Tumor Dormancy and Interplay with Hypoxic Tumor Microenvironment



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Tumor Microenvironment
2.1. Hypoxia in Tumor Microenvironment
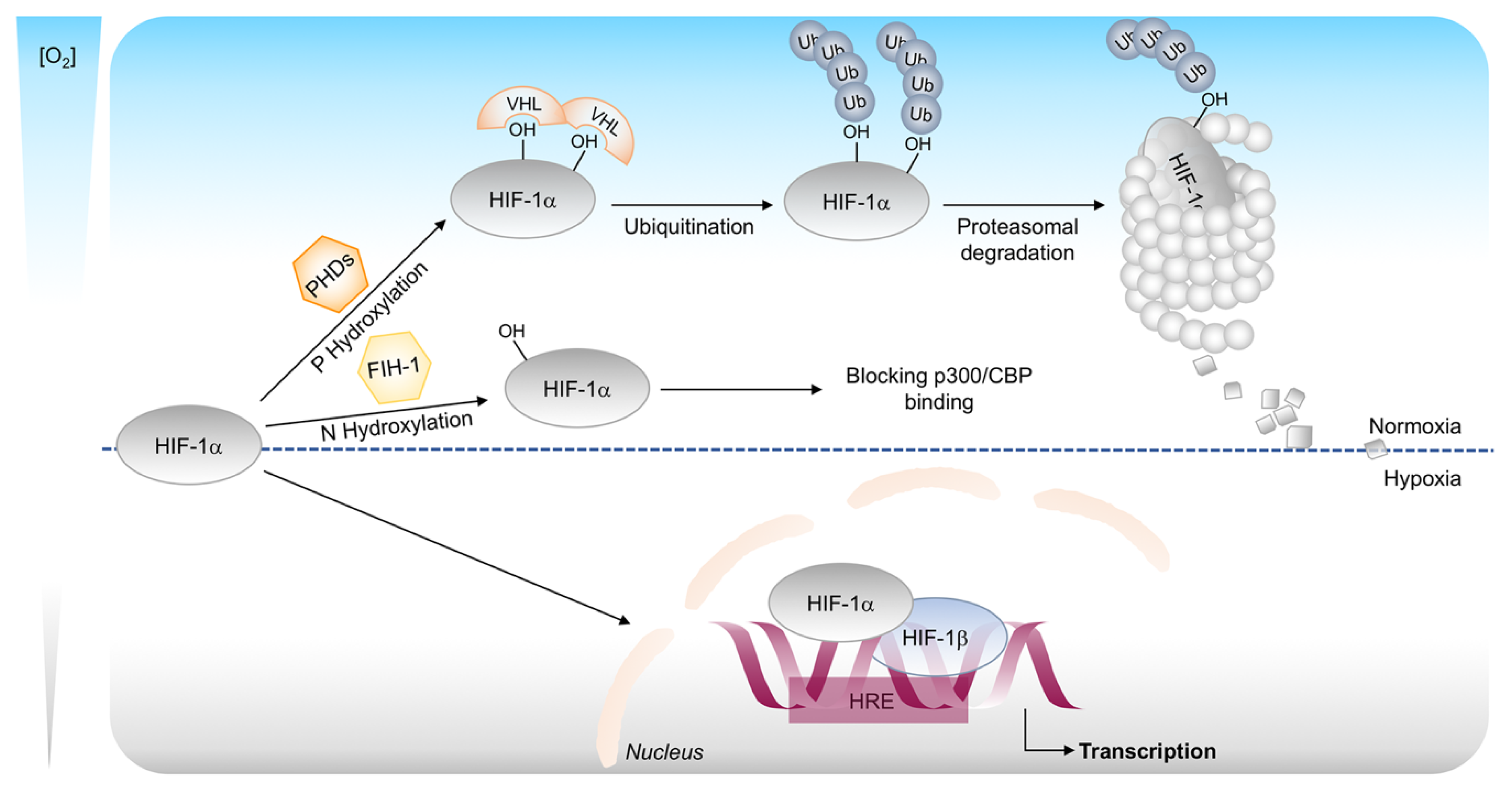
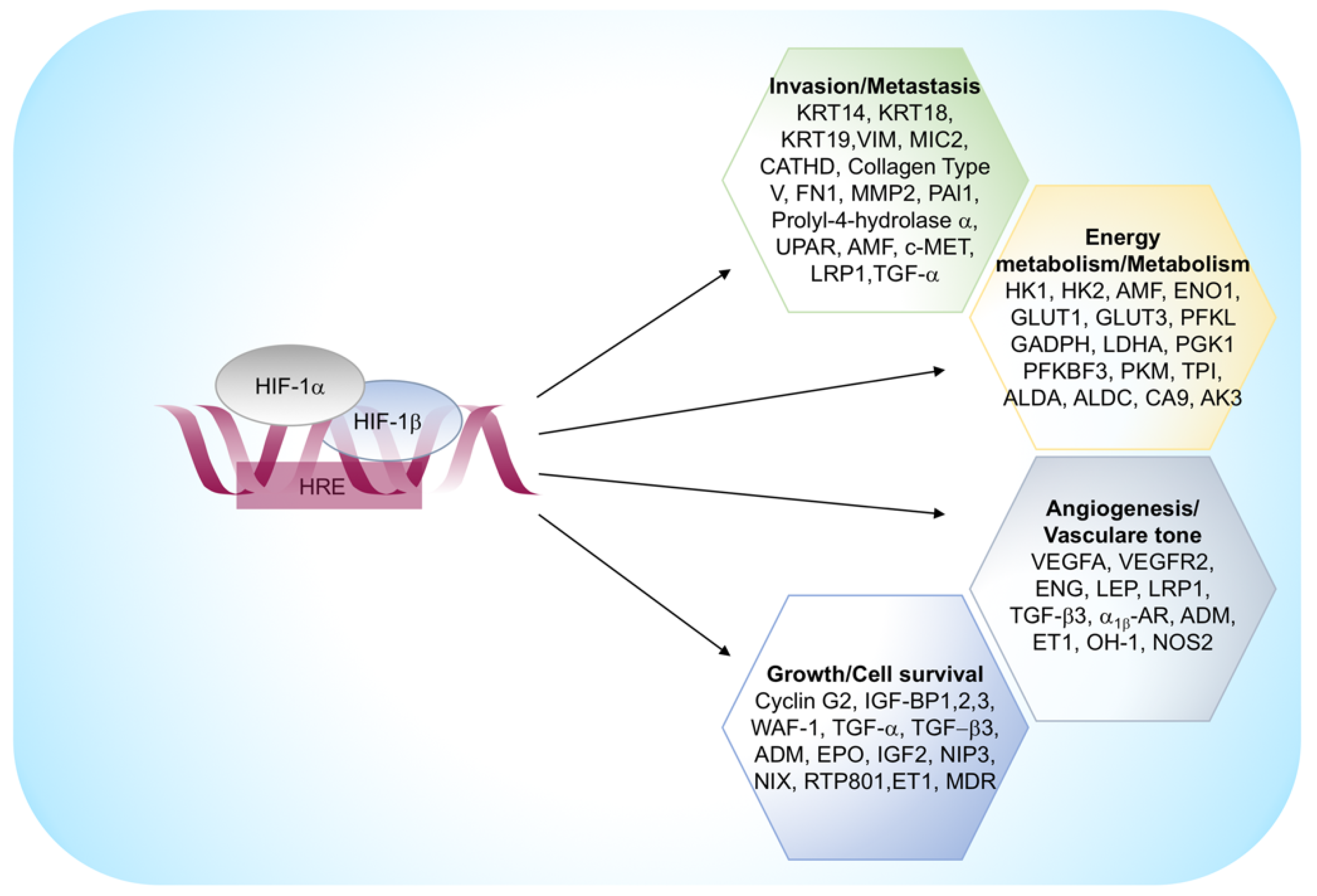
2.2. Hypoxia Inducible Factors in Tumor Microenvironment
2.3. ROS Production in Tumor Microenvironment
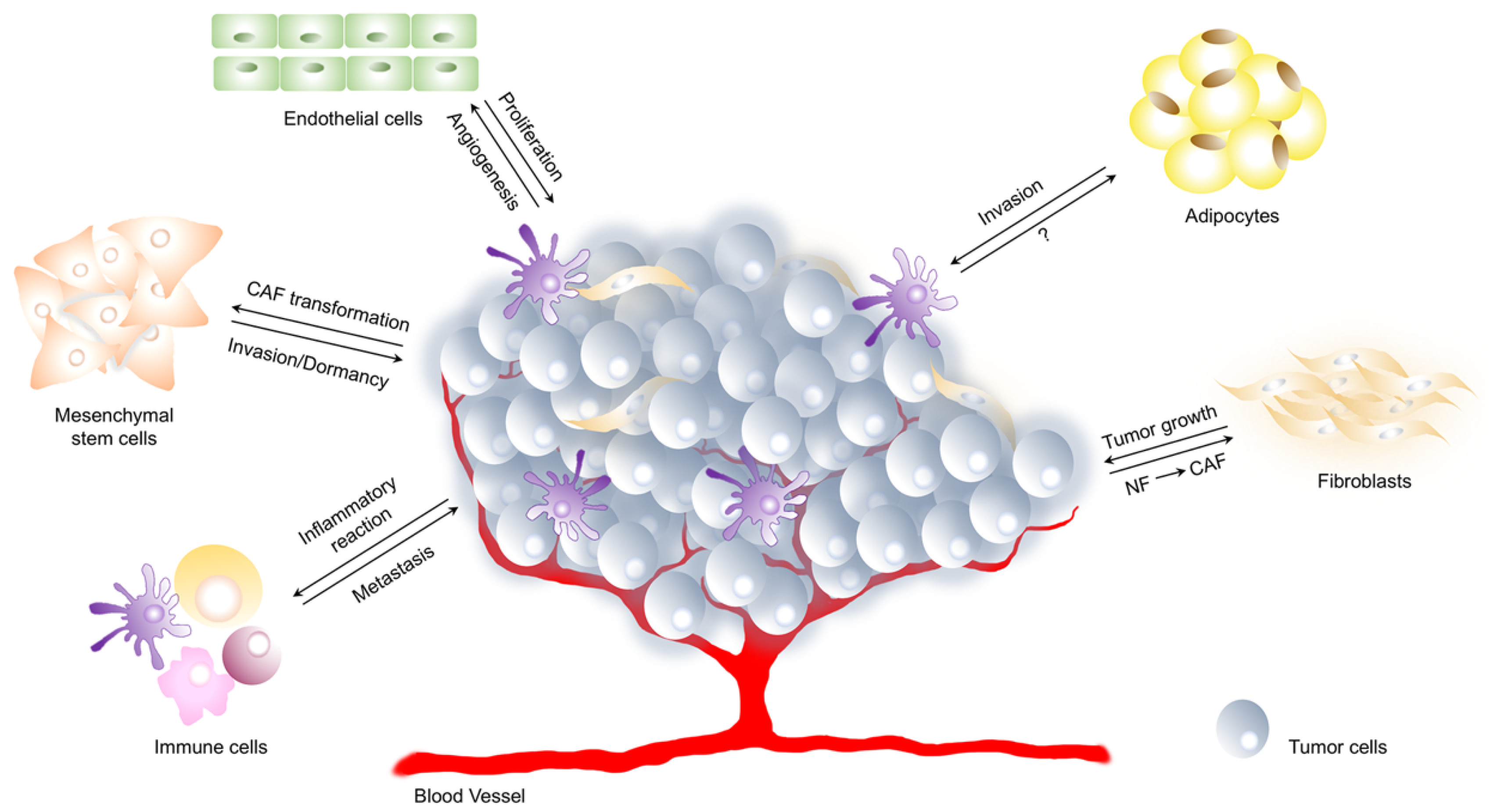
3. Tumor Dormancy and the Interplay with the Tumor Microenvironment
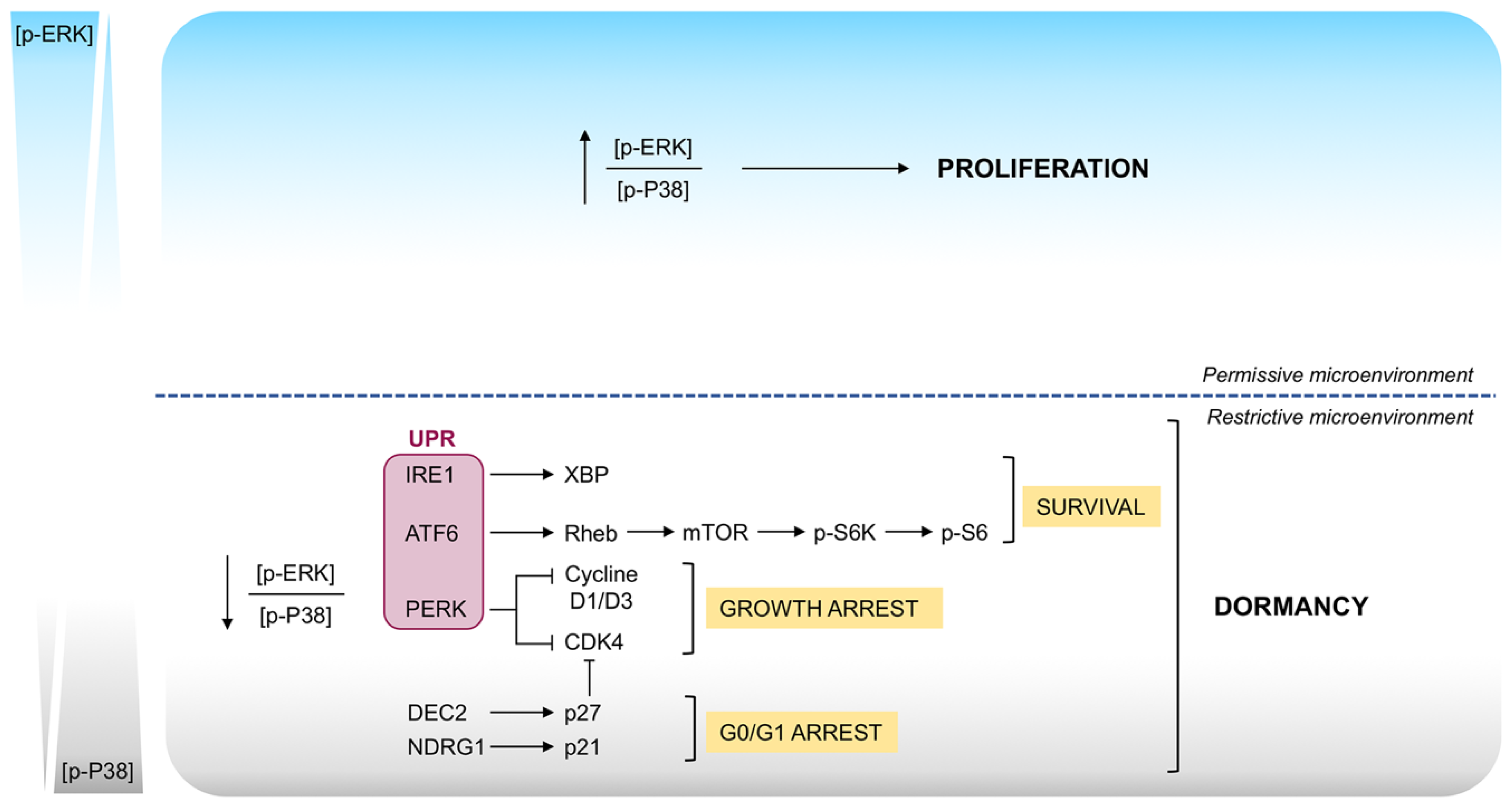
Signaling Mechanisms in Tumor Dormancy
4. Cancer Stem Cells in Tumor Microenvironment
4.1. Cancer Stem Cells Markers
4.2. Tumor Dormancy and Cancer Stem Cells
5. Autophagy in Tumor Microenvironment
Autophagy Sustains Survival of Dormant Cancer Cells
6. Models for the Study of Dormancy
7. Prospective
Author Contributions
Funding
Conflicts of Interest
References
- Klemm, F.; Joyce, J.A. Microenvironmental regulation of therapeutic response in cancer. Trends Cell Biol. 2015, 25, 198–213. [Google Scholar] [CrossRef] [PubMed]
- Pattabiraman, D.R.; Weinberg, R.A. Tackling the cancer stem cells-what challenges do they pose? Nat. Rev. Drug Discov. 2014, 13, 497–512. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Knolhoff, B.L.; Meyer, M.A.; Nywening, T.M.; West, B.L.; Luo, J.; Wang-Gillam, A.; Goedegebuure, S.P.; Linehan, D.C.; DeNardo, D.G. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014, 74, 5057–5069. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Takai, K.; Weaver, V.M.; Werb, Z. Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb. Perspect. Biol. 2011, 3. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, J.; Bae, J.S. ROS homeostasis and metabolism: A critical liaison for cancer therapy. Exp. Mol. Med. 2016, 48, e269. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O.; Franz, W.; Negelein, E. The metabolism of the tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Damaghi, M.; Wojtkowiak, J.W.; Gillies, R.J. pH sensing and regulation in cancer. Front. Physiol. 2013, 4, 370. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Jain, R.K. Angiogenesis in cancer and other diseases. Nature 2000, 407, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Gee, M.S.; Procopio, W.N.; Makonnen, S.; Feldman, M.D.; Yeilding, N.M.; Lee, W.M.F. Tumor Vessel Development and Maturation Impose Limits on the Effectiveness of Anti-Vascular Therapy. Am. J. Pathol. 2003, 162. [Google Scholar] [CrossRef]
- Walsh, J.C.; Lebedev, A.; Aten, E.; Madsen, K.; Marciano, L.; Kolb, H.C. The clinical importance of assessing tumor hypoxia: Relationship of tumor hypoxia to prognosis and therapeutic opportunities. Antioxid. Redox Signal. 2014, 21, 1516–1554. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Wang, Y. Hypoxia Mediates Tumor Malignancy and Therapy Resistance. Adv. Exp. Med. Biol. 2019, 1136, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Eales, K.L.; Hollinshead, K.E.; Tennant, D.A. Hypoxia and metabolic adaptation of cancer cells. Oncogenesis 2016, 5, e190. [Google Scholar] [CrossRef] [PubMed]
- Bayer, C.; Vaupel, P. Acute versus chronic hypoxia in tumors: Controversial data concerning time frames and biological consequences. Strahlenther. Und Onkol. Organ. Der Dtsch. Rontgengesellschaft... [Et Al] 2012, 188, 616–627. [Google Scholar] [CrossRef] [PubMed]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouyssegur, J.; Mazure, N.M. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef] [PubMed]
