Abstract
Progress in epidemiological, molecular and clinical genetics with the development of new techniques has improved knowledge on genetic syndromes associated with autism spectrum disorder (ASD). The objective of this article is to show the diversity of genetic disorders associated with ASD (based on an extensive review of single-gene disorders, copy number variants, and other chromosomal disorders), and consequently to propose a hierarchical diagnostic strategy with a stepwise evaluation, helping general practitioners/pediatricians and child psychiatrists to collaborate with geneticists and neuropediatricians, in order to search for genetic disorders associated with ASD. The first step is a clinical investigation involving: (i) a child psychiatric and psychological evaluation confirming autism diagnosis from different observational sources and assessing autism severity; (ii) a neuropediatric evaluation examining neurological symptoms and developmental milestones; and (iii) a genetic evaluation searching for dysmorphic features and malformations. The second step involves laboratory and if necessary neuroimaging and EEG studies oriented by clinical results based on clinical genetic and neuropediatric examinations. The identification of genetic disorders associated with ASD has practical implications for diagnostic strategies, early detection or prevention of co-morbidity, specific treatment and follow up, and genetic counseling.
1. Introduction
Autism Spectrum Disorder (ASD) is considered as a neurodevelopmental disorder defined in the DSM-5 (the Diagnostic and Statistical Manual of Mental Disorders 5th ed.) [1], the most recent diagnostic classification of mental disorders, by social communication deficits associated with repetitive/stereotyped behaviors or interests with early onset (referenced in the DSM-5 as symptoms present at early developmental period and evident in early childhood). According to the historian Normand J. Carrey, the first description of autism can be traced to Itard in his 1828 report on “mutism produced by a lesion of intellectual functions” [2]. In 1943, Leo Kanner [3], American psychiatrist with Austrian origins, borrowed the term autism from the Swiss psychiatrist Eugen Bleuler to define a specific syndrome observed in 11 children (the term “autism”, derived from the Greek autos which means “oneself”, was used for the first time by Eugen Bleuler in 1911 [4] to describe social withdrawal in adult patients with schizophrenia). The Kanner syndrome was characterized by its early onset (from the first year of life) and symptomatology (social withdrawal, sameness, language impairment, stereotyped motor behaviors, and intellectual disability). It is noteworthy that Kanner did not describe autism in individuals with severe intellectual disability and known brain disorders. The same year, the Austrian psychiatrist Hans Asperger [5] distinguished “personalities with autistic tendencies” which differed from the children that Kanner had described, due to the expression of exceptional isolated talents and conserved linguistic abilities. Currently, autism is viewed as an epistatic and multifactorial disorder involving genetic factors and environmental factors (prenatal, neonatal and/or postnatal environmental factors such as gestational diabetes, neonatal hypoxia conditions, exposure to air pollution, parental immigration or sensory/social deficits; for a review, see Tordjman et al., 2014 [6]).
Several literature reviews underline the important role of genetics in the etiology of autism [7,8,9]. The data come from family and twin studies. Indeed, the concordance rate among monozygotic twins is high (60%–90%) compared to the concordance rate among dizygotic twins (0%–30%) [10,11]. Technological advances in epidemiological and molecular genetics have led recently to new findings in the field of the genetics of neuropsychiatric disorders. These new findings concern also the domain of the genetics of autism and have increased the state of current knowledge on the genetic disorders associated with ASD. Identified genetic causes of ASD can be classified as the cytogenetically visible chromosomal abnormalities, copy number variants (CNVs) (i.e., variations in the number of copies of one segment of DNA, including deletions and duplications), and single-gene disorders [12]. CNVs can have different sizes (small to large deletions or duplications) and therefore concern a variable number of genes according to their size. The number of known genetic disorders associated with ASD has increased with the use of array Comparative Genomic Hybridization (aCGH), also called chromosomal microarrays (CMAs) or high-resolution molecular karyotype, which is one of the most molecular cytogenetic methods used by geneticists. The accuracy of aCGH is 10 to 100 times higher than low-resolution karyotype accuracy and allows detection of small CNVs. Unfortunately, this method does not detect certain genetic anomalies such as balanced chromosome rearrangements (which are rarely involved in developmental disorders) or CNVs present in less than 10% to 20% of cells (somatic mosaicism). CNVs are usually considered to be rare variants but recent techniques have shown that they may be in fact more frequent than expected [13]. In practice, after CNV detection, the geneticist needs to determine the contribution of this variation to the patient’s disorder. Most of the time, parental CNVs are also analyzed for family segregation. Among genetic causes of ASD, besides single specific genetic factors, cumulative effects of Single Nucleotide Variants (SNVs: common small variations in the DNA sequence occurring within a population) also have to be taken into consideration. Single-nucleotide polymorphism (SNP) is the term usually used for SNVs occurring in more than 1% of the general population. In the next few years, it will be probably possible to improve the identification and interpretation of SNPs in ASD using new techniques of High Throughput Sequencing, such as targeted sequencing on known genes (gene panel) or Whole Exome/Genome Sequencing. Finally, it should be highlighted that a genetic model of ASD (including cumulative genetic variance) does not exclude the role of environment (including cumulative environmental variance). It is noteworthy that heritability (h2) is defined as h2 = GV/(GV + EV) where GV is the cumulative genetic variance and EV, the environmental variance [14].
Many susceptibility genes and cytogenetic abnormalities have been reported in ASD and concern almost every chromosome (for a review, see Miles, 2011; available online: http://projects.tcag.ca/autism/) [15]. How can we explain such genetic diversity associated with similar autism cognitive-behavioral phenotypes? Given the current state of knowledge, we can state the hypothesis that the majority of ASD-related genes are involved in brain development and functioning (such as synapse formation and functioning, brain metabolism, chromatin remodeling) [16].
Several studies have reported associations between genetic contribution (such as unbalanced chromosome abnormalities, in particular large chromosomal abnormalities), dysmorphic features and low cognitive functioning [17,18,19]. Indeed, large chromosomal abnormalities are more often found in children with dysmorphic features, whereas small CNVs and de novo SNPs are more often found in individuals without dysmorphic features. The concept of “syndromic autism” or “complex autism” (autism associated with genetic disorders/genetic syndromes) which qualifies individuals with at least one dysmorphic feature/malformation or severe intellectual disability, is opposed to the concept of “non-syndromic autism” or “simplex”/“pure”/idiopatic autism (isolated autism) which qualifies individuals with moderate intellectual disability to normal cognitive functioning and no other associated signs or symptoms (except the presence of seizures) [19,20]. The prevalence of epilepsy in ASD varies from 5% to 40% (compared to 0.5% to 1% in the general population) according to different variables, including syndromic vs. non-syndromic autism (higher rates of epilepsy are observed in syndromic autism compared to non-syndromic autism) [21,22,23,24] and the age of the population (the rate of seizures varies with age and increases especially during the prepubertal period). It is noteworthy that genome-wide association and genetic analyses such as aCGH have revealed hundreds of previously unknown rare mutations and CNVs linked to non-syndromic autism. Some authors [12,19] suggest that syndromic autism (SA), compared to non-syndromic autism (NSA), is associated with a poorer prognosis, a lower male-to-female sex ratio (SA: 3/1; NSA: 6/1), and a lower sibling recurrence risk (SA: 4%–6%; NSA: up to 35%) as well as a lower risk for family history of autism (SA: up to 9%; NSA: up to 20%) given that SA would be more related to accidental mutations than NSA. Similarly, the concept of syndromic intellectual disability (intellectual disability associated with dysmorphism and medical or behavioral signs and symptoms) is widely used as opposed to non-syndromic intellectual disability (intellectual disability without other abnormalities). Intellectual disability and autism spectrum disorder share other similarities given that they are both genetically heterogeneous, and a significant number of genes have been associated with both conditions. It is noteworthy that the concept of non-syndromic autism or non-syndromic intellectual disability depends on technological progress and current state of knowledge. We can foresee, as underlined by Tordjman et al. [25], that individuals with currently and apparently “non-syndromic autism” could become a few years later individuals with “syndromic autism”, if new genetic disorders are identified, allowing a better understanding and more efficient identification of discrete clinical symptoms as minor physical anomalies, malformations and associated biological anomalies. For example, individuals with Smith–Magenis syndrome can show in early childhood ASD associated with subtle minor facial dysmorphic features (these discrete dysmorphic features become more evident later), mild or moderate intellectual disability, disrupted sleep patterns and repetitive self-hugging characteristic of this syndrome. The molecular discovery of Smith–Magenis syndrome is relatively recent (1982) and allowed to better know the characteristic behavioral phenotype combined with physical anomalies of Smith–Magenis syndrome (including short stature and scoliosis, not enough specific to help alone to identify the genetic syndrome), orienting towards the search for certain dysmorphic features and the possible identification of this genetic disorder. Thus, some children with Smith–Magenis syndrome could be considered in early childhood, especially before the discovery of Smith–Magenis syndrome in 1982, as individuals with apparently “non-syndromic autism” (they are more viewed as individuals with syndromic autism over their developmental trajectory). Therefore, taking into account this bias, it would be better to consider autism as a behavioral syndrome related either to known genetic disorders or to currently unknown causes.
The objective of this article is to show the diversity of the genetic disorders associated with ASD (including single-gene disorders, CNVs and other chromosomal disorders), and consequently to propose a hierarchical diagnostic strategy with a stepwise evaluation, helping general practitioners/pediatricians and child psychiatrists to collaborate with geneticists and neuropediatricians, in order to search for genetic disorders associated with ASD.
2. Known Genetic Syndromes Associated with Autism Spectrum Disorder
We chose to limit the review of literature to genetic disorders presented in Table 1. Table 1 includes one part on chromosomal disorders and another part on single-gene disorders. The present descriptions focus on autistic traits encountered in each genetic disorder and other symptoms that may be helpful for the diagnosis. Table 1 summarizes these clinical data and indicates the estimated frequency of each genetic disorder in autism and the frequency of autistic traits in each genetic disorder (when available). This table does not present an exhaustive list of genetic disorders associated with ASD. Furthermore, Table 1 includes only genetic syndromes involving a single-gene disorder or a chromosomal rearrangement, and therefore does not include polygenic causes and possible genetic disorders only related to epigenetic mechanisms. It is noteworthy that some studies suggest that the genetic background might play an important role in many ASD individuals, with in particular cumulative genetic effects related to the load of common risk variants [16].

Table 1.
Main genetic disorders associated with autistic syndrome.
Interestingly, as underlined by Abrahams and Geschwind [7], the descriptive analysis of Table 1 indicates that each of the many known genes or genomic regions associated with ASD, taken separately, accounts usually and on average for less than 2% of autism cases, suggesting high genetic heterogeneity (the maximum average rate is 5% and concerns the Fragile X syndrome). However, taken all together, these single genetic impairments account at least for 10%–25% of ASD individuals [244] and even 35%–40% in more recent studies [245].
3. Recommended Investigations for the Identification of Genetic Disorders Associated with ASD
Facing such a diversity of genetic disorders associated with ASD (see Table 1), it seems important to clarify, as best as possible with regard to the current state of knowledge, the diagnostic steps towards identification of these genetic disorders. A hierarchical diagnostic strategy with a stepwise evaluation towards identification of genetic disorders associated with autism is proposed and developed in Figure 1.
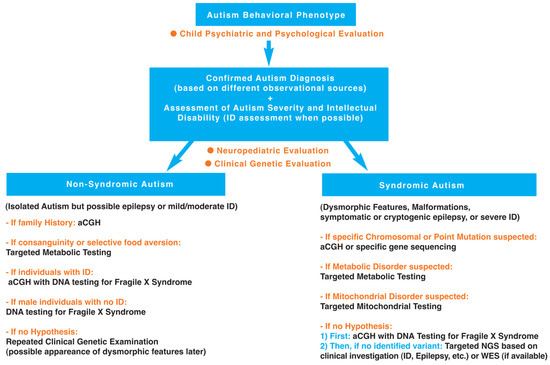
Figure 1.
Hierarchical diagnostic strategy with a stepwise evaluation towards identification of genetic disorders associated with autism.
As it appears in Table 1, the severity of the autistic syndrome is an important variable to take into consideration in the search for genetic disorders associated with ASD. Therefore, the preliminary step in the hierarchical diagnostic strategy is to confirm autism diagnosis and assess autism severity. Furthermore, based on the genetic syndromes listed in Table 1, certain variables are of particular interest such as dysmorphic features, malformations, epilepsy and intellectual disability. It indicates clearly that clinical investigation of behavioral phenotype of autism for identification of genetic disorders associated with ASD requires clinical genetic examination, neuropediatric examination and psychological assessment. In this section, we propose a hierarchal diagnostic strategy with focused investigations for the identification of genetic disorders associated with ASD, based on the history of the individual (including family history and developmental trajectory) and the clinical neurologic and genetic evaluation, but also on up-to-date knowledge and new technology. The first step of the hierarchal diagnostic strategy is a clinical investigation involving: (i) a child psychiatrist’s and psychologist’s evaluation to confirm autism diagnosis using different observational sources (including an extensive parental interview facilitating the study of family history) and assess autism severity (including behavioral and cognitive assessments); (ii) a neuropediatrician’s evaluation to examine neurological symptoms/signs and developmental milestones; and (iii) a geneticist’s evaluation to search for dysmorphic features and malformations based on a clinical physical examination. This clinical step is followed by a second step involving laboratory and if necessary neuroimaging or EEG studies oriented by clinical results based on clinical genetic and neuropediatric examinations. These two steps are described below.
