N. The neuropathology of genetic Parkinson’s disease. Mov. Disord. 2012, 27, 831–842. [Google Scholar] [CrossRef] [PubMed]
- Glasl, L.; Kloos, K.; Giesert, F.; Roethig, A.; Di Benedetto, B.; Kuhn, R.; Zhang, J.; Hafen, U.; Zerle, J.; Hofmann, A.; et al. Pink1-deficiency in mice impairs gait, olfaction and serotonergic innervation of the olfactory bulb. Exp. Neurol. 2012, 235, 214–227. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Dickson, D.W. Parkinson’s disease: Experimental models and reality. Acta Neuropathol. 2018, 135, 13–32. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Cagniard, B.; Mathews, T.; Jones, S.; Koh, H.C.; Ding, Y.; Carvey, P.M.; Ling, Z.; Kang, U.J.; Zhuang, X. Age-dependent motor deficits and dopaminergic dysfunction in DJ-1 null mice. J. Biol. Chem. 2005, 280, 21418–21426. [Google Scholar] [CrossRef]
- Goldberg, M.S.; Pisani, A.; Haburcak, M.; Vortherms, T.A.; Kitada, T.; Costa, C.; Tong, Y.; Martella, G.; Tscherter, A.; Martins, A.; et al. Nigrostriatal dopaminergic deficits and hypokinesia caused by inactivation of the familial Parkinsonism-linked gene DJ-1. Neuron 2005, 45, 489–496. [Google Scholar] [CrossRef]
- Lev, N.; Ickowicz, D.; Melamed, E.; Offen, D. Oxidative insults induce DJ-1 upregulation and redistribution: Implications for neuroprotection. Neurotoxicology 2008, 29, 397–405. [Google Scholar] [CrossRef]
- Kim, R.H.; Smith, P.D.; Aleyasin, H.; Hayley, S.; Mount, M.P.; Pownall, S.; Wakeham, A.; You-Ten, A.J.; Kalia, S.K.; Horne, P.; et al. Hypersensitivity of DJ-1-deficient mice to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyrindine (MPTP) and oxidative stress. Proc. Natl. Acad. Sci. USA 2005, 102, 5215–5220. [Google Scholar] [CrossRef]
- Dave, K.D.; De Silva, S.; Sheth, N.P.; Ramboz, S.; Beck, M.J.; Quang, C.; Switzer, R.C., 3rd; Ahmad, S.O.; Sunkin, S.M.; Walker, D.; et al. Phenotypic characterization of recessive gene knockout rat models of Parkinson’s disease. Neurobiol. Dis. 2014, 70, 190–203. [Google Scholar] [CrossRef]
- Yeo, G.W.; Van Nostrand, E.; Holste, D.; Poggio, T.; Burge, C.B. Identification and analysis of alternative splicing events conserved in human and mouse. Proc. Natl. Acad. Sci. USA 2005, 102, 2850–2855. [Google Scholar] [CrossRef]
- Borghammer, P. How does parkinson’s disease begin? Perspectives on neuroanatomical pathways, prions, and histology. Mov. Disord. 2018, 33, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Heinemann, S.D.; Posimo, J.M.; Mason, D.M.; Hutchison, D.F.; Leak, R.K. Synergistic stress exacerbation in hippocampal neurons: Evidence favoring the dual-hit hypothesis of neurodegeneration. Hippocampus 2016, 26, 980–994. [Google Scholar] [CrossRef] [PubMed]
- Billings, J.L.; Gordon, S.L.; Rawling, T.; Doble, P.A.; Bush, A.I.; Adlard, P.A.; Finkelstein, D.I.; Hare, D.J. l-3,4-dihydroxyphenylalanine (l-DOPA) modulates brain iron, dopaminergic neurodegeneration and motor dysfunction in iron overload and mutant alpha-synuclein mouse models of Parkinson’s disease. J. Neurochem. 2019. [Google Scholar] [CrossRef] [PubMed]
- Campos, F.L.; Carvalho, M.M.; Cristovao, A.C.; Je, G.; Baltazar, G.; Salgado, A.J.; Kim, Y.S.; Sousa, N. Rodent models of Parkinson’s disease: Beyond the motor symptomatology. Front. Behav. Neurosci. 2013, 7, 175. [Google Scholar] [CrossRef]