- Rofstad, E.K.; Gaustad, J.V.; Egeland, T.A.; Mathiesen, B.; Galappathi, K. Tumors exposed to acute cyclic hypoxic stress show enhanced angiogenesis, perfusion and metastatic dissemination. Int. J. Cancer 2010, 127, 1535–1546. [Google Scholar] [CrossRef] [PubMed]
- Michiels, C.; Tellier, C.; Feron, O. Cycling hypoxia: A key feature of the tumor microenvironment. Biochim. Biophys. Acta 2016, 1866, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Martinive, P.; Defresne, F.; Bouzin, C.; Saliez, J.; Lair, F.; Grégoire, V.; Michiels, C.; Dessy, C.; Feron, O. Preconditioning of the Tumor Vasculature and Tumor Cells by Intermittent Hypoxia: Implications for Anticancer Therapies. Cancer Res. 2006, 66, 11736. [Google Scholar] [CrossRef]
- Hsieh, C.-H. Cycling hypoxia increases U87 glioma cell radioresistance via ROS induced higher and long-term HIF-1 signal transduction activity. Oncol. Rep. 2010, 24. [Google Scholar] [CrossRef]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G., Jr. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 2001, 292, 464–468. [Google Scholar] [CrossRef]
- Jaakkola, P.; Mole, D.R.; Tian, Y.M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef]
- Masson, N.; Willam, C.; Maxwell, P.H.; Pugh, C.W.; Ratcliffe, P.J. Independent function of two destruction domains in hypoxia-inducible factor-alpha chains activated by prolyl hydroxylation. Embo J. 2001, 20, 5197–5206. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.J.; Sataur, A.; Wang, L.; Chen, H.; Simon, M.C. The N-terminal transactivation domain confers target gene specificity of hypoxia-inducible factors HIF-1alpha and HIF-2alpha. Mol. Biol. Cell 2007, 18, 4528–4542. [Google Scholar] [CrossRef] [PubMed]
- Heikkila, M.; Pasanen, A.; Kivirikko, K.I.; Myllyharju, J. Roles of the human hypoxia-inducible factor (HIF)-3alpha variants in the hypoxia response. Cell. Mol. Life Sci. CMLS 2011, 68, 3885–3901. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Zhang, Y.; Wang, Z.; Dong, H.; Fu, N.; Han, X. The roles and signaling pathways of prolyl-4-hydroxylase 2 in the tumor microenvironment. Chem. -Biol. Interact. 2019, 303, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Mahon, P.C.; Hirota, K.; Semenza, G.L. FIH-1: A novel protein that interacts with HIF-1alpha and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev. 2001, 15, 2675–2686. [Google Scholar] [CrossRef] [PubMed]
- Lando, D.; Peet, D.J.; Whelan, D.A.; Gorman, J.J.; Whitelaw, M.L. Asparagine hydroxylation of the HIF transactivation domain a hypoxic switch. Science 2002, 295, 858–861. [Google Scholar] [CrossRef]
- Kenneth, N.S.; Rocha, S. Regulation of gene expression by hypoxia. Biochem. J. 2008, 414, 19. [Google Scholar] [CrossRef] [PubMed]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef]
- Ginouves, A.; Ilc, K.; Macias, N.; Pouyssegur, J.; Berra, E. PHDs overactivation during chronic hypoxia “desensitizes” HIFalpha and protects cells from necrosis. Proc. Natl. Acad. Sci. USA 2008, 105, 4745–4750. [Google Scholar] [CrossRef]
- Dewhirst, M.W.; Cao, Y.; Moeller, B. Cycling hypoxia and free radicals regulate angiogenesis and radiotherapy response. Nat. Rev. Cancer 2008, 8, 425–437. [Google Scholar] [CrossRef]
- Pouyssegur, J.; Dayan, F.; Mazure, N.M. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature 2006, 441, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Tong, L.; Chuang, C.C.; Wu, S.; Zuo, L. Reactive oxygen species in redox cancer therapy. Cancer Lett. 2015, 367, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Fiaschi, T.; Chiarugi, P. Oxidative stress, tumor microenvironment, and metabolic reprogramming: A diabolic liaison. Int. J. Cell Biol. 2012, 2012, 762825. [Google Scholar] [CrossRef] [PubMed]