3.1. First Step: Clinical Investigation
3.1.1. Autism Diagnosis Using Different Observational Sources and Autism Severity Assessment (Child Psychiatric and Psychological Evaluation)
The preliminary step requires confirming the diagnosis of autism and assessing the severity of autistic behavioral impairments with validated tools used by trained professionals in different observational situations, such as the Autism Diagnostic Interview-Revised [246] (the ADI-R scale is a parental interview) and the Autism Diagnostic Observation Schedule [247] (the ADOS scale allows a direct observation of the individual through a standardized play situation). The psychiatric appreciation of ICD-10 (International Classification of Diseases, World Health Organization, Geneve, Switzerland, 1993) and DSM-5 (American Psychiatric Association, Washington, D.C., United States, 2013) diagnostic criteria of autism is also necessary and provides a clinical psychiatric judgment. This approach, combining information from multiple sources based on the clinical psychiatric judgment and the administration of the ADI-R completed by the ADOS improves the confidence in the diagnosis of ASD [248]. Interestingly, the ADI-R scale is validated to assess current behavior but also behavior during the 4–5 years old period (the ADI-R algorithm is based on the 4–5 years old period of life which is supposed to correspond to the most severe period of autistic impairments), allowing to see the evolution between these two periods of life. It happens more frequently than expected that children who fulfill the diagnostic criteria for ASD based on the ADI-R algorithm (parental interview for the 4–5 years old period) do not meet the full diagnostic criteria for ASD based on the ADOS algorithm (direct observation of the child for the current period).
The ADI-R scale is the most common assessment used to conduct an extensive semi-structured parental interview searching for medical and psychiatric family history. This clinical interview should be conducted with empathy and allow a three-generation family tree to be constructed, showing, in particular, family psychiatric history (such as, for example, schizophrenia, depression or bipolar disorder) and ASD behaviors (including delayed language development) within the family (mother’s and father’s sides). The construction of the pedigree can be very helpful to get specific hypotheses of genetic disorders associated with autism. More generally, higher rates of CNVs are observed in ASD individuals with a family history of psychiatric disorders or developmental disabilities, highlighting the importance to study family history [249]. It is noteworthy that family history is also investigated and analyzed by the neuropediatrician and the geneticist during their evaluation.
Furthermore, the clinician should indicate in the family tree the existence of consanguineous mating, family malformations, deceased infants and spontaneous abortions occurring in the mother but also in relatives (repeated spontaneous abortions are in favor of chromosomal rearrangement [20,250,251]). It is necessary to specify the trimester of pregnancy when spontaneous abortions occurred given that repeated miscarriages at the first trimester of pregnancy are more specifically in favor of genetic anomalies. Reproductive stoppage is rarely evoked but should be investigated because reproductive curtailment following the birth of a child with ASD has been reported [252]. In addition, the birth order of the child with autism has to be noted in the family tree given that a decrease of ASD in later siblings has been observed [253]. The maternal and paternal ages at the time of procreation should be also systematically noted. Indeed, several studies suggest that advanced parental age, in particular for the father, is a risk factor for ASD [254], but there are some discrepancies in the results [253]. Although advanced parental age may be a risk factor, it orients towards genetic testing but does not lead to definitive conclusions.
Finally, a psychological evaluation is necessary in this hierarchical diagnostic strategy to assess the level of cognitive functioning given that intellectual disability and its severity are associated, as indicated previously, with a risk for several genetic disorders in ASD individuals (see Table 1). In addition, the level of cognitive functioning should be taken into consideration and therefore assessed given that severe intellectual disability can introduce an important bias leading to a misdiagnosis of autism. Indeed, ASD is often misdiagnosed in individuals with severe intellectual disability. It should be noted that the diagnosis of autism might be not valid or relevant for genetic disorders associated with severe intellectual disability due to first, the behavioral overlap between severe intellectual disability and autistic behavioral impairments (social communication deficit as well as repeated behaviors or interests), and second, the absence of validity and reliability of autism diagnostic instruments in the context of very low IQ or mental age of less than 24 months [255]. It is noteworthy that the most currently international instruments used for autism diagnosis, such as the ADI-R and the ADOS scales, were not normed in individuals with severe intellectual disability [255]. Cognitive functioning can be assessed, whenever possible with the child, by a psychologist or a neuropsychologist if available, using the age-appropriate Wechsler intelligence scales (Wechsler Preschool and Primary Scale of Intelligence: WPPSI-IV for children between 2 and 7 years old, Wechsler Intelligence Scale for Children: WISC-5 for children between 6 and 16 years old, Wechsler Adult Intelligence Scale: WAIS-IV for individuals above 16 years old) or the Kaufman Assessment Battery for Children (K-ABC ) [256]. The Kaufman K-ABC is supposed to be more adapted for nonverbal children but cognitive assessment appears in fact easier to perform using the Wechsler intelligence scales with nonverbal subtests for nonverbal ASD children. Some instruments are of particular interest for intellectual disabled children with ASD such as the Raven’s Color Progressive Matrices (CPM) which is a short (20 min) nonverbal intelligence test validated for young children and individuals with intellectual disability [257].
The child psychiatric evaluation and psychological evaluation confirming autism diagnosis from different observational sources and assessing autism severity and intellectual disability are followed by a neuropediatrician’s evaluation and a geneticist’s evaluation (see Figure 1).
3.1.2. Developmental Milestones (Neuropediatric Evaluation)
A neuropediatric examination should be systematically conducted searching for neurological symptoms associated with ASD such as hypotonia, ataxia, abnormal movements or epilepsy. In case of clinical symptoms of epilepsy, the electroencephalogram (EEG) can be a useful tool to detect epileptic brain activity. In complement to this neuropediatric examination, it is necessary to rule out a sensory deficit, such as an auditory or visual deficit, based on ophtamological and ears-nose-throat examination (an audiogram is recommended). In addition, the neuropediatric examination investigates extensively developmental milestones for the following periods (some developmental milestones are often explored also during the child psychiatric evaluation): (i) prenatal period: course of pregnancy, fetal movements, results of ultrasound exams, treatments (in particular, in-utero exposure to thalidomide or valproate, known as teratogenic medications and risk factors for ASD), and other possible prenatal risk factors for ASD (such as gestational diabetes, gestational bleeding or multiple birth [6]); (ii) perinatal period: birth presentation (abnormal presentation in general and breech presentation in particular, have been associated with the development of ASD [258], mode of delivery, clinical status at birth (APGAR, score, weight, body length, and head circumference), premature birth (the risk for autism increased with the severity of preterm birth [259]), conditions related to hypoxia at birth reported as risk factors for ASD (such as umbilical cord complications, meconium aspiration, neonatal anaemia, ABO or Rh incompatibility, hyperbilirubinemia, or birth injury and trauma; for a review, see Gardener et al. [260]), feeding pattern and difficulties, screening tests (hypothyroidism, phenylketonuria), physical examination (apparent dysmorphism and malformations) and neurological examination (neonatal hypotonia); and (iii) postnatal period: psychomotor development, feeding pattern and possible gastrointestinal problems, sleep pattern, epilepsy, sensory dysfunction, language development, early behavioral signs of autism and other atypical behaviors. This clinical evaluation investigates developmental and learning disorders. The study of the height-weight-head circumference growth curve can show a break, decline with regression, or decrease with stagnation. For example, a decrease between 6 and 18 months of head circumference growth should orient the clinician towards the search for Rett syndrome in its first stage (stagnation stage) which occurs before the stage of fast decline of the communication skills (regression stage between 12 and 18 months of age). In addition, a developmental regression should orient the clinician towards the search for metabolic diseases. More generally, neuroimaging is recommended in individuals with ASD and microcephaly [249]. It is noteworthy that parental measures of height-weight-head circumference have also to be considered in order to determine whether the child’s height-weight-head circumference measures are really abnormal compared to the parents’ ones or whether a parent may be mildly affected, orienting towards a congenital or acquired abnormality.
3.1.3. Physical Examination (Genetic Evaluation)
A clinical genetics evaluation is systematically required and involves an extensive physical examination of the whole body. This physical examination should be conducted by a clinical geneticist trained in dysmorphology searching for dysmorphic features or malformations, and includes photos of the head (face and profile) with a good visibility of the ears, hands (palm and dorsal view) and feet. When clinical geneticists are not easily available, alternative options are to send, as a preliminary step, patients’ pictures (face and hands pictures) to a geneticist for a specialized opinion (our group is testing the feasibility and efficiency of such a procedure and is comparing the results obtained based on the analyses of pictures by a group of geneticists to the ones obtained based on the analyses of the same pictures using the genetic search and reference mobile application Face2Gene developed by FDNA: Facial Dysmorphology Novel Analysis). In addition, the skin should be examined with a Wood lamp, in particular when tuberous sclerosis is suspected [251]. Finally, clinical genetics evaluation can greatly benefit, in this strategy of identification of genetic disorders associated with ASD, from examining repeatedly over time the child to follow his/her developmental trajectory and examining at the same time the patient, his/her parents and siblings to compare the findings.
3.2. Second Step: Complementary Investigations (Laboratory Studies)
This secondary investigation includes cytogenetic, molecular analyses, biochemical analyses. Other exams such as neuroimaging and EEG are requested based on the results of the clinical genetic and neuropediatric examinations.
The following hierarchical diagnostic strategy, in line with recent American guidelines [249], can be proposed (see Figure 1).
3.2.1. Non-Syndromic Autism (Isolated ASD)
It should be remembered that non-syndromic autism includes ASD individuals with moderate intellectual disability to normal cognitive functioning (high functioning ASD) and no other associated signs or symptoms (except the possible presence of seizures). To our knowledge, the probability to detect a mutation (CNV or point mutation) is very low in high functioning non-syndromic ASD individuals, and, in this case, we do not recommend molecular analysis. However, in the case of family history of psychiatric disorders or developmental disabilities, it could be of interest to perform aCGH and to collect samples for a research program. In addition, in the case of consanguinity or selective food aversion, it could be useful to perform targeted metabolic testing. In addition, in the case of individuals with intellectual disability or males with no intellectual disability, the search for Fragile X mutation is recommended. If there is no hypothesis, a repeated clinical genetic evaluation over time is proposed given the possible later appearance of dysmorphic features along with the developmental trajectory.
3.2.2. Syndromic Autism (ASD Associated with Dysmorphic Features and/or Malformations and/or Symptomatic or Cryptogenic Epilepsy, and/or Severe Intellectual Disability)
If a specific chromosomal or point mutation is suspected: perform aCGH or specific gene sequencing.
If a metabolic disorder is suspected based on developmental regression (regression observed on the growth curves, including macrocephaly and in particular microcephaly, but also developmental psychomotor regression or neuroregression/neurodegeneration with neurological symptoms), seizures and especially epilepsy (recurrent and refractory seizures), consanguinity and specific dysmorphy or other symptoms described below: perform targeted metabolic testing including measurements of ammonemia and lactatemia, urinary organic acids chromatography, plasmatic amino-acids chromatography, creatine metabolism, iron, vitamin D, glutathione, oxidative stress, and cerebral magnetic resonance spectroscopy combined with standard neuroimaging. A metabolic disease could also be suspected based on certain clinical symptoms, such as digestion-related symptoms (cyclic vomiting, selective eating or gastrointestinal problems), dermatologic and hair changes (rashes, pigmented skin eruptions, hypertrichosis or alopecia), lethargy with hypotonia/extrapyramidal signs (dystonia, abnormal movements), or multiple organ dysfunction (in particular, heart, liver, kidney). In addition, the search for metabolic disorders can be indicated by abnormal biological measures such as anemia or lactic acidosis, and more generally acid/base or electrolyte disturbances. Searching for metabolic disorders is important given the potential availability of treatments such as enzymotherapy for certain metabolic diseases, diet low in phenylalanine for phenylketonuria due to the enzyme phenylalanine hydroxylase deficiency, or supplementation with folinic acid for cerebral folate deficiency. Several metabolic disorders associated with ASD are listed in Table 1 using the superscript #.
If a mitochondrial disorder is suspected based on repeated regressions after three years old, neurological abnormalities (including hypotonia) and multiple organ dysfunction: perform targeted mitochondrial testing including measurement of lactatemia. Abnormal neurologic examination and/ or elevated plasma lactate concentration have been indeed found in mitochonfrial diseases [260]. A mitochondrial disorder could be also suspected in case of ASD associated with loss of speech after a febrile illness or immunization with encephalopathy [261]. Finally, it is noteworthy that mitochondrial disease (rare in ASD) has to be differentiated from mitochondrial dysfunction (common in ASD) [262].
If there is no hypothesis of specific syndrome: perform aCGH and molecular diagnosis of Fragile-X syndrome. Without any identified pathogenic variant, we recommend targeted New Generation Sequencing (NGS) on known genes (for example, panels of genes related to intellectual disability or epilepsy based on the clinical examination) or Whole Exome Sequencing (WES) if available.
