Global Proteome of LonP1+/− Mouse Embryonal Fibroblasts Reveals Impact on Respiratory Chain, but No Interdependence between Eral1 and Mitoribosomes

Abstract
1. Introduction
2. Results
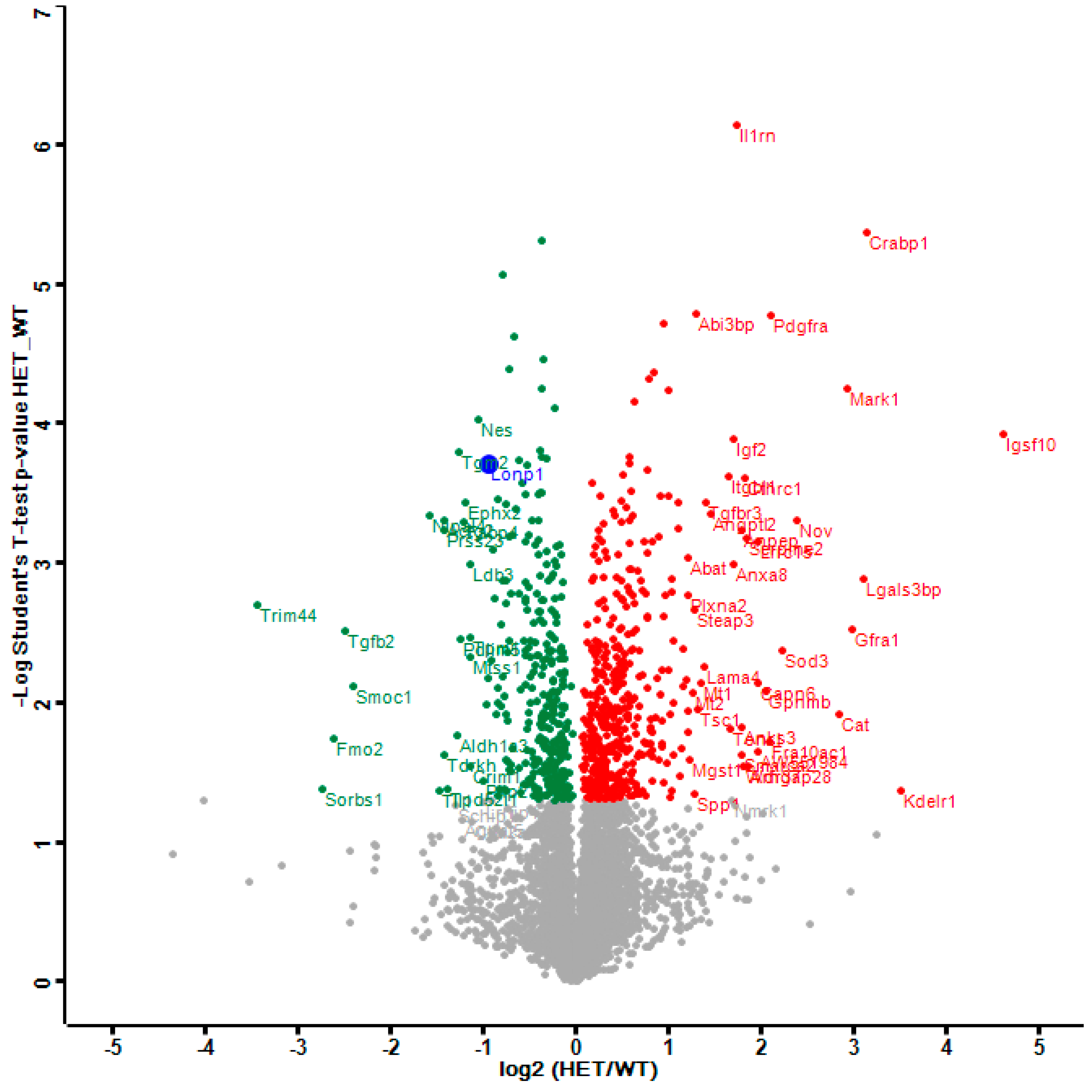
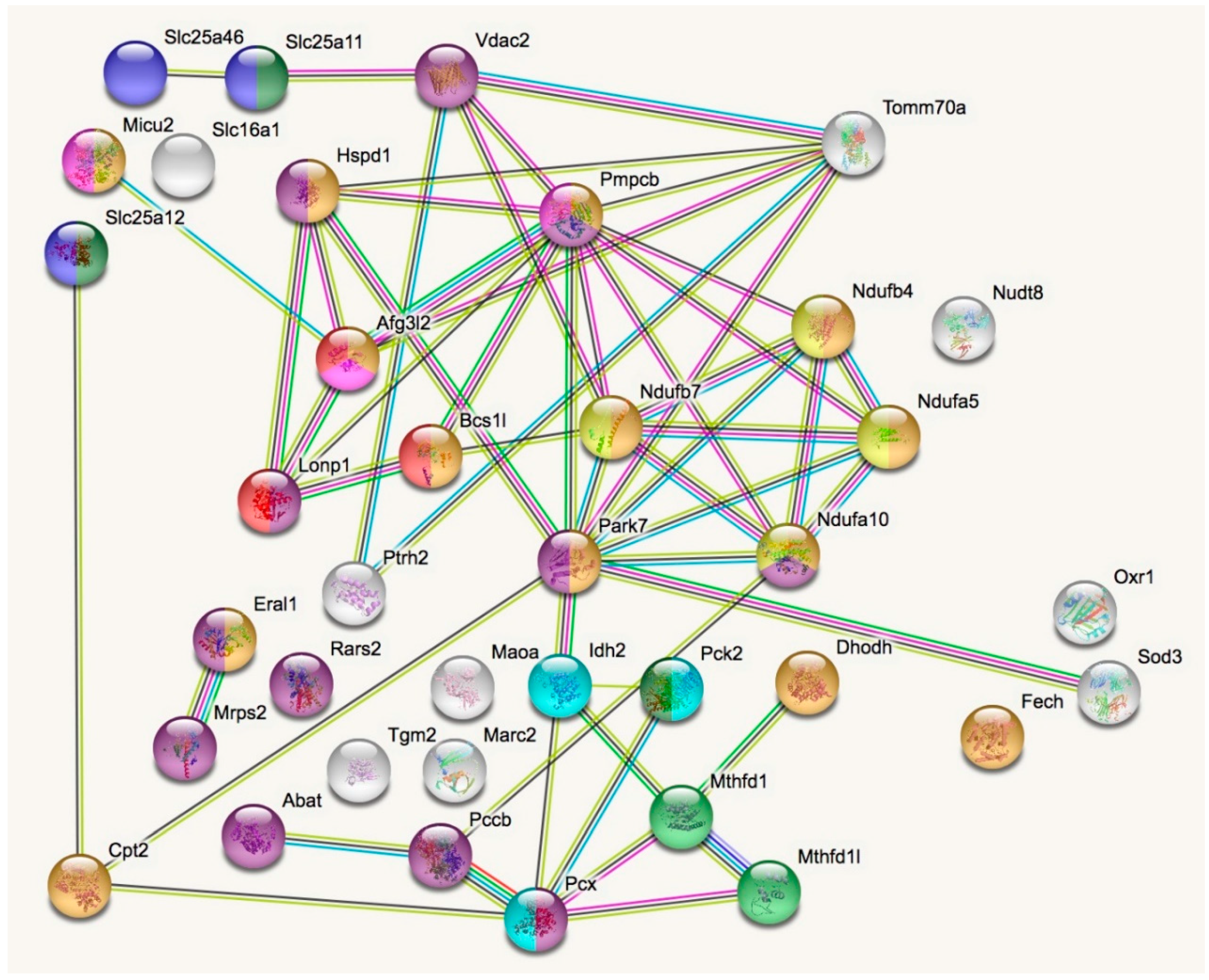
2.1. Dysregulated Mitochondrial Factors in LonP1+/− MEFs are Enriched for Oxidation Processes
2.1.1. Mitochondrial Downregulations in LonP1+/− MEFs
2.1.2. Mitochondrial Upregulations in LonP1+/− MEFs
2.1.3. Eral1 and Mitoribosomal Factors as Substrates of LonP1 Versus ClpXP—Two Meta-Analyses
2.2. Oxidative Stress and Glutathione Pathways in LonP1+/− MEFs
2.3. Activation of the Innate Immune System in LonP1+/− MEFs
2.4. The Strongest Upregulations in LonP1+/− MEFs can be Seen in the Lysosomal Pathway
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Proteomics
4.3. Bioinformatic Analyses
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AAA+ | ATPases Associated with diverse cellular Activities |
| AFG3L2 | AFG3-Like matrix AAA peptidase subunit 2 |
| ATPase | Adenosine Tri-Phosphatase |
| CLPP | Caseinolytic Mitochondrial Matrix Peptidase Proteolytic Subunit |
| CLPX | Caseinolytic Mitochondrial Matrix Peptidase Chaperone Subunit |
| CO2 | Carbon dioxide |
| CoA | Coenzyme-A |
| CODAS | Syndrome with cerebral, ocular, dental, auricular, and skeletal anomalies |
| DNA | Deoxyribonucleic acid |
| dNDPs | Deoxy-Nucleoside Di-Phosphate |
| dNTPs | Deoxy-Nucleoside Tri-Phosphate |
| dsRNA | Double-stranded RNA |
| FDR | False discovery rate |
| g | gram |
| GABA | Gamma-Amino Butyric Acid |
| GO | Gene Ontology |
| HET | Heterozygous mutant |
| HP arm | Membrane hydrophobic part of respiratory complex I |
| INTERPRO | Database of protein families, domains and functional sites |
| IP/FP arm | Iron-sulfur-cluster containing part and FMN containing part of respiratory complex I |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| KO | knockout |
| l | liter |
| LC3 | Light Chain 3 protein, encoded by the MAP1LC3A gene |
| LIM-type | Zinc finger domain named after proteins LIN-11, Isl1 and MEC-3 where they were first found |
| LONP1 | Mitochondrial ATP-Dependent Protease Lon, or PRSS15 for Serine Protease 15 |
| MCU | Mitochondrial Calcium Uniporter |
| MEF | Murine embryonal fibroblasts |
| mAAA | Matrix AAA Peptidase |
| mtDNA | Mitochondrial DNA |
| NOD | Nucleotide-binding oligomerization domain-containing protein |
| PARK2 | Autosomal recessively inherited Parkinson’s disease type 2, caused by mutations in Parkin |
| PARK6 | Autosomal recessively inherited Parkinson’s disease type 6, caused by mutations in PINK1 |
| PARK7 | Autosomal recessively inherited Parkinson’s disease type 7, caused by mutations in DJ-1 |
| PARL | Presenilin-Associated Rhomboid-Like protein, mitochondrial protease |
| PFAM | Database of protein families including their annotations and multiple sequence alignments |
| PINK1 | PTEN-induced Kinase 1, mitochondrial |
| PRLTS3 | Perrault syndrome type 3 due to CLPP mutations |
| REACTOME | Database of reactions, pathways and biological processes |
| RIG-I | Retinoic Acid-Inducible Gene 1 Protein, encoded by the Ddx58 gene |
| RNA | Ribonucleic acid |
| SCA28 | Spinocerebellar Ataxia type 28, caused by mutations in |
| SMART | Simple Modular Architecture Research Tool |
| SMDT1 | Single-pass Membrane protein with Aspartate-rich Tail 1, mitochondrial |
| STING | Stimulator of INterferon Genes protein, encoded by the Tmem173 gene |
| STRING | Search tool for the retrieval of interacting genes |
| TCA cycle | Tricarboxylic acid cycle, Citric acid cycle, Krebs cycle |
| UPRmt | Mitochondrial Unfolded Protein Response |
| WT | Wildtype |
References
- Key, J.; Mueller, A.K.; Gispert, S.; Matschke, L.; Wittig, I.; Corti, O.; Munch, C.; Decher, N.; Auburger, G. Ubiquitylome profiling of Parkin-null brain reveals dysregulation of calcium homeostasis factors ATP1A2, Hippocalcin and GNA11, reflected by altered firing of noradrenergic neurons. Neurobiol. Dis. 2019, 127, 114–130. [Google Scholar] [CrossRef] [PubMed]