- Bablor, M.B. NADPH Oxidase: An Update. Blood 1998, 93, 1464–1476. [Google Scholar]
- Burdon, R.H.; Gill, V.; Rice-Evans, C. Oxidative stress and tumour cell proliferation. Free Radic. Res. Commun. 1990, 11, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Cerutti, P.A. Prooxidant states and tumor promotion. Science 1985, 227, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Deng, J.; Rychahou, P.G.; Qiu, S.; Evers, B.M.; Zhou, B.P. Stabilization of snail by NF-kappaB is required for inflammation-induced cell migration and invasion. Cancer Cell 2009, 15, 416–428. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.H.; Wu, M.Z.; Chiou, S.H.; Chen, P.M.; Chang, S.Y.; Liu, C.J.; Teng, S.C.; Wu, K.J. Direct regulation of TWIST by HIF-1alpha promotes metastasis. Nat. Cell Biol. 2008, 10, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.H.; Wu, K.J. TWIST activation by hypoxia inducible factor-1 (HIF-1): Implications in metastasis and development. Cell Cycle (Georget. Tex.) 2008, 7, 2090–2096. [Google Scholar] [CrossRef]
- Butturini, E.; Carcereri de Prati, A.; Chiavegato, G.; Rigo, A.; Cavalieri, E.; Darra, E.; Mariotto, S. Mild oxidative stress induces S-glutathionylation of STAT3 and enhances chemosensitivity of tumoural cells to chemotherapeutic drugs. Free Radic. Biol. Med. 2013, 65, 1322–1330. [Google Scholar] [CrossRef] [PubMed]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Gomis, R.R.; Gawrzak, S. Tumor cell dormancy. Mol. Oncol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.S.; Avivar-Valderas, A.; Estrada, Y.; Bragado, P.; Sosa, M.S.; Aguirre-Ghiso, J.A.; Segall, J.E. Dormancy signatures and metastasis in estrogen receptor positive and negative breast cancer. PLoS ONE 2012, 7, e35569. [Google Scholar] [CrossRef] [PubMed]
- Tiram, G.; Ferber, S.; Ofek, P.; Eldar-Boock, A.; Ben-Shushan, D.; Yeini, E.; Krivitsky, A.; Blatt, R.; Almog, N.; Henkin, J.; et al. Reverting the molecular fingerprint of tumor dormancy as a therapeutic strategy for glioblastoma. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Naumov, G.N.; Akslen, L.A.; Folkman, J. Role of angiogenesis in human tumor dormancy: Animal models of the angiogenic switch. Cell Cycle 2006, 5, 1779–1787. [Google Scholar] [CrossRef]
- Indraccolo, S.; Favaro, E.; Amadori, A. Dormant tumors awaken by a short-term angiogenic burst: The spike hypothesis. Cell Cycle 2006, 5, 1751–1755. [Google Scholar] [CrossRef]
- Dou, J.; Gu, N. Emerging strategies for the identification and targeting of cancer stem cells. Tumour Biol. J. Int. Soc. Oncodevelopmental Biol. Med. 2010, 31, 243–253. [Google Scholar] [CrossRef]
- Msaki, A.; Pasto, A.; Curtarello, M.; Arigoni, M.; Barutello, G.; Calogero, R.A.; Macagno, M.; Cavallo, F.; Amadori, A.; Indraccolo, S. A hypoxic signature marks tumors formed by disseminated tumor cells in the BALB-neuT mammary cancer model. Oncotarget 2016, 7, 33081–33095. [Google Scholar] [CrossRef]
- Hofstetter, C.P.; Burkhardt, J.K.; Shin, B.J.; Gursel, D.B.; Mubita, L.; Gorrepati, R.; Brennan, C.; Holland, E.C.; Boockvar, J.A. Protein phosphatase 2A mediates dormancy of glioblastoma multiforme-derived tumor stem-like cells during hypoxia. PLoS ONE 2012, 7, e30059. [Google Scholar] [CrossRef]