4. Conclusions
The finality of the hierarchal diagnostic strategy proposed in this article is to identify genetic disorders associated with ASD based on a stepwise evaluation characterized by low invasiveness and cost but high accessibility, feasibility and adaptability of current knowledge and technology to the individual and his/her family. These clinical and laboratory investigations can lead to identify genetic disorders associated with ASD in 35%–40% of individuals [225,262]. The identification of a specific genetic syndrome associated with ASD has practical clinical implications, in terms of diagnostic strategies including early detection and prevention of co-morbidity related to the known medical risks of the genetic disorder, follow up adapted to the genetic disorder with a more precise prognosis, and therapeutic strategies with access to needed services, help provided by specific family associations or specific individualized treatment in some cases. In many cases, identification of genetic disorders does not lead to therapeutic implications but there are a few cases where enzymotherapy, food depletion or supplementation in certain metabolic diseases as well as administration of melatonin associated with acebutolol in Smith–Magenis syndrome have improved considerably the quality of life of patients and their families. It should be noted that sleep problems are frequent in ASD (prevalence of insomnia: 50%–80% [263]) and decreased nocturnal and/or diurnal levels of melatonin (a sleep neurohormone) have been reported in many ASD studies; trial studies support therapeutic benefits of melatonin use in ASD (for a review, see Tordjman et al. [116,264]. Furthermore, the identification of genetic disorders associated with ASD allows unique personalized genetic counseling adapted to each family with regard to the transmission risk to the descendants and the possibility of a prenatal or preimplant diagnosis in the perspective of a future pregnancy. According to the 2013 American guidelines [249], the genetic counselor should note the type of array technology used (e.g., oligonucleotide vs. single-nucleotide polymorphism arrays) and access major databases relevant to this technology in order to provide the best possible and adapted information for the family. Many parents of children with ASD worry about the autism risk for a future pregnancy and knowing that their impaired child has a de novo mutation can be very helpful concerning the decision to have another child. It can be noted that the majority of the found mutations or rearrangements occurs de novo and are therefore not inherited from the parents or relatives [262]. In the same vein, siblings of children with ASD are very often preoccupied by the possible genetic transmission of autism to their future children. This preoccupation is legitimate considering, for instance, that the sisters of a boy with Fragile X Syndrome, even if they express very few or no symptoms, have a risk of 50% to carry the mutation or premutation and therefore a risk of 50% to have a child with Fragile X Syndrome (more severely impaired if it is a male). It is noteworthy that genetic counseling is also offered to families of ASD individuals without identified etiology, using in this case epidemiologic studies, and in particular sibling recurrence-risk updated data [249]. In any case, genetic counseling provides helpful family guidance, and parents of children with ASD are often relieved that biological factors involved in the development of ASD are considered and possibly found when some of them might feel guilty with regard to the role of family environment in ASD. Interestingly, based on our experience and the experience of geneticists or neuropediatricians working with psychoanalysts [265], knowing the underlying genetic cause of a relative’s or patient’s disorder, far from fixing the family or caregiver representations with regard to genetic determinism, rather creates and stimulates new dynamics within the family as well as the caregiver team centered on the ASD individual and probably related to a better known developmental trajectory with therapeutic perspectives and a parental relief. Finally, given all these implications, it is essential that all individuals (children but also adolescents and adults) with ASD can benefit from a clinical genetic examination and neuropediatric examination searching for associated signs or symptoms helping to identify known genetic disorders.
Acknowledgments
Funds from the Laboratory Psychology of Perception (LPP) were receiving for covering the costs to publish in open access.
Author Contributions
Cyrille Robert and Sylvie Tordjman wrote the manuscript; Sylvie Odent, Laurent Pasquier, David Cohen, Mélanie Fradin, Roberto Canitano and Léna Damaj revised the first draft of the paper; Sylvie Tordjman, Sylvie Odent and Laurent Pasquier modified the article according to the reviewers’ comments.
Conflicts of interest
The authors declare no conflict of interest.
References
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM-5), 5th ed.; American Psychiatric Association: Washington, DC, USA, 2013. [Google Scholar]
- Carrey, N.J. Itard’s 1828 Memoire on Mutism caused by a lesion of the intellectual functions: A historical analysis. J. Am. Acad. Child Adolesc. Psychiatry 1995, 34, 1655–1661. [Google Scholar] [CrossRef] [PubMed]
- Kanner, L. Austistic disturbances of affective contact. Nervous Child 1943, 32, 217–253. [Google Scholar]
- Bleuler, E. Dementia Praecox oder Gruppe der Schizophrenien; Handbuch der Psychiatrie; Deuticke: Leipzig, Germany, 1911. [Google Scholar]
- Asperger, H. Die autistischen psychopathen im kindesalter. Arch. Psychiatry Nervenkrandeiten 1944, 117, 73–136. [Google Scholar] [CrossRef]
- Tordjman, S.; Somogyi, E.; Coulon, N.; Kermarrec, S.; Cohen, D.; Bronsard, G.; Bonnot, O.; Weismann-Arcache, C.; Botbol, M.; Lauth, B.; et al. Gene-environment interactions in autism spectrum disorders: Role of epigenetic mechanisms. Front. Psychiatry 2014, 5, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Abrahams, B.S.; Geschwind, D.H. Advances in autism genetics: On the threshold of a new neurobiology. Nat. Rev. Genet. 2008, 9, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Bill, B.R.; Geschwind, D.H. Genetic advances in autism: Heterogeneity and convergence on shared pathways. Curr. Opin. Genet. Dev. 2009, 19, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Muhle, R.; Trentacoste, S.V.; Rapin, I. The genetics of autism. Pediatrics 2004, 113, 472–486. [Google Scholar] [CrossRef]
- Ronald, A.; Hoekstra, R.A. Autism spectrum disorders and autistic traits: A decade of new twin studies. Am. J. Med. Genet. 2011, 156, 255–274. [Google Scholar] [CrossRef] [PubMed]
- Sandin, S.; Lichtenstein, P.; Kuja-Halkola, R.; Larsson, H.; Hultman, C.M.; Reichenberg, A. The familial risk of autism. JAMA 2014, 311, 1770–1777. [Google Scholar] [CrossRef] [PubMed]
- Rosti, R.O.; Sadek, A.A.; Vaux, K.K.; Gleeson, J.G. The genetic landscape of autism spectrum disorders. Dev. Med. Child Neurol. 2014, 56, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Sebat, J.; Lakshmi, B.; Malhotra, D.; Troge, J.; Lese-Martin, C.; Walsh, T. Strong association of de novo copy number mutations with autism. Science 2007, 316, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Hegmann, J.P.; Possidente, B. Estimating genetic correlations from inbred strains. Behav. Genet. 1981, 11, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Miles, J.H. Autism spectrum disorders, a genetics review. Genet. Med. 2011, 13, 278–294. [Google Scholar] [CrossRef] [PubMed]
- Bourgeron, T. From the genetic architecture to synaptic plasticity in autism spectrum disorder. Nat. Rev. Neurosc. 2015, 16, 551–563. [Google Scholar] [CrossRef] [PubMed]
- Miles, J.H.; Hillman, R.E. Value of a clinical morphology examination in autism. Am. J. Med. Genet. 2000, 91, 245–253. [Google Scholar] [CrossRef]
- Jacquemont, M.L.; Sanlaville, D.; Redon, R.; Raoul, O.; Cormier-Daire, V.; Lyonnet, S.; Amiel, J.; Le Merrer, M.; Heron, D.; de Blois, M.C.; et al. Array-based comparative genomic hybridisation identifies high frequency of cryptic chromosomal rearrangements in patients with syndromic autism spectrum disorders. J. Med. Genet. 2006, 43, 843–849. [Google Scholar] [CrossRef] [PubMed]
- Miles, J.H.; Takahashi, T.N.; Bagby, S.; Sahota, P.K.; Vaslow, D.F.; Wang, C.H.; Hillman, R.E.; Farmer, J.E. Essential versus complex autism: Definition of fundamental prognostic subtypes. Am. J. Med. Genet. 2005, 135, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Cohen, D.; Pichard, N.; Tordjman, S.; Baumann, C.; Burglen, L.; Excoffier, E.; Lazar, G.; Mazet, P.; Pinquier, C.; Verloes, A.; et al. Specific genetic disorders and autism: Clinical contribution towards their identification. J. Autism Dev. Disord. 2005, 35, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Amiet, C.; Gourfinkel-An, I.; Bouzamondo, A.; Tordjman, S.; Baulac, M.; Lechat, P.; Cohen, D.J. Epilepsy in autism is associated with intellectual disability and gender: Evidence from a meta-analysis. Biol. Psychiatry 2008, 64, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Amiet, C.; Gourfinkel-An, I.; Lauent, C.; Bodeneau, N.; Genin, B.; Leguern, E.; Tordjman, S.; Cohen, D.J. Does epilepsy in multiplex autism define a different subgroup of clinical characteristics and genetic risk? Mol. Autism 2013, 4, 47. [Google Scholar] [CrossRef] [PubMed]
- Amiet, C.; Gourfinkel-An, I.; Lauent, C.; Carayol, J.; Genin, B.; Leguern, E.; Tordjman, S.; Cohen, D.J. Epilepsy in simplex autism pedigrees is much lower than the rate in multiplex autism pedigrees. Biol. Psychiatry 2013, 74, 3–4. [Google Scholar] [CrossRef] [PubMed]
- Canitano, R. Epilepsy in autism spectrum disorders. Eur. Child Adolesc. Psychiatry 2007, 16, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Tordjman, S.; Cohen, D.; Anderson, G.M.; Canitano, R.; Botbol, M.; Coulon, N.; Roubertoux, P.L. Reframing autism as a behavioral syndrome and not a specific mental disorder: Implications of genetic and phenotypic heterogeneity. Neurosci. Biobehav. Rev. 2017. [Google Scholar] [CrossRef] [PubMed]
- Cook, E.H., Jr.; Lindgren, V.; Leventhal, B.L.; Courchesne, R.; Lincoln, A.; Shulman, C.; Lord, C.; Courchesne, E. Autism or atypical autism in maternally but not paternally derived proximal 15q duplication. Am. J. Hum. Genet. 1997, 60, 928–934. [Google Scholar] [PubMed]
- Bolton, P.F.; Dennis, N.R.; Browne, C.E.; Thomas, N.S.; Veltman, M.W.; Thompson, R.J.; Jacobs, P. The phenotypic manifestations of interstitial duplications of proximal 15q with special reference to the autistic spectrum disorders. Am. J. Hum. Genet. 2001, 105, 675–685. [Google Scholar] [CrossRef] [PubMed]
- Cohen, D.; Martel, C.; Wilson, A.; Déchambre, N.; Amy, C.; Duverger, L.; Guile, J.M.; Pipiras, E.; Benzacken, B.; Cavé, H.; et al. Brief report: visual-spatial deficit in a 16-year-old girl with maternally derived duplication of proximal 15q. J. Autism Dev. Disord. 2007, 37, 1585–1591. [Google Scholar] [CrossRef] [PubMed]