- Rugarli, E.I.; Langer, T. Mitochondrial quality control: A matter of life and death for neurons. EMBO J. 2012, 31, 1336–1349. [Google Scholar] [CrossRef] [PubMed]
- Torres-Odio, S.; Key, J.; Hoepken, H.H.; Canet-Pons, J.; Valek, L.; Roller, B.; Walter, M.; Morales-Gordo, B.; Meierhofer, D.; Harter, P.N.; et al. Progression of pathology in PINK1-deficient mouse brain from splicing via ubiquitination, ER stress, and mitophagy changes to neuroinflammation. J. Neuroinflamm. 2017, 14, 154. [Google Scholar] [CrossRef] [PubMed]
- West, A.P.; Shadel, G.S.; Ghosh, S. Mitochondria in innate immune responses. Nat. Rev. Immunol. 2011, 11, 389–402. [Google Scholar] [CrossRef] [PubMed]
- Bulteau, A.L.; Bayot, A. Mitochondrial proteases and cancer. Biochim. Biophys. Acta 2011, 1807, 595–601. [Google Scholar] [CrossRef] [PubMed]
- Quiros, P.M.; Barcena, C.; Lopez-Otin, C. Lon protease: A key enzyme controlling mitochondrial bioenergetics in cancer. Mol. Cell. Oncol. 2014, 1, e968505. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, S.; Lee, J.; Singh, K.; Lee, I.; Suzuki, C.K. Multitasking in the mitochondrion by the ATP-dependent Lon protease. Biochim. Biophys. Acta 2012, 1823, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Quiros, P.M.; Langer, T.; Lopez-Otin, C. New roles for mitochondrial proteases in health, ageing and disease. Nat. Rev. Mol. Cell Biol. 2015, 16, 345–359. [Google Scholar] [CrossRef]
- Martinelli, P.; Rugarli, E.I. Emerging roles of mitochondrial proteases in neurodegeneration. Biochim. Biophys. Acta 2010, 1797, 1–10. [Google Scholar] [CrossRef]
- Newman, W.G.; Friedman, T.B.; Conway, G.S.; Demain, L.A.M. Perrault Syndrome. In GeneReviews((R)); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Di Bella, D.; Lazzaro, F.; Brusco, A.; Plumari, M.; Battaglia, G.; Pastore, A.; Finardi, A.; Cagnoli, C.; Tempia, F.; Frontali, M.; et al. Mutations in the mitochondrial protease gene AFG3L2 cause dominant hereditary ataxia SCA28. Nat. Genet. 2010, 42, 313–321. [Google Scholar] [CrossRef]
- Strauss, K.A.; Jinks, R.N.; Puffenberger, E.G.; Venkatesh, S.; Singh, K.; Cheng, I.; Mikita, N.; Thilagavathi, J.; Lee, J.; Sarafianos, S.; et al. CODAS syndrome is associated with mutations of LONP1, encoding mitochondrial AAA+ Lon protease. Am. J. Hum. Genet. 2015, 96, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Spinazzi, M.; De Strooper, B. PARL: The mitochondrial rhomboid protease. Semin. Cell Dev. Biol. 2016, 60, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.Y.; McQuibban, G.A. The mitochondrial rhomboid protease: Its rise from obscurity to the pinnacle of disease-relevant genes. Biochim. Biophys. Acta 2013, 1828, 2916–2925. [Google Scholar] [CrossRef] [PubMed]
- Shi, G.; McQuibban, G.A. The Mitochondrial Rhomboid Protease PARL Is Regulated by PDK2 to Integrate Mitochondrial Quality Control and Metabolism. Cell Rep. 2017, 18, 1458–1472. [Google Scholar] [CrossRef] [PubMed]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [PubMed]
- Exner, N.; Treske, B.; Paquet, D.; Holmstrom, K.; Schiesling, C.; Gispert, S.; Carballo-Carbajal, I.; Berg, D.; Hoepken, H.H.; Gasser, T.; et al. Loss-of-function of human PINK1 results in mitochondrial pathology and can be rescued by parkin. J. Neurosci. 2007, 27, 12413–12418. [Google Scholar] [CrossRef] [PubMed]
- Gispert, S.; Ricciardi, F.; Kurz, A.; Azizov, M.; Hoepken, H.H.; Becker, D.; Voos, W.; Leuner, K.; Muller, W.E.; Kudin, A.P.; et al. Parkinson phenotype in aged PINK1-deficient mice is accompanied by progressive mitochondrial dysfunction in absence of neurodegeneration. PLoS ONE 2009, 4, e5777. [Google Scholar] [CrossRef]