- Ratcliffe, P.J. Oxygen sensing and hypoxia signalling pathways in animals: The implications of physiology for cancer. J. Physiol. 2013, 591, 2027–2042. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Oxygen sensing, hypoxia-inducible factors, and disease pathophysiology. Annu. Rev. Pathol. 2014, 9, 47–71. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Harris, A.L. Hypoxia--a key regulatory factor in tumour growth. Nat. Rev. Cancer 2002, 2, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Heredia, J.M.; Felipe-Abrio, B.; Cano, D.A.; Carnero, A. Genetic modification of hypoxia signaling in animal models and its effect on cancer. Clin. Transl. Oncol.: Off. Publ. Fed. Span. Oncol. Soc. Natl. Cancer Inst. Mex. 2015, 17, 90–102. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Engl, W.; Viasnoff, V.; Dufour, S. Biochemical and biophysical origins of cadherin selectivity and adhesion strength. Curr. Opin. Cell Biol. 2012, 24, 614–619. [Google Scholar] [CrossRef] [PubMed]
- Creighton, C.J.; Chang, J.C.; Rosen, J.M. Epithelial-mesenchymal transition (EMT) in tumor-initiating cells and its clinical implications in breast cancer. J. Mammary Gland Biol. Neoplasia 2010, 15, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Settleman, J. EMT, cancer stem cells and drug resistance: An emerging axis of evil in the war on cancer. Oncogene 2010, 29, 4741–4751. [Google Scholar] [CrossRef] [PubMed]
- Bao, B.; Ahmad, A.; Li, Y.; Azmi, A.S.; Ali, S.; Banerjee, S.; Kong, D.; Sarkar, F.H. Targeting CSCs within the tumor microenvironment for cancer therapy: A potential role of mesenchymal stem cells. Expert Opin. Ther. Targets 2012, 16, 1041–1054. [Google Scholar] [CrossRef]
- Bao, B.; Azmi, A.S.; Ali, S.; Ahmad, A.; Li, Y.; Banerjee, S.; Kong, D.; Sarkar, F.H. The biological kinship of hypoxia with CSC and EMT and their relationship with deregulated expression of miRNAs and tumor aggressiveness. Biochim. Biophys. Acta 2012, 1826, 272–296. [Google Scholar] [CrossRef]
- Krishnamachary, B.; Zagzag, D.; Nagasawa, H.; Rainey, K.; Okuyama, H.; Baek, J.H.; Semenza, G.L. Hypoxia-inducible factor-1-dependent repression of E-cadherin in von Hippel-Lindau tumor suppressor-null renal cell carcinoma mediated by TCF3, ZFHX1A, and ZFHX1B. Cancer Res. 2006, 66, 2725–2731. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Bueno, G.; Portillo, F.; Cano, A. Transcriptional regulation of cell polarity in EMT and cancer. Oncogene 2008, 27, 6958–6969. [Google Scholar] [CrossRef] [PubMed]
- Noman, M.Z.; Messai, Y.; Muret, J.; Hasmim, M.; Chouaib, S. Crosstalk between CTC, Immune System and Hypoxic Tumor Microenvironment. Cancer Microenviron. Off. J. Int. Cancer Microenviron. Soc. 2014, 7, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Tinhofer, I.; Saki, M.; Niehr, F.; Keilholz, U.; Budach, V. Cancer stem cell characteristics of circulating tumor cells. Int. J. Radiat. Biol. 2014, 90, 622–627. [Google Scholar] [CrossRef] [PubMed]
- Keith, B.; Simon, M.C. Hypoxia-inducible factors, stem cells, and cancer. Cell 2007, 129, 465–472. [Google Scholar] [CrossRef]
- Bennewith, K.L.; Durand, R.E. Quantifying transient hypoxia in human tumor xenografts by flow cytometry. Cancer Res. 2004, 64, 6183–6189. [Google Scholar] [CrossRef] [PubMed]
- Brurberg, K.G.; Thuen, M.; Ruud, E.B.; Rofstad, E.K. Fluctuations in pO2 in irradiated human melanoma xenografts. Radiat. Res. 2006, 165, 16–25. [Google Scholar] [CrossRef]