- Schroer, R.J.; Phelan, M.C.; Michaelis, R.C.; Crawford, E.C.; Skinner, S.A.; Cuccaro, M.; Simensen, R.J.; Bishop, J.; Skinner, C.; Fender, D.; et al. Autism and maternally derived aberrations of chromosome 15q. Am. J. Hum. Genet. 1998, 76, 327–336. [Google Scholar] [CrossRef]
- Wandstrat, A.E.; Leana-Cox, J.; Jenkins, L.; Schwartz, S. Molecular cytogenetic evidence for a common break-point in the largest inverted duplications for chromosomes 15. Am. J. Hum. Genet. 1998, 62, 925–936. [Google Scholar] [CrossRef] [PubMed]
- Sutcliffe, J.S.; Nurmi, E.L. Genetics of chilhood disorders: XLVII. Autism, Part 6: Duplication and inherited susceptibility of chromosome 15q11–q13 genes in autism. J. Am. Acad. Child Adolesc. Psychiatry 2003, 42, 253–256. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, J.; King, B.H.; Leventhal, B.L.; Christian, S.L.; Ledbetter, D.H.; Cook, E.H., Jr. Molecular screening for proximal 15q abnormalities in a mentally retard population. J. Med. Genet. 1998, 35, 534–538. [Google Scholar] [CrossRef] [PubMed]
- Steffenburg, S.; Gillberg, C.L.; Seffenburg, U.; Kyllerman, M. Autism in Angelman syndrome: A population-based study. Pediatr. Neurol. 1996, 14, 131–136. [Google Scholar] [CrossRef]
- Peters, S.U.; Beaudet, A.L.; Madduri, N.; Bacino, C.A. Autism in Angelman syndrome: Implications for autism research. Clin. Genet. 2004, 66, 530–536. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, S.J.; Chen, P.F.; Ng, K.Y.; Bourgois-Rocha, F.; Lemtiri-Chlieh, F.; Levine, E.S.; Lalande, M. Induced pluripotent stem cell models of the genomic imprinting disorders Angelman and Prader-Willi syndromes. Proc. Natl. Acad. Sci. USA 2010, 107, 17668–17673. [Google Scholar] [CrossRef] [PubMed]
- Rangasamy, S.; D’Mello, S.R.; Narayanan, V. Epigenitics, autism spectrum, and neurodevelopmental disorders. Neurotherapeutics 2013, 10, 742–756. [Google Scholar] [CrossRef] [PubMed]
- Grafodatskaya, D.; Chung, B.; Szatmari, P.; Weksberg, R. Autism spectrum disorders and epigenetics. J. Am. Acad. Child Adolesc. Psychiatry 2010, 49, 794–809. [Google Scholar] [CrossRef] [PubMed]
- Dawson, A.J.; Cox, J.; Hovanes, K.; Spriggs, E. PWS/AS MS-MLPA confirms maternal origin of 15q11.2 microduplication. Case Rep. Genet. 2015, 2015, 474097. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.S.; Allen, J.A.; Mabb, A.M. Topoisomerase inhibitors unsilence the dormant allele of Ube3a in neurons. Nature 2011, 481, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Mabb, A.M.; Judson, M.C.; Zylka, M.J.; Philpot, B.D. Angelman syndrome: Insights into genomic imprinting and neurodevelopmental phenotypes. Trends Neurosci. 2011, 34, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Erickson, C.A.; Wink, L.K.; Baindu, B.; Ray, B.; Schaefer, T.L.; Pedapati, E.V.; Lahiri, D.K. Analysis of peripheral amyloid precursor protein in Angelman syndrome. Am. J. Med. Genet. 2016, 170, 2334–2337. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Yao, A.; Zhi, H.; Kaur, K.; Zhu, Y.; Jia, M.; Wang, Q.; Jin, S.; Zhao, G.; et al. Angelman syndrome protein Ube3a regulates synaptic growth and endocytosis by inhibiting BMP signaling in Drosophila. PLoS Genet. 2016, 12, e1006062. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Person, R.E.; Beaudet, A.L. Ube3a-ATS is an atypical RNA polymerase II transcript that represses the paternal expression of Ube3a. Hum. Mol. Genet. 2012, 21, 3001–3012. [Google Scholar] [CrossRef] [PubMed]
- Kishino, T.; Lalande, M.; Wasgstaff, J. UBE3A/E6-AP mutations cause Angelman syndrome. Nat. Genet. 1997, 15, 70–73. [Google Scholar] [CrossRef] [PubMed]
- Petit, E.; Hérault, J.; Martineau, J.; Perrot, A.; Barthélémy, C.; Hameury, L.; Sauvage, D.; Lelord, G.; Müh, J.P. Association study with two markers of a human homeogene in infantile autism. J. Med. Genet. 1995, 32, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Clayton-Smith, J. Clinical research on Angelman syndrome in the United Kingdom: Observations on 82 affected individuals. Am. J. Med. Genet. 1993, 46, 12–15. [Google Scholar] [CrossRef] [PubMed]
- Summers, J.A.; Allison, D.B.; Lynch, P.S.; Sandler, L. Behaviour problems in Angelman syndrome. J. Intellect. Disabil. Res. 1995, 39, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Walz, N.C. Parent report of stereotyped behaviors, social interaction, and developmental disturbances in individuals with Angelman syndrome. J. Autism Dev. Disord. 2007, 37, 940–947. [Google Scholar] [CrossRef] [PubMed]
- Dykens, E.M.; Cassidy, S.B. Correlates of maladaptive behavior in children and adults with Prader-Willi syndrome. Am. J. Med. Genet. 1995, 60, 546–549. [Google Scholar] [CrossRef] [PubMed]
- Demb, H.B.; Papola, P. PDD and Prader-Willi syndrome. J. Am. Acad. Child Adolesc. Psychiatry 1995, 34, 539–540. [Google Scholar] [CrossRef] [PubMed]
- Dykens, E.M.; Cassidy, S.B.; King, B.H. Maladaptive behavior differences in Prader-Willi syndrome due to paternal deletion versus maternal uniparental disomy. Am. J. Ment. Retard. 1995, 104, 67–77. [Google Scholar] [CrossRef]
- Veltman, M.W.; Thompson, R.J.; Roberts, S.E.; Thomas, N.S.; Whittington, J.; Bolton, P.F. Prader-Willi syndrome—A study comparing deletion and uniparental disomy cases with reference to autism spectrum disorders. Eur. Child Adolesc. Psychiatry 2004, 13, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Descheemaeker, M.J.; Govers, V.; Vermeulen, P.; Fryns, J.P. Pervasive developmental disorders in Prader-Willi syndrome: The Leuven experience in 59 subjects and controls. Am. J. Med. Genet. 2006, 140, 1136–1142. [Google Scholar] [CrossRef] [PubMed]
- Hogart, A.; Wu, D.; LaSalle, J.M.; Schanen, N.C. The comorbidity of autism with the genomic disorders of chromosome 15q11.2–q13. Neurobiol. Dis. 2010, 38, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Horsthemke, B.; Wagstaff, J. Mechanisms of imprinting of the Prader-Willi/Angelman region. Am. J. Med. Genet. 2008, 146, 2041–2052. [Google Scholar] [CrossRef] [PubMed]
- Buiting, K. Prader-Willi and Angelman syndrome. Am. J. Med. Genet. 2010, 154, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, S.B.; Scwartz, S.; Miller, H.; Driscoll, D.J. Prader-Willi syndrome. Genet. Med. 2012, 14, 10–26. [Google Scholar] [CrossRef] [PubMed]
- Bennett, J.A.; Germani, T.; Haqq, A.M.; Zwaigenbaum, L. Autism spectrum disorder in Prader-Willi syndrome: A systematic review. Am. J. Med. Genet. 2015, 167, 2936–2944. [Google Scholar] [CrossRef] [PubMed]
- Veltman, M.W.; Thompson, R.J.; Craig, E.E.; Dennis, N.R.; Roberts, S.E.; Moore, V.; Brown, J.A.; Bolton, P.F. A paternally inherited duplication in the Prader-Willi/Angelman syndrome critical region: A case and family study. J. Autism Dev. Disord. 2015, 35, 117–127. [Google Scholar] [CrossRef]
- Bolton, P.F.; Roobol, M.; Allsopp, L.; Pickles, A. Association between idiopathic infantile macrocephaly and autism spectrum disorders. Lancet 2001, 358, 726–727. [Google Scholar] [CrossRef]
- Roberts, S.E.; Dennis, N.R.; Browne, C.E.; Willatt, L.; Woods, G.; Cross, I.; Jacobs, P.A.; Thomas, S. Characterisation of interstitial duplications and triplications of chromosome 15q11–q13. Hum. Genet. 2002, 110, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Goizet, C.; Excoffier, E.; Taine, L.; Taupiac, E.; El Moneim, A.A.; Arveiler, B.; Bouvard, M.; Lacombe, D. Case with autistic syndrome and chromosome 22q13.3 deletion detected by FISH. Am. J. Hum. Genet. 2000, 96, 839–844. [Google Scholar] [CrossRef]
- Manning, M.A.; Cassidy, S.B.; Clericuzio, C.; Cherry, A.M.; Schwartz, S.; Hudgins, L.; Enns, G.M.; Hoyme, H.E. Terminal 22q deletion syndrome: A newly recognized cause of speech and language disability in the autism spectrum. Pediatrics 2004, 114, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Prasad, C.; Prasad, A.N.; Chodirker, B.N.; Lee, C.; Dawson, A.K.; Jocelyn, L.J.; Chudley, A.E. Genetic evaluation of pervasive developmental disorders: The terminal 22q13 deletion syndrome may represent a recognizable phenotype. Clin.Genet. 2000, 57, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Phelan, M.C.; Rogers, R.C.; Saul, R.A.; Stapleton, G.A.; Sweet, K.; McDermid, H.; Shaw, S.R.; Claytor, J.; Willis, J.; Kelly, D.P. 22q13 Deletion Syndrome. Am. J. Hum. Genet. 2001, 101, 91–99. [Google Scholar] [CrossRef]
- Soorya, L.; Kolevzon, A.; Zweifach, J.; Lim, T.; Dobry, Y.; Schwartz, L.; Frank, Y.; Wang, A.T.; Cai, G.; Parkhomenko, E.; et al. Prospective investigation of autism and genotype-phenotype correlations in 22q13 deletion syndrome and SHANK3 deficiency. Mol. Autism 2013, 4, 18. [Google Scholar] [CrossRef] [PubMed]
- Shcheglovitov, A.; Shcheglovitova, O.; Yazawa, M.; Portmann, T.; Shu, R.; Sebastiano, V.; Krawisz, A.; Froehlich, W.; Bernstein, J.A.; Hallmayer, J.F.; et al. SHANK3 and IGF1 restore synaptic deficits in neurons from 22q13 deletion syndrome patients. Nature 2013, 503, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Phelan, K.; McDermid, H.E. The 22q13.3 deletion syndrome (Phelan-McDermid syndrome). Mol. Syndromol. 2012, 2, 186–201. [Google Scholar] [CrossRef] [PubMed]
- Peca, J.; Feliciano, C.; Ting, J.T.; Wang, W.; Wells, M.F.; Venkatraman, T.N.; Lascola, C.D.; Fu, Z.; Feng, G. SHANK3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature 2011, 472, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Jafri, F.; Fink, J.; Higgins, R.R.; Tervo, R. 22q13.32 deletion and duplication and inversion in the same family: A rare occurrence. ISRN Pediatr. 2011, 2011, 829825. [Google Scholar] [CrossRef] [PubMed]
- Zwanenburg, R.J.; Ruiter, S.A.J.; van Den Heuvel, E.R.; Flapper, B.C.T.; van Ravenswaaij-Arts, C.M.A. Developmental phenotype in Phelan-McDermid (22q13.3 deletion) syndrome: A systematic and prospective study in 34 children. J. Neurodev. Disord. 2016, 8, 16. [Google Scholar] [CrossRef] [PubMed]
- Marshall, C.R.; Noor, A.; Vincent, J.B.; Lionel, A.C.; Feuk, L.; Skaug, J.; Shago, M.; Moessner, R.; Pinto, D.; Ren, Y.; et al. Structural variation of chromosomes in autism spectrum disorder. Am. J. Hum. Genet. 2008, 82, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Hanson, E.; Nasir, R.H.; Fong, A.; Lian, A.; Hundley, R.; Shen, Y.; Wu, B.L.; Holm, I.A.; Miller, D.T. 16p11.2 Study group clinicians cognitive and behavioral characterization of 16p11.2 deletion syndrome. J. Dev. Behav. Pediatr. 2010, 31, 649–657. [Google Scholar] [CrossRef] [PubMed]
- Weiss, L.A.; Shen, Y.; Korn, J.M.; Arking, D.E.; Miller, D.T.; Fossdal, R.; Saemundsen, E.; Stefansson, H.; Ferreira, M.A.; Green, T.; et al. Consortium association between microdeletion and microduplication at 16p11.2 and autism. New Engl. J. Med. 2008, 358, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.A.; KaraMohamed, S.; Sudi, J.; Conrad, D.F.; Brune, C.; Badner, J.A.; Gilliam, T.C.; Nowak, N.J.; Cook, E., Jr.; Dobyns, W.B.; et al. Recurrent 16p11.2 microdeletions in autism. Hum. Mol. Genet. 2008, 17, 628–638. [Google Scholar] [CrossRef] [PubMed]
- Fombonne, E.; Du Mazaubrun, C.; Cans, C.; Grandjean, H. Autism and associated medical disorders in a French epidemiological survey. J. Am. Child Adolesc. Psychiatry 1997, 36, 1561–1569. [Google Scholar]