- Gispert, S.; Brehm, N.; Weil, J.; Seidel, K.; Rub, U.; Kern, B.; Walter, M.; Roeper, J.; Auburger, G. Potentiation of neurotoxicity in double-mutant mice with Pink1 ablation and A53T-SNCA overexpression. Hum. Mol. Genet. 2015, 24, 1061–1076. [Google Scholar] [CrossRef]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef]
- Wai, T.; Saita, S.; Nolte, H.; Muller, S.; Konig, T.; Richter-Dennerlein, R.; Sprenger, H.G.; Madrenas, J.; Muhlmeister, M.; Brandt, U.; et al. The membrane scaffold SLP2 anchors a proteolytic hub in mitochondria containing PARL and the i-AAA protease YME1L. EMBO Rep. 2016, 17, 1844–1856. [Google Scholar] [CrossRef]
- Sekine, S.; Wang, C.; Sideris, D.P.; Bunker, E.; Zhang, Z.; Youle, R.J. Reciprocal Roles of Tom7 and OMA1 during Mitochondrial Import and Activation of PINK1. Mol. Cell 2019, 73, 1028–1043. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, L.; Chin, L.S. Parkinson disease protein DJ-1 converts from a zymogen to a protease by carboxyl-terminal cleavage. Hum. Mol. Genet. 2010, 19, 2395–2408. [Google Scholar] [CrossRef]
- Ariga, H.; Takahashi-Niki, K.; Kato, I.; Maita, H.; Niki, T.; Iguchi-Ariga, S.M. Neuroprotective function of DJ-1 in Parkinson’s disease. Oxid. Med. Cell. Longev. 2013, 2013, 683920. [Google Scholar] [CrossRef] [PubMed]
- Shendelman, S.; Jonason, A.; Martinat, C.; Leete, T.; Abeliovich, A. DJ-1 is a redox-dependent molecular chaperone that inhibits alpha-synuclein aggregate formation. PLoS Biol. 2004, 2, e362. [Google Scholar] [CrossRef]
- Baker, M.J.; Tatsuta, T.; Langer, T. Quality control of mitochondrial proteostasis. Cold Spring Harb. Perspect. Biol. 2011, 3, a007559. [Google Scholar] [CrossRef] [PubMed]
- Hwang, A.B.; Jeong, D.E.; Lee, S.J. Mitochondria and organismal longevity. Curr. Genom. 2012, 13, 519–532. [Google Scholar] [CrossRef]
- Aerts, A.M.; Zabrocki, P.; Govaert, G.; Mathys, J.; Carmona-Gutierrez, D.; Madeo, F.; Winderickx, J.; Cammue, B.P.; Thevissen, K. Mitochondrial dysfunction leads to reduced chronological lifespan and increased apoptosis in yeast. FEBS Lett. 2009, 583, 113–117. [Google Scholar] [CrossRef]
- Fischer, F.; Weil, A.; Hamann, A.; Osiewacz, H.D. Human CLPP reverts the longevity phenotype of a fungal ClpP deletion strain. Nat. Commun. 2013, 4, 1397. [Google Scholar] [CrossRef]
- Bota, D.A.; Ngo, J.K.; Davies, K.J. Downregulation of the human Lon protease impairs mitochondrial structure and function and causes cell death. Free Radic. Biol. Med. 2005, 38, 665–677. [Google Scholar] [CrossRef]
- Bota, D.A.; Davies, K.J. Lon protease preferentially degrades oxidized mitochondrial aconitase by an ATP-stimulated mechanism. Nat. Cell Biol. 2002, 4, 674–680. [Google Scholar] [CrossRef]
- Fu, G.K.; Markovitz, D.M. The human LON protease binds to mitochondrial promoters in a single-stranded, site-specific, strand-specific manner. Biochemistry 1998, 37, 1905–1909. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Liu, T.; Crosby, J.A.; Thomas-Wohlever, J.; Lee, I.; Suzuki, C.K. The ATP-dependent Lon protease of Mus musculus is a DNA-binding protein that is functionally conserved between yeast and mammals. Gene 2003, 306, 45–55. [Google Scholar] [CrossRef]
- Gur, E.; Sauer, R.T. Recognition of misfolded proteins by Lon, a AAA(+) protease. Genes Dev. 2008, 22, 2267–2277. [Google Scholar] [CrossRef] [PubMed]
- Peter, B.; Waddington, C.L.; Olahova, M.; Sommerville, E.W.; Hopton, S.; Pyle, A.; Champion, M.; Ohlson, M.; Siibak, T.; Chrzanowska-Lightowlers, Z.M.A.; et al. Defective mitochondrial protease LonP1 can cause classical mitochondrial disease. Hum. Mol. Genet. 2018, 27, 1743–1753. [Google Scholar] [CrossRef] [PubMed]
- Dikoglu, E.; Alfaiz, A.; Gorna, M.; Bertola, D.; Chae, J.H.; Cho, T.J.; Derbent, M.; Alanay, Y.; Guran, T.; Kim, O.H.; et al. Mutations in LONP1, a mitochondrial matrix protease, cause CODAS syndrome. Am. J. Med. Genet. Part A 2015, 167, 1501–1509. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, R.; Zhang, H.; Kim, J.W.; Shimoda, L.; Dang, C.V.; Semenza, G.L. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell 2007, 129, 111–122. [Google Scholar] [CrossRef]