- Yang, W.; Li, S.; Li, X.J. A CRISPR monkey model unravels a unique function of PINK1 in primate brains. Mol. Neurodegener. 2019, 14, 17. [Google Scholar] [CrossRef]
- Lee, E.J.; Yoon, H.H.; Park, E.S.; Min, J.; Jeon, S.R. A Novel Animal Model of Parkinson’s Disease Using Optogenetics: Representation of Various Disease Stages by Modulating the Illumination Parameter. Stereotact. Funct. Neurosurg. 2018, 96, 22–32. [Google Scholar] [CrossRef]
- Luk, K.C.; Kehm, V.; Carroll, J.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 2012, 338, 949–953. [Google Scholar] [CrossRef]
- Luk, K.C.; Kehm, V.M.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M. Intracerebral inoculation of pathological alpha-synuclein initiates a rapidly progressive neurodegenerative alpha-synucleinopathy in mice. J. Exp. Med. 2012, 209, 975–986. [Google Scholar] [CrossRef]
- Kim, A.; Nigmatullina, R.; Zalyalova, Z.; Soshnikova, N.; Krasnov, A.; Vorobyeva, N.; Georgieva, S.; Kudrin, V.; Narkevich, V.; Ugrumov, M. Upgraded Methodology for the Development of Early Diagnosis of Parkinson’s Disease Based on Searching Blood Markers in Patients and Experimental Models. Mol. Neurobiol. 2018. [Google Scholar] [CrossRef]
- Lee, K.S.; Lee, J.K.; Kim, H.G.; Kim, H.R. Differential Effects of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine on Motor Behavior and Dopamine Levels at Brain Regions in Three Different Mouse Strains. Korean J. Physiol. Pharmacol. 2013, 17, 89–97. [Google Scholar] [CrossRef]
- Kaur, D.; Peng, J.; Chinta, S.J.; Rajagopalan, S.; Di Monte, D.A.; Cherny, R.A.; Andersen, J.K. Increased murine neonatal iron intake results in Parkinson-like neurodegeneration with age. Neurobiol. Aging 2007, 28, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Giasson, B.I.; Duda, J.E.; Quinn, S.M.; Zhang, B.; Trojanowski, J.Q.; Lee, V.M. Neuronal alpha-synucleinopathy with severe movement disorder in mice expressing A53T human alpha-synuclein. Neuron 2002, 34, 521–533. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Animal Model | The Main Difference Between Human PD and Animal PD Model | |
|---|---|---|
| Neurotoxin model | 6-OHDA |
|
| MPTP |
| |
| Pesticides |
| |
| Genetic model |
| |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kin, K.; Yasuhara, T.; Kameda, M.; Date, I. Animal Models for Parkinson’s Disease Research: Trends in the 2000s. Int. J. Mol. Sci. 2019, 20, 5402. https://doi.org/10.3390/ijms20215402
Kin K, Yasuhara T, Kameda M, Date I. Animal Models for Parkinson’s Disease Research: Trends in the 2000s. International Journal of Molecular Sciences. 2019; 20(21):5402. https://doi.org/10.3390/ijms20215402
Chicago/Turabian StyleKin, Kyohei, Takao Yasuhara, Masahiro Kameda, and Isao Date. 2019. "Animal Models for Parkinson’s Disease Research: Trends in the 2000s" International Journal of Molecular Sciences 20, no. 21: 5402. https://doi.org/10.3390/ijms20215402
APA StyleKin, K., Yasuhara, T., Kameda, M., & Date, I. (2019). Animal Models for Parkinson’s Disease Research: Trends in the 2000s. International Journal of Molecular Sciences, 20(21), 5402. https://doi.org/10.3390/ijms20215402