- Steinman, K.J.; Spence, S.J.; Ramocki, M.B.; Proud, M.B.; Kessler, S.K.; Marco, E.J.; Green Snyder, L.; D’Angelo, D.; Chen, Q.; Chung, W.K.; et al. 16p11.2 Deletion and duplication: Characterizing neurologic phenotypes in a large clinically ascertained cohort. Am. J. Med. Genet. 2016, 9999, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Green Snyder, L.; D’Angelo, D.; Chen, Q.; Bernier, R.; Goin-Kochel, R.P.; Wallace, A.S.; Gerdts, J.; Kanne, S.; Berry, L.; Blaskey, L.; et al. Autism spectrum disorder, developmental and psychiatric features in 16p11.2 duplication. J. Autism Dev. Disord. 2016, 46, 2734–2748. [Google Scholar] [CrossRef] [PubMed]
- Fisch, G.S.; Grossfeld, P.; Falk, R.; Battaglia, A.; Youngblom, J.; Simensen, R. Cognitive-behavioral features of Wolf-Hirschhorn syndrome and other subtelomeric microdeletions. Am. J. Med. Genet. 2010, 154, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Fisch, G.S.; Davis, R.; Youngblom, J.; Gregg, J. Genotype-phenotype association studies of chromosome 8p inverted duplication deletion syndrome. Behav. Genet. 2011, 41, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Bregmen, J.D.; Volkmar, F.R. Autism spectrum disorders in genetic syndromes: Implications for diagnosis, intervention and understanding the wider autism spectrum disorder population. J. Intellect. Disabil. Res. 1988, 53, 852–873. [Google Scholar]
- Mariner, R.; Jackson, A.W.; Levitas, A.; Hagerman, R.J.; Braden, M.; McBogg, P.M.; Smith, A.C.; Berry, R. Autism, mental retardation, and chromosomal abnormalities. J. Autism Dev. Disord. 1986, 16, 425–440. [Google Scholar] [CrossRef] [PubMed]
- Ghaziuddin, M. Autism in down syndrome: Family history correlates. J. Intellect. Disabil. Res. 1997, 41, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Moss, J.; Howlin, P. Autism spectrum disorders in genetic syndromes: Implications for diagnosis, intervention and understanding the wider autism spectrum disorder population. J. Intellect. Disabil. Res. 2009, 53, 852–873. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Jing, Y.; Cost, G.J.; Chiang, J.C.; Kolpa, H.J.; Cotton, A.M.; Carone, D.M.; Carone, B.R.; Shivak, D.A.; Guschin, D.Y.; et al. Translating dosage compensation to trisomy 21. Nature 2013, 500, 296–300. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.C.; McGavran, L.; Robinson, J.; Waldstein, G.; Macfarlane, J.; Zonona, J.; Reiss, J.; Lahr, M.; Allen, L.; Magenis, E. Interstitial deletion of (17) (P11.2p11.2) in nine patients. Am. J. Hum. Genet. 1986, 24, 393–414. [Google Scholar] [CrossRef] [PubMed]
- Vostanis, P.; Harrington, R.; Prendergast, M.; Farndon, P. Case reports of autism with interstitial deletion of chromosome 17 (p11.2 p11.2) and monosomy of chromosome 5 (5pter > 5p15.3). Psychiatr. Genet. 1994, 4, 109–111. [Google Scholar] [CrossRef] [PubMed]
- Laje, G.; Morse, R.; Richter, W.; Ball, J.; Pao, M.; Smith, A.C.M. Autism spectrum features in smith-magenis syndrome. Am. J. Med. Genet. 2010, 154, 456–462. [Google Scholar] [CrossRef] [PubMed]
- Potocki, L.; Bi, W.; Treadwell-Deering, D.; Carvalho, C.M.; Eifert, A.; Friedman, E.M.; Glaze, D.; Krull, K.; Lee, J.A.; Lewis, R.A.; et al. Characterization of Potocki-Lupski syndrome (dup(17) (p11.2p11.2)) and delineation of a dosage-sensitive critical interval that can convey an autism phenotype. Am. J. Med. Genet. 2007, 80, 633–649. [Google Scholar] [CrossRef]
- Ghaziuddin, M.; Burmeister, M. Deletion of chromosome 2q37 and autism: A distinct subtype? J. Autism Dev. Disord. 1999, 29, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Devillard, F.; Guinchat, V.; Moreno-De-Luca, D.; Tabet, A.C.; Gruchy, N.; Guillem, P.; Nguyen Morel, M.A.; Leporrier, N.; Leboyer, M.; Jouk, P.S.; et al. Paracentric inversion of chromosome 2 associated with cryptic duplication of 2q14 and deletion of 2q37 in a patient with autism. Am. J. Med. Genet. 2010, 152, 2346–2354. [Google Scholar] [CrossRef] [PubMed]
- Galasso, C.; Lo-Castro, A.; Lalli, C.; Nardone, A.M.; Gullotta, F.; Curatolo, P. Deletion 2q37: An identifiable clinical syndrome with mental retardation and autism. J. Child Neurol. 2008, 23, 802–806. [Google Scholar] [CrossRef] [PubMed]
- Falk, R.E.; Casas, K.A. Chromosome 2q37 deletion: Clinical and molecular aspects. Am. J. Med. Genet. 2007, 145, 357–371. [Google Scholar] [CrossRef] [PubMed]
- Fisch, G.S.; Falk, R.E.; Carey, J.C.; Imitola, J.; Sederberg, M.; Caravalho, K.S.; South, S. Deletion 2q37 syndrome: Cognitive-Behavioral trajectories and autistic features related to breakpoint and deletion size. Am. J. Med. Genet. 2016, 170, 2282–2291. [Google Scholar] [CrossRef] [PubMed]
- Lo-Castro, A.; Galasso, C.; Cerminara, C.; El-Malhany, N.; Benedetti, S.; Nardone, A.M.; Curatolo, P. Association of syndromic mental retardation and autism with 22q11.2 duplication. Neuropediatrics 2009, 40, 137–140. [Google Scholar] [CrossRef] [PubMed]
- Lo-Castro, A.; Benvenuto, A.; Galasso, C.; Porfirio, C.; Curatolo, P. Autism spectrum disorders associated with chromosomal abnormalities. Res. Autism Spectr. Disord. 2010, 4, 319–327. [Google Scholar] [CrossRef]
- Portnoï, M.F. Microduplication 22q11.2: A new chromosomal syndrome. Eur. J. Med. Genet. 2009, 52, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Baker, K.D.; Skuse, D.H. Adolescents and young adults with 22q11 deletion syndrome: Psychopathology in an at-risk group. Br. J. Psychiatry 2005, 186, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Wenger, T.L.; Miller, J.S.; DePolo, L.M.; de Marchena, A.B.; Clements, C.C.; Emanuel, B.S.; Zackai, E.H.; McDonald-McGinn, D.M.; Schultz, R.T. 22q11.2 duplication syndrome: Elevated rate of autism spectrum disorder and need for medical screening. Mol. Autism 2016, 7, 27. [Google Scholar] [CrossRef] [PubMed]
- Hidding, E.; Swaab, H.; de Sonneville, L.M.J.; van Engeland, H.; Vorstman, J.A.S. The role of COMT and plasma proline in the variable penetrance of autistic spectrum symptoms in 22q11.2 deletion syndrome. Clin. Genet. 2016, 90, 420–427. [Google Scholar] [CrossRef] [PubMed]
- Mefford, H.C.; Sharp, A.J.; Baker, C.; Itsara, A.; Jiang, Z.; Buysse, K.; Huang, S.; Maloney, V.K.; Crolla, J.A.; Baralle, D.; et al. Recurrent rearrangements of chromosome 1q21.1 and variable pediatric phenotypes. N. Engl. J. Med. 2008, 359, 1685–1699. [Google Scholar] [CrossRef] [PubMed]
- Brunetti-Pierri, N.; Berg, J.S.; Scaglia, F.; Belmont, J.; Bacino, C.A.; Sahoo, T.; Lalani, S.R.; Graham, B.; Lee, B.; Shinawi, M.; et al. Recurrent reciprocal 1q21.1 deletions and duplications associated with microcephaly or macrocephaly and developmental and behavioral abnormalities. Nat. Genet. 2008, 40, 1466–1471. [Google Scholar] [CrossRef] [PubMed]
- Bernier, R.; Steinman, K.J.; Reilly, B.; Wallace, A.S.; Sherr, E.H.; Pojman, N.; Mefford, H.C.; Gerdts, J.; Earl, R.; Hanson, E.; et al. Clinical phenotype of the recurrent 1q21.1 copy-number variant. Gen. Med. 2016, 18, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Torres, F.; Barbosa, M.; Maciel, P. Recurrent copy number variations as risk factors for neurodevelopmental disorders: Critical overview and analysis of clinical implications. J. Med. Genet. 2016, 53, 73–90. [Google Scholar] [CrossRef] [PubMed]
- Berg, J.S.; Brunetti-Pierri, N.; Peters, S.U.; Kang, S.H.; Fong, C.T.; Salamone, J.; Freedenberg, D.; Hannig, V.L.; Prock, L.A.; Miller, D.T.; et al. Speech delay and autism spectrum behaviors are frequently associated with duplication of the 7q11.23 Williams-Beuren syndrome region. Genet. Med. 2007, 9, 427–441. [Google Scholar] [CrossRef] [PubMed]
- Depienne, C.; Heron, D.; Betancur, C.; Benyahia, B.; Trouillard, O.; Bouteiller, D.; Verloes, A.; LeGuern, E.; Leboyer, M.; Brice, A. Autism, language delay and mental retardation in a patient with 7q11 duplication. J. Med. Genet. 2007, 44, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Edelmann, L.; Prosnitz, A.; Pardo, S.; Bhatt, J.; Cohen, N.; Lauriat, T.; Ouchanov, L.; González, P.J.; Manghi, E.R.; Bondy, P.; et al. An atypical deletion of the Williams-Beuren syndrome interval implicates genes associated with defective visuospatial processing and autism. J. Med. Genet. 2007, 44, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Gagliardi, C.; Bonaglia, M.C.; Selicorni, A.; Borgatti, R.; Giorda, R. Unusual cognitive and behavioural profile in a Williams syndrome patient with atypical 7q11.23 deletion. J. Med. Genet. 2003, 40, 526–530. [Google Scholar] [CrossRef] [PubMed]
- Lincoln, A.J.; Searcy, Y.M.; Jones, W.; Lord, C. Social interaction behaviors discriminate young children with autism and Williams syndrome. J. Am. Acad. Child Adolesc. Psychiatry 2007, 46, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Sanders, S.J.; Ercan-Sencicek, A.G.; Hus, V.; Luo, R.; Murtha, M.T.; Moreno-de-Luca, D.; Chu, S.H.; Moreau, M.P.; Gupta, A.R.; Thomson, S.A.; et al. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron 2011, 70, 863–885. [Google Scholar] [CrossRef] [PubMed]
- Somerville, M.J.; Mervis, C.B.; Young, E.J.; Seo, E.J.; del Campo, M.; Bamforth, S.; Peregrine, E.; Loo, W.; Lilley, M.; Pérez-Jurado, L.A.; et al. Severe expressive-language delay related to duplication of the Williams-Beuren locus. N. Engl. J. Med. 2005, 353, 1694–1701. [Google Scholar] [CrossRef] [PubMed]
- Van der Aa, N.; Rooms, L.; Vandeweyer, G.; van den Ende, J.; Reyniers, E.; Fichera, M.; Romano, C.; Delle Chiaie, B.; Mortier, G.; Menten, B.; et al. Fourteen new cases contribute to the characterization of the 7q11.23 microduplication syndrome. Eur. J. Hum. Genet. 2009, 52, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Makeyev, A.V.; Enkhamandakh, B.; Hong, S.H.; Joshi, P.; Shin, D.G.; Bayarsaihan, D. Diversity and complexity in chromatin recognition by TFII-I transcription factors in pluripotent embryonic stem cells and embryonic tissues. PLoS ONE 2012, 7, e44443. [Google Scholar] [CrossRef] [PubMed]
- Tordjman, S.; Anderson, G.M.; Botbol, M.; Toutain, A.; Sarda, P.; Carlier, M.; Saugier-Veber, P.; Baumann, C.; Cohen, D.; Lagneaux, C.; et al. Autistic disorder in patients with Williams-Beuren syndrome: A reconsideration of the Williams-Beuren syndrome phenotype. PLoS ONE 2012, 7, e30778. [Google Scholar] [CrossRef] [PubMed]
- Tordjman, S.; Anderson, G.M.; Cohen, D.; Kermarrec, S.; Carlier, M.; Touitou, Y.; Saugier-Veber, P.; Lagneaux, C.; Chevreuil, C.; Verloes, A. Presence of autism, hyperserotonemia, and severe expressive language impairment in Williams-Beuren syndrome. Mol. Autism 2013, 4, 29. [Google Scholar] [CrossRef] [PubMed]
- Tordjman, S.; Najjar, I.; Bellissant, E.; Anderson, G.M.; Barburoth, M.; Cohen, D.; Jaafari, N.; Schischmanoff, O.; Fagard, R.; Lagdas, I.; et al. Advances in the research of melatonin in autism spectrum disorders: Literature review and new perspectives. Int. J. Mol. Sci. 2013, 14, 20508–20542. [Google Scholar] [CrossRef] [PubMed]
- Richards, C.; Jones, C.; Groves, L.; Moss, J.; Oliver, C. Prevalence of autism spectrum disorder phenomenology in genetic disorders: A systematic review and meta-analysis. Lancet Psychiatry 2015, 2, 909–916. [Google Scholar] [CrossRef]