- Granot, Z.; Kobiler, O.; Melamed-Book, N.; Eimerl, S.; Bahat, A.; Lu, B.; Braun, S.; Maurizi, M.R.; Suzuki, C.K.; Oppenheim, A.B.; et al. Turnover of mitochondrial steroidogenic acute regulatory (StAR) protein by Lon protease: The unexpected effect of proteasome inhibitors. Mol. Endocrinol. 2007, 21, 2164–2177. [Google Scholar] [CrossRef] [PubMed]
- Tian, Q.; Li, T.; Hou, W.; Zheng, J.; Schrum, L.W.; Bonkovsky, H.L. Lon peptidase 1 (LONP1)-dependent breakdown of mitochondrial 5-aminolevulinic acid synthase protein by heme in human liver cells. J. Biol. Chem. 2011, 286, 26424–26430. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Lee, J.; Nie, X.; Li, M.; Morozov, Y.I.; Venkatesh, S.; Bogenhagen, D.F.; Temiakov, D.; Suzuki, C.K. Phosphorylation of human TFAM in mitochondria impairs DNA binding and promotes degradation by the AAA+ Lon protease. Mol. Cell 2013, 49, 121–132. [Google Scholar] [CrossRef]
- Baker, T.A.; Sauer, R.T. ClpXP, an ATP-powered unfolding and protein-degradation machine. Biochim. Biophys. Acta 2012, 1823, 15–28. [Google Scholar] [CrossRef]
- Al-Furoukh, N.; Ianni, A.; Nolte, H.; Holper, S.; Kruger, M.; Wanrooij, S.; Braun, T. ClpX stimulates the mitochondrial unfolded protein response (UPRmt) in mammalian cells. Biochim. Biophys. Acta 2015, 1853, 2580–2591. [Google Scholar] [CrossRef] [PubMed]
- Haynes, C.M.; Petrova, K.; Benedetti, C.; Yang, Y.; Ron, D. ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Dev. Cell 2007, 13, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Andersson, F.I.; Tryggvesson, A.; Sharon, M.; Diemand, A.V.; Classen, M.; Best, C.; Schmidt, R.; Schelin, J.; Stanne, T.M.; Bukau, B.; et al. Structure and function of a novel type of ATP-dependent Clp protease. J. Biol. Chem. 2009, 284, 13519–13532. [Google Scholar] [CrossRef] [PubMed]
- Jenkinson, E.M.; Rehman, A.U.; Walsh, T.; Clayton-Smith, J.; Lee, K.; Morell, R.J.; Drummond, M.C.; Khan, S.N.; Naeem, M.A.; Rauf, B.; et al. Perrault syndrome is caused by recessive mutations in CLPP, encoding a mitochondrial ATP-dependent chambered protease. Am. J. Hum. Genet. 2013, 92, 605–613. [Google Scholar] [CrossRef] [PubMed]
- Gispert, S.; Parganlija, D.; Klinkenberg, M.; Drose, S.; Wittig, I.; Mittelbronn, M.; Grzmil, P.; Koob, S.; Hamann, A.; Walter, M.; et al. Loss of mitochondrial peptidase Clpp leads to infertility, hearing loss plus growth retardation via accumulation of CLPX, mtDNA and inflammatory factors. Hum. Mol. Genet. 2013, 22, 4871–4887. [Google Scholar] [CrossRef] [PubMed]
- Szczepanowska, K.; Maiti, P.; Kukat, A.; Hofsetz, E.; Nolte, H.; Senft, K.; Becker, C.; Ruzzenente, B.; Hornig-Do, H.T.; Wibom, R.; et al. CLPP coordinates mitoribosomal assembly through the regulation of ERAL1 levels. EMBO J. 2016, 35, 2566–2583. [Google Scholar] [CrossRef]
- Jin, S.M.; Youle, R.J. The accumulation of misfolded proteins in the mitochondrial matrix is sensed by PINK1 to induce PARK2/Parkin-mediated mitophagy of polarized mitochondria. Autophagy 2013, 9, 1750–1757. [Google Scholar] [CrossRef]
- Horner, S.M.; Wilkins, C.; Badil, S.; Iskarpatyoti, J.; Gale, M., Jr. Proteomic analysis of mitochondrial-associated ER membranes (MAM) during RNA virus infection reveals dynamic changes in protein and organelle trafficking. PLoS ONE 2015, 10, e0117963. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, G.; Xu, Z.G.; Tu, H.; Hu, F.; Dai, J.; Chang, Y.; Chen, Y.; Lu, Y.; Zeng, H.; et al. Lactate Is a Natural Suppressor of RLR Signaling by Targeting MAVS. Cell 2019, 178, 176–189. [Google Scholar] [CrossRef]
- Key, J.; Maletzko, A.; Kohli, A.; Gispert, S.; Torres-Odio, S.; Wittig, I.; Heidler, J.; Bárcena, C.; López-Otín, C.; Lei, Y.; et al. Loss of mitochondrial peptidase ClpP triggers transcriptional induction of Rnf213, a susceptibility factor for Moyamoya disease. in submission.