- Sosa, M.S.; Avivar-Valderas, A.; Bragado, P.; Wen, H.C.; Aguirre-Ghiso, J.A. ERK1/2 and p38alpha/beta signaling in tumor cell quiescence: Opportunities to control dormant residual disease. Clin. Cancer Res: Off. J. Am. Assoc. Cancer Res. 2011, 17, 5850–5857. [Google Scholar] [CrossRef]
- Sosa, M.S.; Bragado, P.; Aguirre-Ghiso, J.A. Mechanisms of disseminated cancer cell dormancy: An awakening field. Nat. Rev. Cancer 2014, 14, 611–622. [Google Scholar] [CrossRef]
- Aguirre Ghiso, J.A.; Liu, D.; Mignatti, A.; Kovalski, K.; Ossowski, L. Urokinase receptor and fibronectin regulate the ERK(MAPK) to p38(MAPK) activity ratios that determine carcinoma cell proliferation or dormancy in vivo. Mol. Biol. Cell 2001, 12, 863–879. [Google Scholar] [CrossRef]
- Koul, H.K.; Pal, M.; Koul, S. Role of p38 MAP Kinase Signal Transduction in Solid Tumors. Genes Cancer 2013, 4, 342–359. [Google Scholar] [CrossRef] [PubMed]
- Aguirre Ghiso, J.A.; Ossowski, L.; SK, R. Green fluorescent protein tagging of extracellular signal-regulated kinase and p38 pathways reveals novel dynamics of pathway activation during primary and metastastic growth. Cancer Res. 2004, 64, 7335–7345. [Google Scholar] [CrossRef] [PubMed]
- Bragado, P.; Estrada, Y.; Parikh, F.; Krause, S.; Capobianco, C.; Farina, H.G.; Schewe, D.M.; Aguirre-Ghiso, J.A. TGF-beta2 dictates disseminated tumour cell fate in target organs through TGF-beta-RIII and p38alpha/beta signalling. Nat. Cell Biol. 2013, 15, 1351–1361. [Google Scholar] [CrossRef] [PubMed]
- Aguirre-Ghiso, J.A. Models, mechanisms and clinical evidence for cancer dormancy. Nat. Rev. Cancer 2007, 7, 834–846. [Google Scholar] [CrossRef] [PubMed]
- Aguirre Ghiso, J.A. Inhibition of FAK signaling activated by urokinase receptor induces dormancy in human carcinoma cells in vivo. Oncogene 2002, 21, 2513–2524. [Google Scholar] [CrossRef] [PubMed]
- Aguirre-Ghiso, J.A.; Estrada, Y.D.; Ossowsky, L. ERK (MAPK) Activity as a Determinant of Tumor Growth and Dormancy; Regulation by p38 (SAPK). Cancer Res. 2003, 63, 1684–1695. [Google Scholar] [CrossRef] [PubMed]
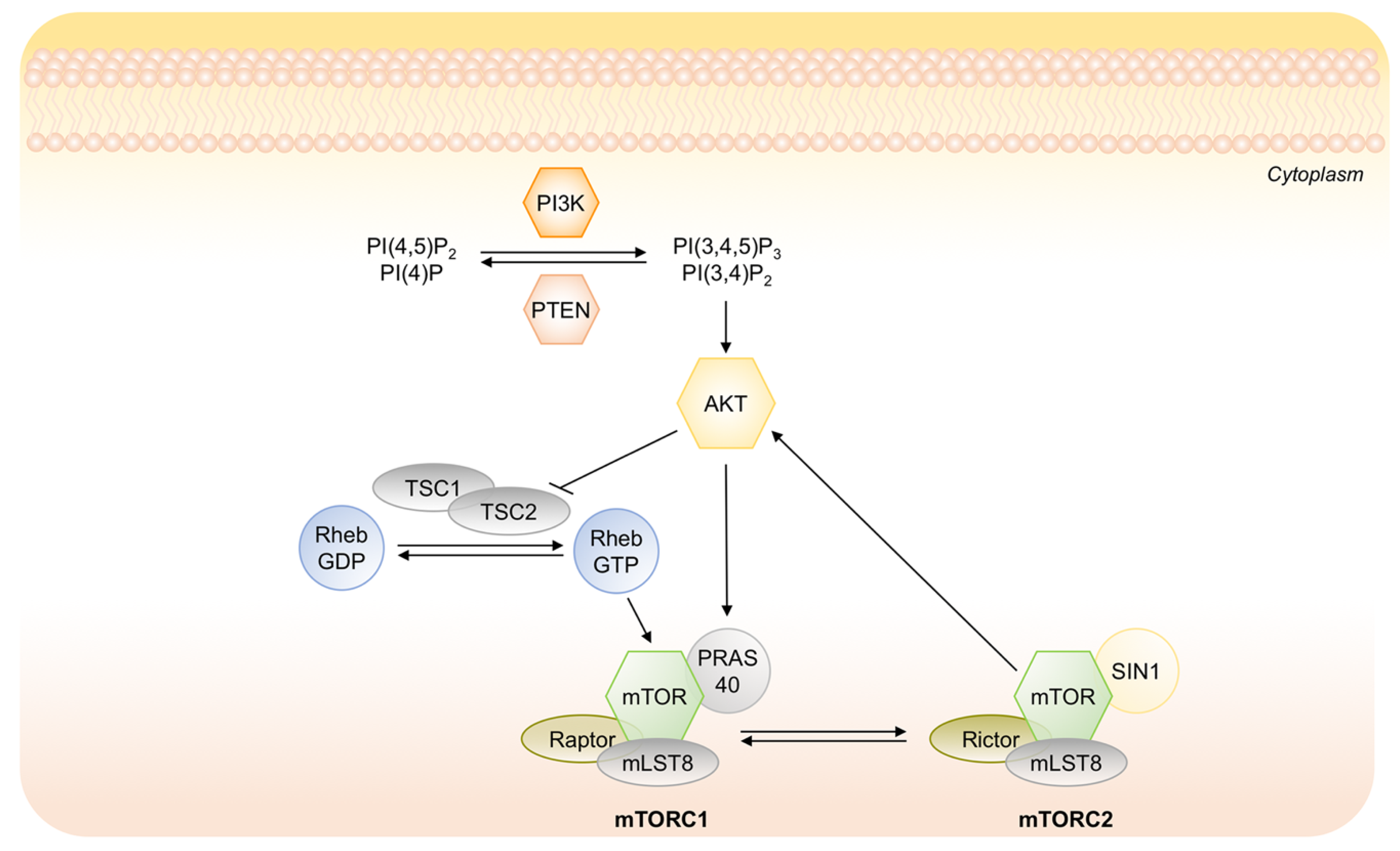
- Schewe, D.M.; Aguirre-Ghiso, J.A. ATF6alpha-Rheb-mTOR signaling promotes survival of dormant tumor cells in vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 10519–10524. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A. Targeting PI3K signalling in cancer: Opportunities, challenges and limitations. Nat. Rev. Cancer 2009, 9, 550. [Google Scholar] [CrossRef] [PubMed]
- Endo, H.; Okuyama, H.; Ohue, M.; Inoue, M. Dormancy of cancer cells with suppression of AKT activity contributes to survival in chronic hypoxia. PLoS ONE 2014, 9, e98858. [Google Scholar] [CrossRef]
- Memmott, R.M.; Dennis, P.A. Akt-dependent and -independent mechanisms of mTOR regulation in cancer. Cell. Signal. 2009, 21, 656–664. [Google Scholar] [CrossRef]
- Lobo, N.A.; Shimono, Y.; Qian, D.; Clarke, M.F. The biology of cancer stem cells. Annu. Rev. Cell Dev. Biol. 2007, 23, 675–699. [Google Scholar] [CrossRef] [PubMed]
- Haubner, S.; Perna, F.; Kohnke, T.; Schmidt, C.; Berman, S.; Augsberger, C.; Schnorfeil, F.M.; Krupka, C.; Lichtenegger, F.S.; Liu, X.; et al. Coexpression profile of leukemic stem cell markers for combinatorial targeted therapy in AML. Leukemia 2019, 33, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Skoda, J.; Hermanova, M.; Loja, T.; Nemec, P.; Neradil, J.; Karasek, P.; Veselska, R. Co-Expression of Cancer Stem Cell Markers Corresponds to a Pro-Tumorigenic Expression Profile in Pancreatic Adenocarcinoma. PLoS ONE 2016, 11, e0159255. [Google Scholar] [CrossRef] [PubMed]