- Skuse, D.H.; James, R.S.; Bishop, D.V.; Coppin, B.; Dalton, P.; Aamodt-Leeper, G.; Bacarese-Hamilton, M.; Creswell, C.; McGurk, R.; Jacobs, P.A. Evidence from Turner’s syndrome of an imprinted X-linked locus affecting cognitive function. Nature 1997, 387, 705–708. [Google Scholar] [CrossRef] [PubMed]
- Sybert, V.P.; McCauley, E. Turner’ syndrome. N. Engl. J. Med. 2004, 351, 1227–1238. [Google Scholar] [CrossRef] [PubMed]
- Kent, L.; Bowdin, S.; Kirby, G.A.; Cooper, W.N.; Maher, E.R. Beckwith Wiedemann syndrome: A behavorial phenotype-genotype study. Am. Med. Genet. 2008, 147, 1295–1297. [Google Scholar] [CrossRef] [PubMed]
- DeBaun, M.R.; Niemitz, E.L.; Feinberg, A.P. Association of in vitro fertilization with Beckwith-Wiedemann syndrome and epigenetic alterations of LIT1 and H19. Am. J. Hum. Genet. 2003, 72, 156–160. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.C.; Choufani, S.; Ferreira, J.C.; Weksberg, R. Growth regulation, imprinted genes, and chromosome 11p15.5. Pediatr. Res. 2007, 61, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Weksberg, R.; Shuman, C.; Beckwith, J.B. Beckwith-Wiedemann syndrome. Eur. J. Hum. Genet. 2010, 18, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Wolpert, C.M.; Menold, M.M.; Bass, M.P.; Qumsiyeh, M.B.; Donnelly, S.L.; Ravan, S.A.; Vance, J.M.; Gilbert, J.R.; Abramson, R.K.; Wright, H.H.; et al. Three probands with autistic disorder and isodicentric chromosome 15. Am. J. Med. Genet. 2000, 96, 365–372. [Google Scholar] [CrossRef]
- Wolpert, C.; Pericak-Vance, M.A.; Abramson, R.K.; Wright, H.H.; Cuccaro, M.L. Autistic symptoms among children and young adults with isodicentric chromosome 15. Am. J. Med. Genet. 2000, 96, 128–129. [Google Scholar] [CrossRef]
- Parisi, L.; di Filippo, T.; Roccella, M. Hypomelanosis of ito: Neurological and psychiatric pictures in developmental age. Minerva Pediatr. 2012, 64, 65–70. [Google Scholar] [PubMed]
- Åkefeldt, A.; Gillberg, C. Hypomelanosis of ito in three cases with autism and autistic-like conditions. Dev. Med. Child Neurol. 1991, 33, 737–743. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.; Gillberg, C.; Råstam, M. Autism spectrum conditions in individuals with Möbius sequence, CHARGE syndrome and oculo-auriculo-vertebral spectrum: Diagnostic aspects. Res. Dev. Disabil. 2010, 31, 9–24. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.; Råstam, M.; Billstedt, E.; Danielsson, S.; Strömland, K.; Miller, M.; Gillberg, C. Autism spectrum disorders and underlying brain pathology in CHARGE association. Dev. Med. Child Neurol. 2006, 48, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Smith, I.M.; Nichols, S.L.; Issekutz, K.; Blake, K. Canadian paediatric surveillance program behavioral profiles and symptoms of autism in CHARGE syndrome: Preliminary Canadian epidemiological data. Am. J. Med. Genet. 2005, 133, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Hartshorne, T.S.; Grialou, T.L.; Parker, K.R. Autistic-like behavior in CHARGE syndrome. Am. J. Med. Genet. 2005, 133, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Patten, S.A.; Jacobs-McDaniels, N.L.; Zaouter, C.; Drapeau, P.; Albertson, R.C.; Moldovan, F. Role of Chd7 in zebrafish: A model for CHARGE syndrome. PLoS ONE 2012, 7, e31650. [Google Scholar] [CrossRef] [PubMed]
- Blake, K.D.; Prasad, C. CHARGE syndrome. Orphanet J. Rare Dis. 2006, 1, 34. [Google Scholar] [CrossRef] [PubMed]
- Bolton, P.F.; Park, R.J.; Higgins, J.N.; Griffiths, P.D.; Pickles, A. Neuro-epileptic determinants of autism spectrum disorders in tuberous sclerosis complex. Brain 2002, 125, 1247–1255. [Google Scholar] [CrossRef] [PubMed]
- Curatolo, P.; Bombardieri, R.; Jozwiak, S. Tuberous sclerosis. Lancet 2008, 372, 657–668. [Google Scholar] [PubMed]
- Jeste, S.S.; Varcin, K.J.; Hellemann, G.S.; Gulsrud, A.C.; Bhatt, R.; Kasari, C.; Wu, J.Y.; Sahin, M.; Nelson, C.A. Symptom profiles of autism spectrum disorder in tuberous sclerosis complex. Neurology 2016. [Google Scholar] [CrossRef] [PubMed]
- McBride, K.L.; Varga, E.A.; Pastore, M.T.; Prior, T.W.; Manickam, K.; Atkin, J.F.; Herman, G.E. Confirmation study of PTEN mutations among individuals with autism or developmental delays/mental retardation and macrocephaly. Autism Res. 2010, 3, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Lintas, C.; Persico, A.M. Autistic phenotypes and genetic testing: State-of-the-art for the clinical geneticist. J. Med. Genet. 2009, 46, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Buxbaum, J.D.; Cai, G.; Chaste, P.; Nygren, G.; Goldsmith, J.; Reichert, J.; Anckarsäter, H.; Rastam, M.; Smith, C.J.; Silverman, J.M.; et al. Mutation screening of the PTEN gene in patients with autism spectrum disorders and macrocephaly. Am. J. Med. Genet. 2007, 144, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Herman, G.E.; Butter, E.; Enrile, B.; Pastore, M.; Prior, T.W.; Sommer, A. Increasing knowledge of PTEN germline mutations: Two additional patients with autism and macrocephaly. Am. J. Med. Genet. 2007, 143, 589–593. [Google Scholar] [CrossRef] [PubMed]
- Goffin, A.; Hoefsloot, L.H.; Bosgoed, E.; Swillen, A.; Fryns, J.P. PTEN mutation in a family with Cowden syndrome and autism. Am. J. Med. Genet. 2001, 105, 521–524. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, L.; Black, F.M.; Berg, J.N.; Eickholt, B.J.; Leslie, N.R. Functionally distinct groups of inherited PTEN mutations in autism and tumour syndromes. J. Med. Genet. 2015, 52, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Amir, R.E.; Van Den Veyver, I.B.; Wan, M.; Tran, C.Q.; Francke, U.; Zoghbi, H.Y. Rett syndrome is cause by mutations in X-linked MECP2, encoding methil-CpG-binding protein 2. Nat. Genet. 1999, 23, 185–188. [Google Scholar] [PubMed]
- Olsonn, B.; Rett, A. A review of the Rett syndrome with a theory of autism. Brain Dev. 1990, 12, 11–15. [Google Scholar] [CrossRef]
- Young, D.J.; Bebbington, A.; Anderson, A.; Ravine, D.; Ellaway, C.; Kulkarni, A.; de Klerk, N.; Kaufmann, W.E.; Leonard, H. The diagnosis of autism in a female: Could it be Rett syndrome? Eur. J. Pediatr. 2008, 167, 661–669. [Google Scholar] [CrossRef] [PubMed]
- Neul, J.L. The relationship of Rett syndrome and MECP2 disorders to autism. Dialogues Clin. Neurosci. 2012, 14, 253–262. [Google Scholar] [PubMed]
- Gadalla, K.; Bailey, M.E.; Cobb, S.R. MeCP2 and Rett syndrome: Reversibility and potential avenues for therapy. Biochem. J. 2011, 439, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Neul, J.L.; Kaufmann, W.E.; Glaze, D.G.; Christodoulou, J.; Angus, J.; Clarke, A.J.; Bahi-Buisson, N.; Leonard, H.; Bailey, M.E.S.; Schanen, C.; et al. Rett syndrome: Revised diagnostic criteria and nomenclature. Ann. Neurol. 2010, 68, 944–950. [Google Scholar] [CrossRef] [PubMed]
- Ramocki, M.B.; Peters, S.U.; Tavyev, Y.J.; Zhang, F.; Carvalho, C.M.; Schaaf, C.P.; Richman, R.; Fang, P.; Glaze, D.G.; Lupski, J.R.; et al. Autism and other neuropsychiatric symptoms are prevalent in individuals with MeCP2 duplication syndrome. Ann. Neurol. 2009, 66, 771–782. [Google Scholar] [CrossRef] [PubMed]
- Nidiffer, F.D.; Kelly, T.E. Developmental and degenerative patterns associated with cognitive, behavioural and motor difficulties in the Sanfilippo syndrome: An epidemiological study. J. Ment. Defic. Res. 1983, 27, 185–203. [Google Scholar] [CrossRef] [PubMed]
- Wraith, J.E. The mucopolysaccharidoses: A clinical review and guide to management. Arch. Dis. Child. 1995, 72, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Ritvo, E.R.; Mason-Brothers, A.; Freeman, B.J.; Pingree, C.; Jenson, W.R.; McMahon, W.M.; Petersen, P.B.; Jorde, L.B.; Mo, A.; Ritvo, A. The UCLA-university of Utah epidemiologic survey of autism: The etiologic role of rare diseases. Am. J. Psychiatry 1990, 147, 1614–1621. [Google Scholar] [PubMed]
- Shapiro, E.; King, K.; Ahmed, A.; Rudser, K.; Rumsey, R.; Yund, B.; Delaney, K.; Nestrasil, I.; Whitley, C.; Potegal, M. The neurobehavioral phenotype in mucopolysaccharidosis type IIIB: An exploratory study. Mol. Genet. Metab. Rep. 2016, 6, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Ramaekers, V.T.; Blau, N. Cerebral folate deficiency. Dev. Med. Child Neurol. 2004, 46, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Moretti, P.; Scaglia, F. Cerebral folate deficiency with developmental delay, autism, and response to folinic acid. Neurology 2005, 64, 1088–1090. [Google Scholar] [CrossRef] [PubMed]
- Gordon, N. Cerebral folate deficiency. Dev. Med. Child Neurol. 2009, 51, 180–182. [Google Scholar] [CrossRef] [PubMed]
- Raidah, S.A.; Mohammed, W.C. Diagnosis and management of cerebral folate deficiency. Neurosciences 2014, 19, 312–316. [Google Scholar]
- Ryan, A.K.; Bartlett, K.; Clayton, P.; Eaton, S.; Mills, L.; Donnai, D.; Winter, R.M.; Burn, J. Smith-Lemli-Opitz syndrome: A variable clinical and biochemical phenotype. J. Med. Genet. 1998, 35, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Kelley, R.I.; Hennekam, R.C.M. The Smith-Lemli-Opitz syndrome. J. Med. Genet. 2000, 37, 321–335. [Google Scholar] [CrossRef] [PubMed]
- Tierny, E.; Nwokoro, N.A.; Porter, F.D.; Freund, L.S.; Ghuman, J.K.; Kelley, R.I. Behavior phenotype in the RSH/ Smith-Lemli-Opitz syndrome. Am. J. Med. Genet. 2001, 98, 191–200. [Google Scholar] [CrossRef]
- Tierny, E.; Nwokoro, N.A.; Kelley, R.I. Behavior phenotype of RSH/Smith-Lemli-Opitz syndrome. Ment. Retard. Dev. Disabil. Res. Rev. 2001, 98, 191–200. [Google Scholar]
- Tint, G.S.; Irons, M.; Elias, E.R.; Batta, A.K.; Frieden, R.; Chen, T.S.; Salen, G. Defective cholesterol biosynthesis associated with the Smith-Lemli-Opitz syndrome. N. Engl. J. Med. 1994, 330, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Thurm, A.; Tierney, E.; Farmer, C.; Albert, P.; Joseph, L.; Swedo, S.; Bianconi, S.; Bukelis, I.; Wheeler, C.; Sarphare, G.; et al. Development, behavior, and biomarker characterization of Smith-Lemli-Opitz syndrome: An update. J. Neurodev. Disord. 2016, 8, 12. [Google Scholar] [CrossRef] [PubMed]
- Baieli, S.; Pavone, L.; Meli, C.; Fiumara, A.; Coleman, M. Autism and phenylketonuria. J. Autism Dev. Disord. 2003, 33, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Miladi, N.; Larnaout, A.; Kaabachi, N.; Helayem, M.; Ben Hamida, M. Phenylketonuria: An underlying etiology of autistic syndrome report. J. Child Neurol. 1992, 7, 22–23. [Google Scholar] [CrossRef] [PubMed]
- Fon, E.A.; Sarrazin, J.; Meunier, C.; Alarcia, J.; Shevell, M.I.; Philippe, A.; Leboyer, M.; Rouleau, G.A. Adenylosuccinate lyase (ADSL) and infantile autism: Absence of previously reported point mutation. Am. J. Hum. Genet. 1995, 18, 554–557. [Google Scholar] [CrossRef] [PubMed]
- Jaeken, J.; van den Berghe, G. An infantile autistic syndrome characterized by the presence of succinylpurines in body fluids. Lancet 1984, 2, 1058–1061. [Google Scholar] [PubMed]
- Race, V.; Marie, S.; Vincent, M.F.; van der Berghe, G. Clinical, biochemical and molecular genetic correlations in adenylosuccinate lyase deficiency. Hum. Mol. Genet. 2000, 9, 2159–2163. [Google Scholar] [CrossRef] [PubMed]