- Gibellini, L.; Pinti, M.; Bartolomeo, R.; De Biasi, S.; Cormio, A.; Musicco, C.; Carnevale, G.; Pecorini, S.; Nasi, M.; De Pol, A.; et al. Inhibition of Lon protease by triterpenoids alters mitochondria and is associated to cell death in human cancer cells. Oncotarget 2015, 6, 25466–25483. [Google Scholar] [CrossRef]
- Li, P.A.; Hou, X.; Hao, S. Mitochondrial biogenesis in neurodegeneration. J. Neurosci. Res. 2017, 95, 2025–2029. [Google Scholar] [CrossRef] [PubMed]
- Villa, E.; Marchetti, S.; Ricci, J.E. No Parkin Zone: Mitophagy without Parkin. Trends Cell Biol. 2018, 28, 882–895. [Google Scholar] [CrossRef] [PubMed]
- Escobar-Henriques, M.; Joaquim, M. Mitofusins: Disease Gatekeepers and Hubs in Mitochondrial Quality Control by E3 Ligases. Front. Physiol. 2019, 10, 517. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Li, W.; Ma, X.; Luan, H. Long Noncoding RNA LINC00265 Promotes Glycolysis and Lactate Production of Colorectal Cancer through Regulating of miR-216b-5p/TRIM44 Axis. Digestion 2019, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Wang, J.; Wang, Y.; Zhou, H.; Wu, X.; Tian, Z.; Sun, B. Novel function of Trim44 promotes an antiviral response by stabilizing VISA. J. Immunol. 2013, 190, 3613–3619. [Google Scholar] [CrossRef]
- Hoshino, A.; Okawa, Y.; Ariyoshi, M.; Kaimoto, S.; Uchihashi, M.; Fukai, K.; Iwai-Kanai, E.; Matoba, S. Oxidative post-translational modifications develop LONP1 dysfunction in pressure overload heart failure. Circ. Heart Fail. 2014, 7, 500–509. [Google Scholar] [CrossRef] [PubMed]
- Leiser, S.F.; Miller, H.; Rossner, R.; Fletcher, M.; Leonard, A.; Primitivo, M.; Rintala, N.; Ramos, F.J.; Miller, D.L.; Kaeberlein, M. Cell nonautonomous activation of flavin-containing monooxygenase promotes longevity and health span. Science 2015, 350, 1375–1378. [Google Scholar] [CrossRef]
- Wirth, C.; Brandt, U.; Hunte, C.; Zickermann, V. Structure and function of mitochondrial complex I. Biochim. Biophys. Acta 2016, 1857, 902–914. [Google Scholar] [CrossRef]
- Bross, P.; Fernandez-Guerra, P. Disease-Associated Mutations in the HSPD1 Gene Encoding the Large Subunit of the Mitochondrial HSP60/HSP10 Chaperonin Complex. Front. Mol. Biosci. 2016, 3, 49. [Google Scholar] [CrossRef]
- Greene, A.W.; Grenier, K.; Aguileta, M.A.; Muise, S.; Farazifard, R.; Haque, M.E.; McBride, H.M.; Park, D.S.; Fon, E.A. Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep. 2012, 13, 378–385. [Google Scholar] [CrossRef]
- Vogtle, F.N.; Brandl, B.; Larson, A.; Pendziwiat, M.; Friederich, M.W.; White, S.M.; Basinger, A.; Kucukkose, C.; Muhle, H.; Jahn, J.A.; et al. Mutations in PMPCB Encoding the Catalytic Subunit of the Mitochondrial Presequence Protease Cause Neurodegeneration in Early Childhood. Am. J. Hum. Genet. 2018, 102, 557–573. [Google Scholar] [CrossRef] [PubMed]
- Besse, A.; Wu, P.; Bruni, F.; Donti, T.; Graham, B.H.; Craigen, W.J.; McFarland, R.; Moretti, P.; Lalani, S.; Scott, K.L.; et al. The GABA transaminase, ABAT, is essential for mitochondrial nucleoside metabolism. Cell Metab. 2015, 21, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Richard, H.T.; Foster, J.W. Acid resistance in Escherichia coli. Adv. Appl. Microbiol. 2003, 52, 167–186. [Google Scholar] [PubMed]
- Nilsson, G.E.; Lutz, P.L. Anoxia tolerant brains. J. Cereb. Blood Flow Metab. 2004, 24, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Wagener, N.; Neupert, W. Bcs1, a AAA protein of the mitochondria with a role in the biogenesis of the respiratory chain. J. Struct. Biol. 2012, 179, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Hornig-Do, H.T.; Tatsuta, T.; Buckermann, A.; Bust, M.; Kollberg, G.; Rotig, A.; Hellmich, M.; Nijtmans, L.; Wiesner, R.J. Nonsense mutations in the COX1 subunit impair the stability of respiratory chain complexes rather than their assembly. EMBO J. 2012, 31, 1293–1307. [Google Scholar] [CrossRef] [PubMed]
- Konig, T.; Troder, S.E.; Bakka, K.; Korwitz, A.; Richter-Dennerlein, R.; Lampe, P.A.; Patron, M.; Muhlmeister, M.; Guerrero-Castillo, S.; Brandt, U.; et al. The m-AAA Protease Associated with Neurodegeneration Limits MCU Activity in Mitochondria. Mol. Cell 2016, 64, 148–162. [Google Scholar] [CrossRef]
- Dhir, A.; Dhir, S.; Borowski, L.S.; Jimenez, L.; Teitell, M.; Rotig, A.; Crow, Y.J.; Rice, G.I.; Duffy, D.; Tamby, C.; et al. Mitochondrial double-stranded RNA triggers antiviral signalling in humans. Nature 2018, 560, 238–242. [Google Scholar] [CrossRef]
- West, A.P.; Khoury-Hanold, W.; Staron, M.; Tal, M.C.; Pineda, C.M.; Lang, S.M.; Bestwick, M.; Duguay, B.A.; Raimundo, N.; MacDuff, D.A.; et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature 2015, 520, 553–557. [Google Scholar] [CrossRef]
- Uchiumi, T.; Ohgaki, K.; Yagi, M.; Aoki, Y.; Sakai, A.; Matsumoto, S.; Kang, D. ERAL1 is associated with mitochondrial ribosome and elimination of ERAL1 leads to mitochondrial dysfunction and growth retardation. Nucleic Acids Res. 2010, 38, 5554–5568. [Google Scholar] [CrossRef]
- Herias, V.; Biessen, E.A.; Beckers, C.; Delsing, D.; Liao, M.; Daemen, M.J.; Pham, C.C.; Heeneman, S. Leukocyte cathepsin C deficiency attenuates atherosclerotic lesion progression by selective tuning of innate and adaptive immune responses. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Campden, R.I.; Zhang, Y. The role of lysosomal cysteine cathepsins in NLRP3 inflammasome activation. Arch. Biochem. Biophys. 2019, 670, 32–42. [Google Scholar] [CrossRef]