- Carcereri de Prati, A.; Butturini, E.; Rigo, A.; Oppici, E.; Rossin, M.; Boriero, D.; Mariotto, S. Metastatic Breast Cancer Cells Enter Into Dormant State and Express Cancer Stem Cells Phenotype Under Chronic Hypoxia. J. Cell. Biochem. 2017, 118, 3237–3248. [Google Scholar] [CrossRef] [PubMed]
- Bar, E.E.; Lin, A.; Mahairaki, V.; Matsui, W.; Eberhart, C.G. Hypoxia increases the expression of stem-cell markers and promotes clonogenicity in glioblastoma neurospheres. Am. J. Pathol 2010, 177, 1491–1502. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Rich, J.N. Hypoxia and hypoxia inducible factors in cancer stem cell maintenance. Curr. Top. Microbiol. Immunol. 2010, 345, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.; Chen, E.I. Cancer stem cells, tumor dormancy, and metastasis. Front. Endocrinol. 2012, 3, 125. [Google Scholar] [CrossRef]
- Giancotti, F.G. Mechanisms governing metastatic dormancy and reactivation. Cell 2013, 155, 750–764. [Google Scholar] [CrossRef]
- Lin, W.C.; Rajbhandari, N.; Wagner, K.U. Cancer cell dormancy in novel mouse models for reversible pancreatic cancer: A lingering challenge in the development of targeted therapies. Cancer Res. 2014, 74, 2138–2143. [Google Scholar] [CrossRef]
- Zhou, N.; Wu, X.; Yang, B.; Yang, X.; Zhang, D.; Qing, G. Stem cell characteristics of dormant cells and cisplatininduced effects on the stemness of epithelial ovarian cancer cells. Mol. Med. Rep. 2014, 10, 2495–2504. [Google Scholar] [CrossRef]
- Plaks, V.; Kong, N.; Werb, Z. The cancer stem cell niche: How essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 2015, 16, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Axelson, H.; Fredlund, E.; Ovenberger, M.; Landberg, G.; Pahlman, S. Hypoxia-induced dedifferentiation of tumor cells--a mechanism behind heterogeneity and aggressiveness of solid tumors. Semin. Cell Dev. Biol. 2005, 16, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Kleffel, S.; Schatton, T. Tumor dormancy and cancer stem cells: Two sides of the same coin? Adv. Exp. Med. Biol. 2013, 734, 145–179. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Wu, Q.; Sathornsumetee, S.; Hao, Y.; Li, Z.; Hjelmeland, A.B.; Shi, Q.; McLendon, R.E.; Bigner, D.D.; Rich, J.N. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res. 2006, 66, 7843–7848. [Google Scholar] [CrossRef] [PubMed]
- Frank, N.Y.; Schatton, T.; Kim, S.; Zhan, Q.; Wilson, B.J.; Ma, J.; Saab, K.R.; Osherov, V.; Widlund, H.R.; Gasser, M.; et al. VEGFR-1 expressed by malignant melanoma-initiating cells is required for tumor growth. Cancer Res. 2011, 71, 1474–1485. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Setoguchi, T.; Taga, T. Persistence of a small subpopulation of cancer stem-like cells in the C6 glioma cell line. Proc. Natl. Acad. Sci. USA 2004, 101, 781–786. [Google Scholar] [CrossRef] [PubMed]
- Hirschmann-Jax, C.; Foster, A.E.; Wulf, G.G.; Nuchtern, J.G.; Jax, T.W.; Gobel, U.; Goodell, M.A.; Brenner, M.K. A distinct “side population” of cells with high drug efflux capacity in human tumor cells. Proc. Natl. Acad. Sci. USA 2004, 101, 14228–14233. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.; Fojo, T.; Bates, S. Tumour stem cells and drug resistance. Nat. Rev. Cancer 2005, 5, 275–284. [Google Scholar] [CrossRef] [PubMed]
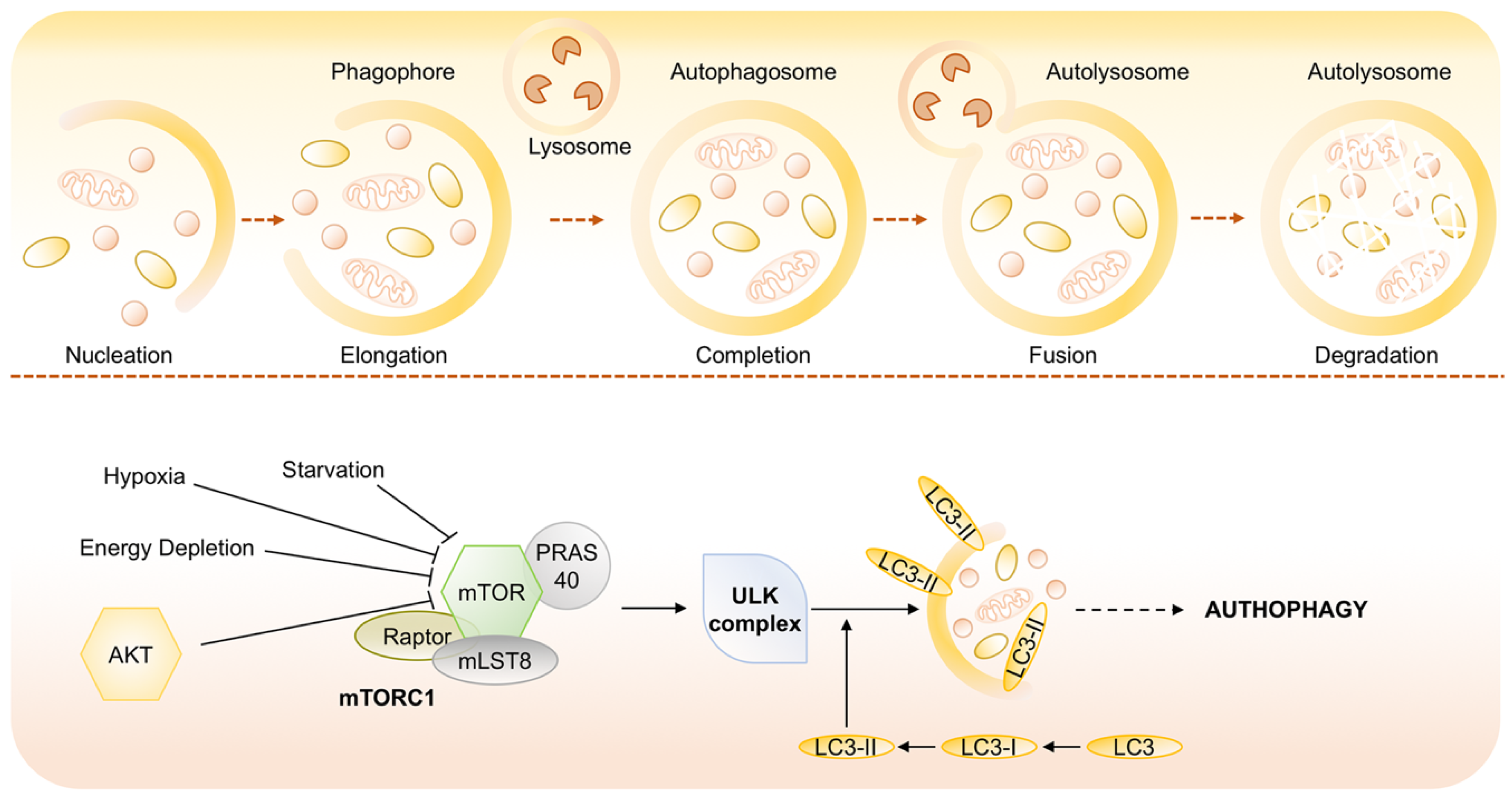
- Mizushima, N.; Yoshimori, T.; Levine, B. Methods in mammalian autophagy research. Cell 2010, 140, 313–326. [Google Scholar] [CrossRef]