- Stone, R.L.; Aimi, J.; Barshop, B.A.; Jaeken, J.; van den Berghe, G.; Zalkin, H.; Dixon, J.E. A mutation in adenylosuccinate lyase associated with mental retardation autistic features. Nat. Genet. 1992, 1, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Jinnah, H.A.; Sabina, R.L.; Van Den Berghe, G. Metabolic disorders of purine metabolism affecting the nervous system. Handb. Clin. Neurol. 2013, 113, 1827–1836. [Google Scholar] [PubMed]
- Nasrallah, F.; Feki, M.; Kaabachi, N. Creatine and creatine deficiency syndromes: Biochemical and clinical aspects. Pediatr. Neurol. 2010, 42, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Póo-Argüelles, P.; Arias, A.; Vilaseca, M.A.; Ribes, A.; Artuch, R.; Sans-Fito, A.; Moreno, A.; Jakobs, C.; Salomons, G. X-linked creatine transporter deficiency in two patients with severe mental retardation and autism. J. Inherit. Metab. Dis. 2006, 29, 220–223. [Google Scholar] [CrossRef] [PubMed]
- Schulze, A.; Bauman, M.; Tsai, A.C.; Reynolds, A.; Roberts, W.; Anagnostou, E.; Cameron, J.; Nozzolillo, A.A.; Chen, S.; Kyriakopoulou, L.; et al. Prevalence of creatine deficiency syndromes in children with nonsyndromic autism. Pediatrics 2016. [Google Scholar] [CrossRef] [PubMed]
- Campistol, J.; Díez-Juan, M.; Callejón, L.; Fernandez-De Miguel, A.; Casado, M.; Garcia Cazorla, A.; Lozano, R.; Artuch, R. Inborn error metabolic screening in individuals with nonsyndromic autism spectrum disorders. Dev. Med. Child Neurol. 2016, 58, 842–847. [Google Scholar] [CrossRef] [PubMed]
- Durand, C.M.; Betancur, C.; Boeckers, T.M.; Bockmann, J.; Chaste, P.; Fauchereau, F.; Nygren, G.; Rastam, M.; Gillberg, I.C.; Anckarsäter, H.; et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat. Genet. 2007, 39, 25–27. [Google Scholar] [CrossRef] [PubMed]
- Moessner, R.; Marshall, C.R.; Sutcliffe, J.S.; Skaug, J.; Pinto, D.; Vincent, J.; Zwaigenbaum, L.; Fernandez, B.; Roberts, W.; Szatmari, P.; et al. Contribution of SHANK3 mutations to autism spectrum disorder. Am. J. Hum. Genet. 2007, 81, 1289–1297. [Google Scholar] [CrossRef] [PubMed]
- Guiltmatre, A.; Huquet, G.; Delorme, R.; Bourgeron, T. The emerging role of SHANK genes in neuropsychiatric disorders. Dev. Neurobiol. 2014, 74, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Schroer, R.; Yan, J.; Song, W.; Yang, C.; Bockholt, A.; Cook, E.H., Jr.; Skinner, C.; Schwartz, C.E.; Sommer, S.S. High frequency of neurexin 1β signal peptide structural variants in patients with autism. Neurosci. Lett. 2006, 27, 10–13. [Google Scholar] [CrossRef] [PubMed]
- Ching, M.S.; Shen, Y.; Tan, W.H.; Jeste, S.S.; Morrow, E.M.; Chen, X.; Mukaddes, N.M.; Yoo, S.Y.; Hanson, E.; Hundley, R.; et al. Deletions of NRXN1 (neurexin-1) predispose to a wide spectrum of developmental disorders. Am. J. Med. Genet. 2010, 153, 937–947. [Google Scholar] [CrossRef] [PubMed]
- Autism Genome Project Consortium. Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat. Genet. 2007, 39, 319–328. [Google Scholar]
- Viñas-Jornet, M.; Esteba-Castillo, S.; Gabau, E.; Ribas-Vidal, N.; Baena, N.; San, J.; Ruiz, A.; Coll, M.D.; Novell, R.; Guitart, M. A common cognitive, psychiatric and dysmorphic phenotype in carriers of NRXN1 deletion. Mol. Genet. Genom. Med. 2014, 2, 512–521. [Google Scholar] [CrossRef] [PubMed]
- Dabell, M.P.; Rosenfeld, J.A.; Bader, P.; Escobar, L.F.; El-Khechen, D.; Vallee, S.E.; Dinulos, M.B.P.; Curry, C.; Fisher, J.; Tervo, R.; et al. Investigation of NRXN1 deletions: Clinical and molecular characterization. Am. J. Med. Genet. 2013, 161, 717–731. [Google Scholar] [CrossRef] [PubMed]
- Alarcon, M.; Abrahams, B.S.; Stone, J.L. Linkage, association, and gene-expression analyses identify CNTNAP2 as an autism-susceptibility gene. Am. J. Med. Genet. 2008, 82, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Werling, A.M.; Bobrowski, E.; Taurines, R.; Gundelfinger, R.; Romanos, M.; Grünblatt, E.; Walitza, S. CNTNAP2 gene in high functioning autism: No association according to family and meta-analysis approaches. J. Neural Transm. 2016, 123, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, J.D.; Gupta, A.R.; Sanders, S.J.; Walker, M.F.; Keaney, J.; Fernandez, T.V.; Murtha, M.T.; Anyanwu, S.; Ober, G.T.; Raubeson, M.J.; et al. No evidence for association of autism with rare heterozygous point mutations in contactin-associated protein-like 2 (CNTNAP2), or in other contactin-associated proteins or contactins. PLoS Genet. 2015, 11, e1004852. [Google Scholar] [CrossRef] [PubMed]
- Bakkaloglu, B.; O’Roak, B.J.; Louvi, A.; Gupta, A.R.; Abelson, J.F.; Morgan, T.M.; Chawarska, K.; Klin, A.; Ercan-Sencicek, A.G.; Stillman, A.A.; et al. Molecular cytogenetic analysis and resequencing of contactin associated protein-like 2 in autism spectrum disorders. Am. J. Hum. Genet. 2008, 82, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, T.; Morgan, T.; Davis, N.; Klin, A.; Morris, A.; Farhi, A.; Lifton, R.P.; State, M.W. Disruption of contactin 4 (CNTN4) results in developmental delay and other features of 3p deletion syndrome. Am. J. Hum. Genet. 2004, 74, 1286–1293. [Google Scholar] [CrossRef] [PubMed]
- Roohi, J.; Montagna, C.; Tegay, D.H.; Palmer, L.E.; DeVincent, C.; Pomeroy, J.C.; Christian, S.L.; Nowak, N.; Hatchwell, E. Disruption of contactin 4 in three subjects with autism spectrum disorder. J. Med. Genet. 2008, 46, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Karayannis, T.; Au, E.; Patel, J.C.; Kruglikov, I.; Markx, S.; Delorme, R.; Héron, D.; Salomon, D.; Glessner, J.; Restituito, S.; et al. Cntnap4 differentially contributes to GABAergic and dopaminergic synaptic transmission. Nature 2014, 511, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Zhiling, Y.; Fujita, E.; Tanabe, Y. Mutations in the gene encoding CADM1 are associated with autism spectrum disorder. Biochem. Biophys. Res. Commun. 2008, 377, 926–929. [Google Scholar] [CrossRef] [PubMed]
- Fujita, E.; Tanabe, Y.; Imhof, B.A.; Mamsi, M.Y.; Momoi, T. A complex of synaptic adhesion molecule CADM1, a molecule related to autism spectrum disorder, with MUPP1 in the cerebellum. J. Neurochem. 2012, 123, 886–894. [Google Scholar] [CrossRef] [PubMed]
- Kitagishi, Y.; Minami, A.; Nakanishi, A.; Ogura, Y.; Matsuda, S. Neuron membrane trafficking and protein kinases involved in autism and ADHD. Int. J. Mol. Sci. 2015, 16, 3095–3115. [Google Scholar] [CrossRef] [PubMed]
- Morrow, E.M.; Yoo, S.Y.; Flavell, S.W.; Kim, T.K.; Lin, Y.; Hill, R.S.; Mukaddes, N.M.; Balkhy, S.; Gascon, G.; Hashmi, A.; et al. Identifying autism loci and genes by tracing recent shared ancestry. Science 2008, 321, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Jamain, S.; Quach, H.; Betancur, C.; Rastam, M.; Colineaux, C.; Gillberg, I.C.; Soderstrom, H.; Giros, B.; Leboyer, M.; Gillberg, C.; et al. Mutations of the X-linked genes encodage neuroligins NLGN3 and NLGN4 are associated with autism. Nat. Genet. 2003, 34, 27–29. [Google Scholar] [CrossRef] [PubMed]
- Laumonnier, F.; Bonnet-Brilhault, F.; Gomot, M.; Blanc, R.; David, A.; Moizard, M.P.; Raynaud, M.; Ronce, N.; Lemonnier, E.; Calvas, P.; et al. X-linked mental retardation and autism are associated with a mutation in the NLGN4 gene, a member of the neuroligin family. Am. J. Hum. Genet. 2004, 74, 552–557. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Oliveira, G.; Coutinho, A.; Yang, C.; Feng, J.; Katz, C.; Sram, J.; Bockholt, A.; Jones, I.R.; Craddock, N.; et al. Analysis of the neuroligin 3 and 4 genes in autism and other neuropsychiatric patients. Mol. Psychiatry 2005, 10, 329–332. [Google Scholar] [CrossRef] [PubMed]
- Fisch, G.S. Is autism associated with the fragile X syndrome? Am. J. Med. Genet. 1992, 43, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Fisch, G.S. What is associated with the fragile X syndrome? Am. J. Med. Genet. 1993, 48, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Rogers, S.J.; Wehner, D.E.; Hagerman, R. The behavioral phenotype in fragile X: Symptoms of autism in very young children with fragile X syndrome, idiopathic autism, and other developmental disorders. J. Dev. Behav. Pediatr. 2001, 22, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Bailey, D.B., Jr.; Hatton, D.D.; Skinner, M.; Mesibov, G. Autistic behavior, FMR1 protein, and developmental trajectories in young males with fragile X syndrome. J. Autism Dev. Disord. 2001, 31, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Belmonte, M.K.; Bourgeron, T. Fragile X syndrome and autism at the intersection of genetic and neural netwoks. Nat. Neurosci. 2006, 9, 1221–1225. [Google Scholar] [CrossRef] [PubMed]
- Kau, A.S.; Tierney, E.; Bukelis, I.; Stump, M.H.; Kates, W.R.; Trescher, W.H.; Kaufmann, W.E. Social behavior profile in young males with fragile X syndrome: Characteristics and specificity. Am. J. Med. Genet. 2004, 126, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Loesch, D.Z.; Bui, Q.M.; Dissanayake, C.; Clifford, S.; Gould, E.; Bulhak-Paterson, D.; Tassone, F.; Taylor, A.K.; Hessl, D.; Hagerman, R.; et al. Molecular and cognitive predictors of the continuum of autistic behaviours in fragile X. Neurosci. Biobehav. Rev. 2007, 31, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Clifford, S.; Dissanayake, C.; Bui, Q.M.; Huggins, R.; Taylor, A.K.; Loesch, D.Z. Autism spectrum phenotype in males and females with fragike X full mutation and premutation. J. Autism Dev. Disord. 2007, 337, 738–747. [Google Scholar] [CrossRef] [PubMed]
- Harris, S.W.; Hessl, D.; Goodlin-Jones, B.; Ferranti, J.; Bacalman, S.; Barbato, I.; Tassone, F.; Hagerman, P.J.; Herman, H.; Hagerman, R.J. Autism profiles of males with fragile X syndrome. Am. J. Ment. Retard. 2008, 113, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Dölen, G.; Carpenter, R.L.; Ocain, T.D.; Bear, M.F. Mechanism-based approaches to treating fragile X. Pharmacol. Ther. 2010, 127, 78–93. [Google Scholar] [CrossRef] [PubMed]
- McLennan, Y.; Polussa, J.; Tassone, F.; Hagerman, R. Fragile X syndrome. Curr. Genom. 2011, 12, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Bar-Nur, O.; Caspi, I.; Benvenisty, N. Molecular analysis of FMR 1 reactivation in fragile-X induced pluripotent stem cells and their neuronal derivatives. J. Mol. Cell. Biol. 2012, 4, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Hagerman, R.; Lauterborn, J.; Au, J.; Berry-Kravis, E. Fragile X syndrome and targeted treatment trials. Results Probl. Cell Differ. 2012, 54, 297–335. [Google Scholar] [PubMed]
- Tabolacci, E.; Chiurazzi, P. Epignetics, fragile X syndrome and transcriptional therapy. Am. J. Med. Genet. 2013, 161, 2797–2808. [Google Scholar] [CrossRef] [PubMed]
- De Vries, B.B.; Halley, D.J.; Oostra, B.A.; Niermeijer, C. The fragile X syndrome. Am. J. Med. Genet. 1998, 35, 579–589. [Google Scholar] [CrossRef]
- Tolmie, J. The genetics of mental retardation. Curr. Opin. Psychiatry 1998, 11, 507–513. [Google Scholar] [CrossRef]
- Hagerman, R.J. Fragile X syndrome. Child Adolesc. Psychiatr. Clin. N. Am. 1996, 5, 895–911. [Google Scholar] [CrossRef]