- Pareek, G.; Pallanck, L.J. Inactivation of Lon protease reveals a link between mitochondrial unfolded protein stress and mitochondrial translation inhibition. Cell Death Dis. 2018, 9, 1168. [Google Scholar] [CrossRef] [PubMed]
- Bezawork-Geleta, A.; Brodie, E.J.; Dougan, D.A.; Truscott, K.N. LON is the master protease that protects against protein aggregation in human mitochondria through direct degradation of misfolded proteins. Sci. Rep. 2015, 5, 17397. [Google Scholar] [CrossRef]
- Hum, D.W.; Bell, A.W.; Rozen, R.; MacKenzie, R.E. Primary structure of a human trifunctional enzyme. Isolation of a cDNA encoding methylenetetrahydrofolate dehydrogenase-methenyltetrahydrofolate cyclohydrolase-formyltetrahydrofolate synthetase. J. Biol. Chem. 1988, 263, 15946–15950. [Google Scholar]
- Loffler, M.; Jockel, J.; Schuster, G.; Becker, C. Dihydroorotat-ubiquinone oxidoreductase links mitochondria in the biosynthesis of pyrimidine nucleotides. Mol. Cell. Biochem. 1997, 174, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Rohrdanz, E.; Schmuck, G.; Ohler, S.; Kahl, R. The influence of oxidative stress on catalase and MnSOD gene transcription in astrocytes. Brain Res. 2001, 900, 128–136. [Google Scholar] [CrossRef]
- Wang, Y.; Branicky, R.; Noe, A.; Hekimi, S. Superoxide dismutases: Dual roles in controlling ROS damage and regulating ROS signaling. J. Cell Biol. 2018, 217, 1915–1928. [Google Scholar] [CrossRef]
- Sies, H. Glutathione and its role in cellular functions. Free Radic. Biol. Med. 1999, 27, 916–921. [Google Scholar] [CrossRef]
- Cole, A.; Wang, Z.; Coyaud, E.; Voisin, V.; Gronda, M.; Jitkova, Y.; Mattson, R.; Hurren, R.; Babovic, S.; Maclean, N.; et al. Inhibition of the Mitochondrial Protease ClpP as a Therapeutic Strategy for Human Acute Myeloid Leukemia. Cancer Cell 2015, 27, 864–876. [Google Scholar] [CrossRef]
- Seo, J.H.; Rivadeneira, D.B.; Caino, M.C.; Chae, Y.C.; Speicher, D.W.; Tang, H.Y.; Vaira, V.; Bosari, S.; Palleschi, A.; Rampini, P.; et al. The Mitochondrial Unfoldase-Peptidase Complex ClpXP Controls Bioenergetics Stress and Metastasis. PLoS Biol. 2016, 14, e1002507. [Google Scholar] [CrossRef]
- Lu, B.; Shangguan, F.; Huang, D.; Gong, S.; Shi, Y.; Song, Z.; Jia, L.; Xu, J.; Yan, C.; Chen, T.; et al. LonP1 Orchestrates UPRmt and UPRER and Mitochondrial Dynamics to Regulate Heart Function. BioRXiv Prepr. Serv. Biol. 2019. [Google Scholar] [CrossRef]
- Venkatesh, S.; Li, M.; Saito, T.; Tong, M.; Rashed, E.; Mareedu, S.; Zhai, P.; Barcena, C.; Lopez-Otin, C.; Yehia, G.; et al. Mitochondrial LonP1 protects cardiomyocytes from ischemia/reperfusion injury in vivo. J. Mol. Cell. Cardiol. 2019, 128, 38–50. [Google Scholar] [CrossRef]
- Stroud, D.A.; Surgenor, E.E.; Formosa, L.E.; Reljic, B.; Frazier, A.E.; Dibley, M.G.; Osellame, L.D.; Stait, T.; Beilharz, T.H.; Thorburn, D.R.; et al. Accessory subunits are integral for assembly and function of human mitochondrial complex I. Nature 2016, 538, 123–126. [Google Scholar] [CrossRef]
- Luce, K.; Osiewacz, H.D. Increasing organismal healthspan by enhancing mitochondrial protein quality control. Nat. Cell Biol. 2009, 11, 852–858. [Google Scholar] [CrossRef]
- Ngo, J.K.; Davies, K.J. Importance of the lon protease in mitochondrial maintenance and the significance of declining lon in aging. Ann. N. Y. Acad. Sci. 2007, 1119, 78–87. [Google Scholar] [CrossRef]
- Yang, W.; Hekimi, S. Two modes of mitochondrial dysfunction lead independently to lifespan extension in Caenorhabditis elegans. Aging Cell 2010, 9, 433–447. [Google Scholar] [CrossRef]
- Altmann, C.; Hardt, S.; Fischer, C.; Heidler, J.; Lim, H.Y.; Haussler, A.; Albuquerque, B.; Zimmer, B.; Moser, C.; Behrends, C.; et al. Progranulin overexpression in sensory neurons attenuates neuropathic pain in mice: Role of autophagy. Neurobiol. Dis. 2016, 96, 294–311. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef]
- Franceschini, A.; Szklarczyk, D.; Frankild, S.; Kuhn, M.; Simonovic, M.; Roth, A.; Lin, J.; Minguez, P.; Bork, P.; von Mering, C.; et al. STRING v9.1: Protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013, 41, D808–D815. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Protein Names | Gene Names | p-Value | q-Value | Fold Change |
|---|---|---|---|---|
| Lon protease homolog, mitochondrial | Lonp1 | 1.920 × 10−4 | 0.011 | −1.921 |
| Dihydroorotate dehydrogenase (quinone), mitochondrial | Dhodh | 0.022 | 0.147 | −1.438 |
| Amine oxidase [flavin-containing] A | Maoa | 0.008 | 0.102 | −1.381 |
| Solute carrier family 25 member 46 | Slc25a46 | 0.044 | 0.355 | −1.209 |
| Mitochondrial-processing peptidase subunit beta | Pmpcb | 0.012 | 0.290 | −1.193 |
| 28S ribosomal protein S2, mitochondrial | Mrps2 | 0.042 | 0.379 | −1.183 |
| 60 kDa heat shock protein, mitochondrial | Hspd1 | 0.004 | 0.251 | −1.178 |
| Voltage-dependent anion-selective channel protein 2 | Vdac2 | 0.046 | 0.485 | −1.123 |
| NADH dehydrogenase [ubiquinone] 1 alpha subcomplex subunit 5 | Ndufa5 | 0.031 | 0.576 | −1.086 |
| Mitochondrial import receptor subunit TOM70 | Tomm70a | 0.020 | 0.578 | −1.081 |