- Cecconi, F.; Levine, B. The role of autophagy in mammalian development: Cell makeover rather than cell death. Dev. Cell 2008, 15, 344–357. [Google Scholar] [CrossRef]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef] [PubMed]
- Longatti, A.; Tooze, S.A. Vesicular trafficking and autophagosome formation. Cell Death Differ. 2009, 16, 956–965. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yamamoto, A.; Matsui, M.; Yoshimori, T.; Ohsumi, Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol. Biol. Cell 2004, 15, 1101–1111. [Google Scholar] [CrossRef] [PubMed]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Noda, T.; Fujita, N.; Yoshimori, T. The late stages of autophagy: How does the end begin? Cell Death Differ. 2009, 16, 984. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; Rodriguez-Enriquez, S.; Lemasters, J.J. Selective degradation of mitochondria by mitophagy. Arch. Biochem. Biophys. 2007, 462, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Thomas, S.M.; Woodside, D.G.; Rose, D.M.; Kiosses, W.B.; Pfaff, M.; Ginsberg, a.M.H. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999, 402, 672–676. [Google Scholar]
- Jung, C.H.; Ro, S.-H.; Cao, J.; Otto, N.M.; Kim, D.-H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295. [Google Scholar] [CrossRef]
- Kroemer, G.; Levine, B. Autophagic cell death: The story of a misnomer. Nat. Rev. Mol. Cell Biol. 2008, 9, 1004–1010. [Google Scholar] [CrossRef]
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gelinas, C.; Fan, Y.; et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006, 10, 51–64. [Google Scholar] [CrossRef]
- Lock, R.; Roy, S.; Kenific, C.M.; Su, J.S.; Salas, E.; Ronen, S.M.; Debnath, J. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol. Biol. Cell 2011, 22, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Perera, R.M.; Stoykova, S.; Nicolay, B.N.; Ross, K.N.; Fitamant, J.; Boukhali, M.; Lengrand, J.; Deshpande, V.; Selig, M.K.; Ferrone, C.R.; et al. Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature 2015, 524, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Maycotte, P.; Thorburn, A. Autophagy and cancer therapy. Cancer Biol. Ther. 2014, 11, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Meléndez, A.; Tallóczy, Z.; Seaman, M.; Eskelinen, E.-L.; Hall, D.H.; Levine, A.B. Autophagy Genes Are Essential for Dauer Development and Life-Span Extension in C. elegans. Science 2003, 301, 1387–1391. [Google Scholar] [CrossRef] [PubMed]
- Lock, R.; Debnath, J. Extracellular matrix regulation of autophagy. Curr. Opin. Cell Biol. 2008, 20, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Campos, T.; Ziehe, J.; Fuentes-Villalobos, F.; Riquelme, O.; Pena, D.; Troncoso, R.; Lavandero, S.; Morin, V.; Pincheira, R.; Castro, A.F. Rapamycin requires AMPK activity and p27 expression for promoting autophagy-dependent Tsc2-null cell survival. Biochim. Biophys. Acta 2016, 1863, 1200–1207. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Luo, R.Z.; Lu, Y.; Zhang, X.; Yu, Q.; Khare, S.; Kondo, S.; Kondo, Y.; Yu, Y.; Mills, G.B.; et al. The tumor suppressor gene ARHI regulates autophagy and tumor dormancy in human ovarian cancer cells. J. Clin. Investig. 2008, 118, 3917–3929. [Google Scholar] [CrossRef]
- Sosa, M.S.; Bragado, P.; Debnath, J.; Aguirre-Ghiso, J.A. Regulation of tumor cell dormancy by tissue microenvironments and autophagy. Adv. Exp. Med. Biol. 2013, 734, 73–89. [Google Scholar] [CrossRef]
- Adjei, I.M.; Blanka, S. Modulation of the tumor microenvironment for cancer treatment: A biomaterials approach. J. Funct. Biomater. 2015, 6, 81–103. [Google Scholar] [CrossRef]