| C-1-tetrahydrofolate synthase, | Mthfd1 | 0.043 | 0.610 | −1.078 |
| Monofunctional C1-tetrahydrofolate synthase, mitochondrial | Mthfd1l | 0.050 | 0.729 | −1.051 |
| 4-aminobutyrate aminotransferase, mitochondrial | Abat | 0.001 | 0.012 | 2.322 |
| Calcium uptake protein 2, mitochondrial | Micu2 | 0.004 | 0.021 | 2.239 |
| Monocarboxylate transporter 1 | Slc16a1 | 0.002 | 0.021 | 1.718 |
| Pyruvate carboxylase;Pyruvate carboxylase, mitochondrial | Pcx;Pc | 0.007 | 0.087 | 1.428 |
| Carnitine O-palmitoyltransferase 2, mitochondrial | Cpt2 | 0.003 | 0.075 | 1.393 |
| Oxidation resistance protein 1 | Oxr1 | 0.005 | 0.092 | 1.360 |
| Mitochondrial chaperone BCS1 | Bcs1l | 0.013 | 0.157 | 1.320 |
| Mitochondrial 2-oxoglutarate/malate carrier protein | Slc25a11 | 0.011 | 0.170 | 1.285 |
| Nucleoside diphosphate-linked moiety X motif 8, mitochondrial | Nudt8 | 0.047 | 0.323 | 1.245 |
| Propionyl-CoA carboxylase beta chain, mitochondrial | Pccb | 0.004 | 0.164 | 1.242 |
| Ferrochelatase;Ferrochelatase, mitochondrial | Fech | 0.005 | 0.184 | 1.230 |
| Probable arginine--tRNA ligase, mitochondrial | Rars2 | 0.023 | 0.295 | 1.217 |
| AFG3-like protein 2 | Afg3l2 | 0.013 | 0.272 | 1.206 |
| NADH dehydrogenase [ubiquinone] 1 alpha subcomplex subunit 10, mitochondrial | Ndufa10 | 0.031 | 0.335 | 1.199 |
| GTPase Era, mitochondrial | Eral1 | 0.002 | 0.216 | 1.185 |
| Peptidyl-tRNA hydrolase 2, mitochondrial | Ptrh2 | 0.036 | 0.376 | 1.177 |
| NADH dehydrogenase [ubiquinone] 1 beta subcomplex subunit 7 | Ndufb7 | 0.018 | 0.380 | 1.149 |
| NADH dehydrogenase [ubiquinone] 1 beta subcomplex subunit 4 | Ndufb4 | 0.025 | 0.456 | 1.120 |
| Calcium-binding mitochondrial carrier protein Aralar1 | Slc25a12 | 0.016 | 0.443 | 1.119 |
| Mitochondrial amidoxime reducing component 2 | Marc2 | 0.019 | 0.452 | 1.118 |
| Isocitrate dehydrogenase [NADP], mitochondrial | Idh2 | 0.049 | 0.519 | 1.109 |
| Phosphoenolpyruvate carboxykinase [GTP], mitochondrial | Pck2 | 0.004 | 0.486 | 1.092 |
| Protein Names | Gene Names | p-Value | q-Value | Fold Change |
|---|---|---|---|---|
| Catalase | Cat | 0.012 | 0.021 | 7.218 |
| Superoxide dismutase [Cu-Zn]; Extracellular superoxide dismutase [Cu-Zn] | Sod3 | 0.004 | 0.015 | 4.672 |
| Aminopeptidase N | Anpep | 0.001 | 0.000 | 3.443 |
| Microsomal glutathione S-transferase 1 | Mgst1 | 0.026 | 0.079 | 2.352 |
| Glutathione S-transferase A4 | Gsta4 | 0.012 | 0.048 | 2.016 |
| Lactoylglutathione lyase | Glo1 | 0.043 | 0.155 | 1.653 |
| Nicotinate phosphoribosyltransferase | Naprt | 0.047 | 0.192 | 1.498 |
| Glutathione S-transferase omega-1 | Gsto1 | 0.001 | 0.047 | 1.415 |
| Glutathione S-transferase Mu 5 | Gstm5 | 0.036 | 0.197 | 1.391 |
| Serine/threonine-protein kinase 24; Serine/threonine-protein kinase 24 35 kDa subunit; Serine/threonine-protein kinase 24 12 kDa subunit | Stk24 | 0.005 | 0.167 | 1.246 |
| Isocitrate dehydrogenase [NADP]; Isocitrate dehydrogenase [NADP] cytoplasmic | Idh1 | 0.015 | 0.252 | 1.227 |
| Maleylacetoacetate isomerase | Gstz1 | 0.012 | 0.295 | 1.190 |
| Glutathione S-transferase Mu 2 | Gstm2 | 0.004 | 0.294 | 1.160 |
| Isocitrate dehydrogenase [NADP], mitochondrial | Idh2 | 0.049 | 0.519 | 1.109 |
| Protein deglycase DJ-1 | Park7;Dj1 | 0.021 | 0.509 | 1.098 |
| Glutathione synthetase | Gss | 0.015 | 0.229 | −1.248 |
| Spermine synthase | Sms | 0.014 | 0.187 | −1.286 |
| Egl nine homolog 1 | Egln1 | 0.029 | 0.297 | −1.228 |
| Catalase | Cat | 0.022 | 0.318 | −1.197 |
| Protein Names | Gene Names | p-Value | q-Value | Fold Change |
|---|---|---|---|---|
| Transmembrane glycoprotein NMB | Gpnmb | 0.008 | 0.021 | 4.168 |
| Interleukin-1 receptor antagonist protein | Il1rn | 7.280 × 10−7 | 0.000 | 3.323 |
| Osteopontin | Spp1 | 0.045 | 0.108 | 2.422 |
| Lymphocyte antigen 6A-2/6E-1 | Ly6a | 0.008 | 0.033 | 2.235 |
| Atypical chemokine receptor 3 | Ackr3 | 0.002 | 0.021 | 1.919 |
| Coxsackievirus and adenovirus receptor homolog | Cxadr | 0.006 | 0.035 | 1.913 |
| Interferon-activable protein 204 | Ifi204 | 0.025 | 0.091 | 1.856 |
| H-2 class I histocompatibility antigen, D-B alpha chain | H2-D1;H-2D;H2-L | 0.013 | 0.057 | 1.820 |
| H-2 class I histocompatibility antigen, K-K alpha chain; | H2-K1;H2-K;H2-D1 | 0.013 | 0.058 | 1.808 |
| Tetraspanin;CD82 antigen | Cd82 | 4.349 × 10−5 | 0.014 | 1.799 |
| Signal transducer and activator of transcription;Signal transducer and activator of transcription 2 | Stat2 | 0.024 | 0.094 | 1.768 |
| Interferon-induced 35 kDa protein homolog | Ifi35 | 0.037 | 0.125 | 1.767 |
| Interferon-induced helicase C domain-containing protein 1 | Ifih1 | 0.023 | 0.107 | 1.613 |
| Tetraspanin;Tetraspanin-6 | Tspan6 | 0.013 | 0.083 | 1.597 |
| Gamma-interferon-inducible lysosomal thiol reductase | Ifi30 | 0.001 | 0.021 | 1.591 |
| Signal transducer and activator of transcription;Signal transducer and activator of transcription 1 | Stat1 | 0.027 | 0.136 | 1.529 |
| Stimulator of interferon genes protein, STING | Tmem173 | 0.016 | 0.119 | 1.440 |
| Interferon-induced, double-stranded RNA-activated protein kinase, PKR | Eif2ak2 | 0.021 | 0.155 | 1.395 |
| Interferon-activable protein 205-B; Interferon-activable protein 205-A | Ifi205b;Mnda; Ifi205a | 0.007 | 0.113 | 1.347 |
| E3 ubiquitin/ISG15 ligase TRIM25 | Trim25 | 0.011 | 0.138 | 1.345 |
| Lymphocyte-specific protein 1 | Lsp1 | 0.016 | 0.162 | 1.342 |
| Interferon regulatory factor 2-binding protein 2 | Irf2bp2 | 0.007 | 0.114 | 1.341 |
| Interleukin-6 receptor subunit beta | Il6st;il6st | 0.011 | 0.202 | 1.249 |
| Signal transducer and activator of transcription; Signal transducer and activator of transcription 3 | Stat3 | 0.026 | 0.374 | 1.167 |
| Tripartite motif-containing protein 44 | Trim44 | 0.002 | 0.014 | −10.823 |
| CD97 antigen | Adgre5;Cd97 | 0.002 | 0.050 | −1.435 |
| CD44 antigen | Cd44 | 0.012 | 0.105 | −1.431 |
| CD302 antigen | Cd302 | 0.041 | 0.255 | −1.319 |
| CD109 antigen | Cd109 | 0.001 | 0.081 | −1.311 |
| Poliovirus Receptor | Pvr | 0.012 | 0.194 | −1.262 |
| Interferon regulatory factor 2-binding protein-like | Irf2bpl | 0.016 | 0.283 | −1.208 |
| Protein Names | Gene Names | p-Value | q-Value | Fold Change |
|---|---|---|---|---|
| Dipeptidyl peptidase 1; Dipeptidyl peptidase 1 exclusion domain chain; Dipeptidyl peptidase 1 heavy chain; Dipeptidyl peptidase 1 light chain | Ctsc | 0.001 | 0.013 | 2.148 |
| Transmembrane protein 59 | Tmem59 | 0.016 | 0.080 | 1.783 |
| Cathepsin Z | Ctsz | 0.015 | 0.092 | 1.550 |
| Alpha-mannosidase; Lysosomal alpha-mannosidase | Man2b1 | 0.010 | 0.082 | 1.530 |
| Beta-hexosaminidase; Beta-hexosaminidase subunit alpha | Hexa | 0.003 | 0.046 | 1.527 |
| Sulfatase-modifying factor 1 | Sumf1 | 0.033 | 0.157 | 1.508 |
| Alpha-galactosidase A | Gla | 0.012 | 0.094 | 1.484 |
| Beta-glucuronidase | Gusb | 0.003 | 0.053 | 1.470 |
| Type 1 phosphatidylinositol 4,5-bisphosphate 4-phosphatase | Tmem55b | 0.005 | 0.079 | 1.430 |
| Cathepsin D | Ctsd | 3.585 × 10−4 | 0.033 | 1.415 |
| Prosaposin | Psap | 0.021 | 0.149 | 1.413 |
| Lysosomal thioesterase PPT2 | Ppt2 | 0.046 | 0.228 | 1.399 |
| Putative phospholipase B-like 2; Putative phospholipase B-like 2 28 kDa form; Putative phospholipase B-like 2 40 kDa form; Putative phospholipase B-like 2 15 kDa form | Plbd2 | 0.046 | 0.230 | 1.397 |
| Carboxypeptidase; Lysosomal protective protein; Lysosomal protective protein 32 kDa chain; Lysosomal protective protein 20 kDa chain | Ctsa | 0.048 | 0.233 | 1.393 |
| Granulins; Acrogranin; Granulin-1; Granulin-2; Granulin-3; Granulin-4; Granulin-5; Granulin-6; Granulin-7 | Grn | 0.017 | 0.144 | 1.388 |
| N-Acetyl-Alpha-Glucosaminidase | Naglu | 0.007 | 0.093 | 1.387 |
| Lysosomal alpha-glucosidase | Gaa | 0.007 | 0.099 | 1.385 |
| Arylsulfatase B | Arsb | 0.024 | 0.170 | 1.376 |
| WD repeat-containing protein 59 | Wdr59 | 0.011 | 0.120 | 1.375 |
| Beta-hexosaminidase; Beta-hexosaminidase subunit beta | Hexb | 0.015 | 0.148 | 1.366 |
| Glucosylceramidase | Gba | 0.006 | 0.097 | 1.360 |
| Gamma-glutamyl hydrolase | Ggh | 0.011 | 0.133 | 1.355 |
| Beta-galactosidase | Glb1 | 0.004 | 0.093 | 1.343 |
| Cation-independent mannose-6-phosphate receptor | Igf2r | 0.002 | 0.087 | 1.326 |
| Ganglioside GM2 activator | Gm2a | 4.257× 10−4 | 0.053 | 1.318 |
| Lysosomal Pro-X carboxypeptidase | Prcp | 0.031 | 0.286 | 1.249 |
| Transmembrane protein 106B | Tmem106b | 0.029 | 0.281 | 1.248 |
| Dipeptidyl peptidase 2 | Dpp7 | 0.022 | 0.268 | 1.238 |
| Beta-mannosidase | Manba | 0.022 | 0.274 | 1.232 |
| N(4)-(beta-N-acetylglucosaminyl)-L-asparaginase; Glycosylasparaginase alpha chain; Glycosylasparaginase beta chain | Aga | 0.012 | 0.323 | 1.171 |
| V-type proton ATPase subunit H | Atp6v1h | 0.038 | 0.394 | 1.167 |
| Ragulator complex protein LAMTOR1 | Lamtor1 | 0.040 | 0.443 | 1.140 |
| AP-1 complex subunit beta-1; AP complex subunit beta | Ap1b1 | 0.018 | 0.411 | 1.134 |
| Pro-cathepsin H; Cathepsin H mini chain; Cathepsin H; Cathepsin H heavy chain; Cathepsin H light chain | Ctsh | 8.684× 10−6 | 0.000 | −1.734 |
| Sequestosome-1 | Sqstm1 | 0.009 | 0.212 | −1.232 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Key, J.; Kohli, A.; Bárcena, C.; López-Otín, C.; Heidler, J.; Wittig, I.; Auburger, G. Global Proteome of LonP1+/− Mouse Embryonal Fibroblasts Reveals Impact on Respiratory Chain, but No Interdependence between Eral1 and Mitoribosomes. Int. J. Mol. Sci. 2019, 20, 4523. https://doi.org/10.3390/ijms20184523
Key J, Kohli A, Bárcena C, López-Otín C, Heidler J, Wittig I, Auburger G. Global Proteome of LonP1+/− Mouse Embryonal Fibroblasts Reveals Impact on Respiratory Chain, but No Interdependence between Eral1 and Mitoribosomes. International Journal of Molecular Sciences. 2019; 20(18):4523. https://doi.org/10.3390/ijms20184523
Chicago/Turabian StyleKey, Jana, Aneesha Kohli, Clea Bárcena, Carlos López-Otín, Juliana Heidler, Ilka Wittig, and Georg Auburger. 2019. "Global Proteome of LonP1+/− Mouse Embryonal Fibroblasts Reveals Impact on Respiratory Chain, but No Interdependence between Eral1 and Mitoribosomes" International Journal of Molecular Sciences 20, no. 18: 4523. https://doi.org/10.3390/ijms20184523
APA StyleKey, J., Kohli, A., Bárcena, C., López-Otín, C., Heidler, J., Wittig, I., & Auburger, G. (2019). Global Proteome of LonP1+/− Mouse Embryonal Fibroblasts Reveals Impact on Respiratory Chain, but No Interdependence between Eral1 and Mitoribosomes. International Journal of Molecular Sciences, 20(18), 4523. https://doi.org/10.3390/ijms20184523