- Unger, C.; Kramer, N.; Walzl, A.; Scherzer, M.; Hengstschlager, M.; Dolznig, H. Modeling human carcinomas: Physiologically relevant 3D models to improve anti-cancer drug development. Adv. Drug Deliv. Rev. 2014, 79–80, 50–67. [Google Scholar] [CrossRef]
- Wang, C.; Tang, Z.; Zhao, Y.; Yao, R.; Li, L.; Sun, W. Three-dimensional in vitro cancer models: A short review. Biofabrication 2014, 6, 022001. [Google Scholar] [CrossRef] [PubMed]
- Costa, E.C.; Moreira, A.F.; de Melo-Diogo, D.; Gaspar, V.M.; Carvalho, M.P.; Correia, I.J. 3D tumor spheroids: An overview on the tools and techniques used for their analysis. Biotechnol. Adv. 2016, 34, 1427–1441. [Google Scholar] [CrossRef] [PubMed]
- Villasante, A.; Vunjak-Novakovic, G. Tissue-engineered models of human tumors for cancer research. Expert Opin. Drug Discov. 2015, 10, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Leong, D.T.; Ng, K.W. Probing the relevance of 3D cancer models in nanomedicine research. Adv. Drug Deliv. Rev. 2014, 79-80, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Haycock, J.W. 3D cell culture: A review of current approaches and techniques. Methods Mol. Biol. 2011, 695, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Pampaloni, F.; Reynaud, E.G.; Stelzer, E.H. The third dimension bridges the gap between cell culture and live tissue. Nat. Rev. Mol. Cell Biol. 2007, 8, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Lovitt, C.J.; Shelper, T.B.; Avery, V.M. Advanced cell culture techniques for cancer drug discovery. Biology 2014, 3, 345–367. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, S.; Sperduto, J.L.; Farino, C.J.; Slater, J.H. Engineered In Vitro Models of Tumor Dormancy and Reactivation. J. Biol. Eng. 2018, 12, 37. [Google Scholar] [CrossRef]
- Louie, E.; Nik, S.; Chen, J.S.; Schmidt, M.; Song, B.; Pacson, C.; Chen, X.F.; Park, S.; Ju, J.; Chen, E.I. Identification of a stem-like cell population by exposing metastatic breast cancer cell lines to repetitive cycles of hypoxia and reoxygenation. Breast Cancer Res. BCR 2010, 12, R94. [Google Scholar] [CrossRef]
- Lee, H.R.; Leslie, F.; Azarin, S.M. A facile in vitro platform to study cancer cell dormancy under hypoxic microenvironments using CoCl2. J. Biol. Eng. 2018, 12, 12. [Google Scholar] [CrossRef]
- Fang, J.Y.; Tan, S.J.; Yang, Z.; Tayag, C.; Han, B. Tumor bioengineering using a transglutaminase crosslinked hydrogel. PLoS ONE 2014, 9, e105616. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Butturini, E.; Carcereri de Prati, A.; Boriero, D.; Mariotto, S. Tumor Dormancy and Interplay with Hypoxic Tumor Microenvironment. Int. J. Mol. Sci. 2019, 20, 4305. https://doi.org/10.3390/ijms20174305
Butturini E, Carcereri de Prati A, Boriero D, Mariotto S. Tumor Dormancy and Interplay with Hypoxic Tumor Microenvironment. International Journal of Molecular Sciences. 2019; 20(17):4305. https://doi.org/10.3390/ijms20174305
Chicago/Turabian StyleButturini, Elena, Alessandra Carcereri de Prati, Diana Boriero, and Sofia Mariotto. 2019. "Tumor Dormancy and Interplay with Hypoxic Tumor Microenvironment" International Journal of Molecular Sciences 20, no. 17: 4305. https://doi.org/10.3390/ijms20174305
APA StyleButturini, E., Carcereri de Prati, A., Boriero, D., & Mariotto, S. (2019). Tumor Dormancy and Interplay with Hypoxic Tumor Microenvironment. International Journal of Molecular Sciences, 20(17), 4305. https://doi.org/10.3390/ijms20174305