- Hagerman, R.J.; Chudley, A.E.; Knoll, J.H.; Jackson, A.W.; Kemper, M.; Ahmad, R. Autism in fragile X females. Am. J. Med. Genet. 1986, 23, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Hatton, D.D.; Sideris, J.; Skinner, M.; Mankowski, J.; Bailey, D.B., Jr.; Roberts, J.; Mirrett, P. Autistic behavior in children with fragile X syndrome: Prevalence, stability, and the impact of FMRP. Am. J. Med. Genet. 2006, 140, 1804–1813. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.E.; Tonnsen, B.L.; McCary, L.M.; Caravella, K.E.; Shinkareva, S.V. Brief report: Autism symptoms in infants with fragile X syndrome. J. Autism Dev. Disord. 2016, 46, 3830–3837. [Google Scholar] [CrossRef] [PubMed]
- Einfeld, S.; Molony, H.; Hall, W. Autism is not associated with the fragile X syndrome. Am. J. Med. Genet. 1989, 34, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Reiss, A.L.; Freund, L. Behavioral phenotype of fragile X syndrome; DSM-III-R autistic behavior in male children. Am. J. Med. Genet. 1992, 43, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Todd, R.D.; Hudziak, J.J. Genetics of autism. Curr. Opin. Psychiatry 1993, 6, 486–488. [Google Scholar] [CrossRef]
- Farzin, F.; Perry, H.; Hessl, D.; Loesch, D.; Cohen, J.; Bacalman, S.; Gane, L.; Tassone, F.; Hagerman, P.; Hagerman, R. Autism spectrum disorders and attention-deficit/hyperactivity disorder in boys with the fragile X premutation. J. Dev. Behav. Pediatr. 2006, 27, 137–144. [Google Scholar] [CrossRef]
- Hagerman, R.J.; Berry-Kravis, E.; Kaufmann, W.E.; Ono, M.Y.; Tartaglia, N.; Lachiewicz, A.; Kronk, R.; Delahunty, C.; Hessl, D.; Visootak, J.; et al. Advances in the treatment of fragile X syndrome. Pediatrics 2009, 123, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Hallmayer, J.; Hebert, J.M.; Spiker, D.; Lotspeich, L.; McMahon, W.M.; Petersen, P.B.; Nicholas, P.; Pingree, C.; Lin, A.A.; Cavalli-Sforza, L.L.; et al. Autism and the X chromosome. Multipoint sib-pair analysis. Arch. Gen. Psychiatry 1996, 53, 985–989. [Google Scholar] [CrossRef] [PubMed]
- Constantino, J.N.; Zhang, Y.; Holzhauer, K.; Sant, S.; Long, K.; Vallorani, A.; Malik, L.; Gutmann, D.H. Distribution and Within-Family specificity of quantitative autistic traits in patients with neurofibromatosis type I. J. Pediatr. 2015, 167, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Garg, S.; Plasschaert, E.; Descheemaeker, M.J.; Huson, S.; Borghgraef, M.; Vogels, A.; Evans, D.G.; Legius, E.; Green, J. Autism spectrum disorder profile in neurofibromatosis type I. J. Autism Dev. Disord. 2015, 45, 1649–1657. [Google Scholar] [CrossRef] [PubMed]
- Plasschaert, E.; Descheemaeker, M.J.; Van Eylen, L.; Noens, I.; Steyaert, J.; Legius, E. Prevalence of autism spectrum disorder symptoms in children with neurofibromatosis type 1. Am. J. Med. Genet. 2014, 168, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Timonen-Soivio, L.; Vanhala, R.; Malm, H.; Hinkka-Yli-Salomäki, S.; Gissler, M.; Brown, A.; Sourander, A. Brief report: Syndromes in autistic children in a Finnish birth cohort. J. Autism Dev. Disord. 2016, 46, 2780–2784. [Google Scholar] [CrossRef] [PubMed]
- Sheth, K.; Moss, J.; Hyland, S.; Stinton, C.; Cole, T.; Oliver, C. The behavioral characteristics of Sotos syndrome. Am. J. Med. Genet. 2015, 167, 2945–2956. [Google Scholar] [CrossRef] [PubMed]
- Assumpcao, F.; Santos, R.C.; Rosario, M.; Mercadante, M. Brief report: Autism and Aarskog syndrome. J. Autism Dev. Disord. 1999, 29, 179–181. [Google Scholar] [CrossRef] [PubMed]
- De Wolf, V.; Crepel, A.; Schuit, F.; van Lommel, L.; Ceulemans, B.; Steyaert, J.; Seuntjens, E.; Peeters, H.; Devriendt, K. A complex Xp11.22 deletion in a patient with syndromic autism: Exploration of FAM120C as a positional candidate gene for autism. Am. J. Med. Genet. 2014, 164, 3035–3041. [Google Scholar] [CrossRef] [PubMed]
- Parisi, L.; Di Filippo, T.; Roccella, M. Behavioral phenotype and autism spectrum disorders in Cornelia de Lange syndrome. Ment. Illn. 2015, 7, 5988. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Landy-Schmitt, C.; Clark, B.; Kline, A.D.; Specht, M.; Grados, M.A. Autism traits in children and adolescents with Cornelia de Lange syndrome. Am. J. Med. Genet. 2014, 321, 1400–1410. [Google Scholar] [CrossRef] [PubMed]
- Alvarez Retuerto, A.I.; Cantor, R.M.; Gleeson, J.G.; Ustaszewska, A.; Schackwitz, W.S.; Pennacchio, L.A.; Geschwind, D.H. Association of common variants in the Joubert syndrome gene (AHI1) with autism. Hum. Mol. Genet. 2008, 17, 3887–3896. [Google Scholar] [CrossRef] [PubMed]
- Ionita-Laza, I.; Capanu, M.; De Rubeis, S.; McCallum, K.; Buxbaum, J.D. Identification of rare causal variants in sequence-based studies: Methods and applications to VPS13B, a gene involved in Cohen syndrome and Autism. PLoS Genet. 2014, 10, e1004729. [Google Scholar] [CrossRef] [PubMed]
- Howlin, P.; Karpf, J.; Turk, J. Behavioural characteristics and autistic features in individuals with Cohen syndrome. Eur. Child Adolesc. Psychiatry 2005, 14, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Chandler, K.E.; Moffett, M.; Clayton-Smith, J.; Baker, G.A. Neuropsychological assessment of a group of UK patients with Cohen syndrome. Neuropediatrics 2003, 34, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Lerma-Carrillo, I.; Molina, J.D.; Cuevas-Duran, T.; Julve-Correcher, C.; Espejo-Saavedra, J.M.; Andrade-Rosa, C.; Lopez-Muñoz, F. Psychopathology in the Lujan-Fryns syndrome: Report of two patients and review. Am. J. Med. Genet. 2006, 140, 2807–2811. [Google Scholar] [CrossRef] [PubMed]
- Alfieri, P.; Piccini, G.; Caciolo, C.; Perrino, F.; Gambardella, M.L.; Mallardi, M.; Cesarini, L.; Leoni, C.; Leone, D.; Fossati, C.; et al. Behavioral profile in RASopathies. Am. J. Med. Genet. 2014, 164, 934–942. [Google Scholar] [CrossRef] [PubMed]
- Adviento, B.; Corbin, I.L.; Widjaja, F.; Desachy, G.; Enrique, N.; Rosser, T.; Risi, S.; Marco, E.J.; Hendren, R.L.; Bearden, C.E.; et al. Autism traits in the RASopathies. J. Med. Genet. 2014, 51, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Ghaziuddin, M.; Bolyard, B.; Alessi, N. Autistic disorder in Noonan syndrome. J. Intellect. Disabil. Res. 1994, 38, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.; Wentz, E.; Fernell, E.; Strömland, K.; Miller, M.T.; Gillberg, C. Autistic spectrum disorders in Moëbius sequence: a comprehensive study of 25 cases. Dev. Med. Child Neurol. 2001, 43, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Briegel, W.; Schimek, M.; Kamp-Becker, I.; Hofmann, C.; Schwab, K.O. Autism spectrum disorders in children and adolescents with Moebius sequence. Eur. Child Adolesc. Psychiatry. 2009, 18, 515–519. [Google Scholar] [CrossRef] [PubMed]
- Helsmoortel, C.; Vulto-van Silfhout, A.T.; Coe, B.P.; Vandeweyer, G.; Rooms, L.; van den Ende, J.; Schuurs-Hoeijmakers, J.H.; Marcelis, C.L.; Willemsen, M.H.; Vissers, L.E.; et al. A SWI/SNF-related autism syndrome caused by de novo mutations in ADNP. Nat. Genet. 2014, 46, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Splawski, I.; Timothy, K.W.; Sharpe, L.M.; Decher, N.; Kumar, P.; Bloise, R.; Napolitano, C.; Schwartz, P.J.; Joseph, R.M.; Condouris, K.; et al. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 2004, 119, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Huguet, G.; Ey, E.; Bourgeron, T. The genetic landscapes of autism spectrum disorders. Annu. Rev. Genom. Hum. Genet. 2013, 14, 191–213. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, G.B. Clinical genetic aspects of autism spectrum sisorders. Int. J. Mol. Sci. 2016, 17, 180. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.; Rutter, M.; Le Couteur, A. Autism diagnostic interview-revised: A revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. J. Autism Dev. Disord. 1994, 24, 659–685. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.; Risi, S.; Lambrecht, L.; Cook, E.; Leventhal, B.; DiValore, P.; Pickles, A.; Rutter, M. The autism diagnostic observation schedule-generic: A standard measures of social and communication deficits associated with the spectrum of autism. J. Autism Dev. Disord. 2000, 30, 205–223. [Google Scholar] [CrossRef] [PubMed]
- Risi, S.; Lord, C.; Gotham, K.; Corsello, C.; Chrysler, C.; Szatmari, P.; Cook, E.H., Jr.; Leventhal, B.L.; Pickles, A. Combining information from multiple sources in the diagnosis of autism spectrum disorders. J. Am. Acad. Child Adolesc. Psychiatry 2006, 45, 1094–1103. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, G.B.; Mendelson, N.J. Clinical genetics evaluation in identifying the etiology of autism spectrum disorders: 2013 guideline revisions. Genet. Med. 2013, 15, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Ponsot, O.; Moutard, M.L.; Villeneuve, N. Orientation Diagnostique en Neurologie Pédiatrique. In Neurologie Pédiatrique, 2nd ed.; Flammarion Médecine-Sciences: Paris, France, 1998; pp. 75–87. [Google Scholar]
- Simonoff, E.; Bolton, P.; Rutter, M. Mental retardation: Genetics findings, clinical implications and research agenda. J. Child Psychol. Psychiatry 1996, 37, 259–280. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, T.J.; Windham, G.C.; Anderson, M.; Croen, L.A.; Grether, J.K.; Risch, N. Evidence of reproductive stoppage in families with autism spectrum disorder: A large, population-based cohort study. JAMA Psychiatry 2014, 71, 943–951. [Google Scholar] [CrossRef] [PubMed]
- Ozonoff, S.; Young, G.S.; Carter, A.; Messinger, D.; Yirmiya, N.; Zwaigenbaum, L.; Bryson, S.; Carver, L.J.; Constantino, J.N.; Dobkins, K.; et al. Recurrence risk for autism spectrum disorders: A baby siblings research consortium study. Pediatrics 2011, 128, 488–495. [Google Scholar] [CrossRef] [PubMed]
- Durkin, M.S.; Maenner, M.J.; Newschaffer, C.J.; Lee, L.C.; Cunniff, C.M.; Daniels, J.L.; Kirby, R.S.; Leavitt, L.; Miller, L.; Zahorodny, W.; et al. Advanced parental age and the risk of autism spectrum disorder. Am. J. Epidemiol. 2008, 168, 1268–1276. [Google Scholar] [CrossRef] [PubMed]
- Lord, C. Diagnostic Instruments in Autism Spectrum Disorders. In Handbook of Autism and Pervasive Developmental Disorders, 2nd ed.; Cohen, D.J., Volkmar, F.R., Eds.; John, Wiley and Sons Inc.: New York, NY, USA, 1997; pp. 460–483. [Google Scholar]
- Anastasi, A. Psychological Testing, 6th ed.; Macmillan Publishing Co Inc.: New York, NY, USA, 1988. [Google Scholar]
- Manual for the Raven’s Color Progressive Matrices (CPM); Les Editions Centre de Psychologie Appliquée (ECPA): Paris, France, 2016.
- Guinchat, V.; Thorsen, P.; Laurent, C.; Cans, C.; Bodeau, N.; Cohen, D. Pre-peri-and neonatal risk factors for autism. Acta Obstet. Gynecol. Scand. 2012, 91, 287–300. [Google Scholar] [CrossRef] [PubMed]
- Gardener, H.; Spiegelman, D.; Buka, S.L. Perinatal and neonatal risk factors for autism: A comprehensive meta-analysis. Pediatrics 2011, 128, 344–355. [Google Scholar] [CrossRef] [PubMed]
- Weissman, J.R.; Kelley, R.I.; Bauman, M.L.; Cohen, B.H.; Murray, K.F.; Mitchell, R.L.; Kern, R.L.; Natowicz, M.R. Mitochondrial disease in autism spectrum disorder patients: A cohort analysis. PLoS ONE 2008, 3, e3815. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, D.A.; Frye, R.E. Mitochondrial dysfunction in autism spectrum disorders: A systematic review and meta-analysis. Mol. Psychiatry 2012, 17, 290–314. [Google Scholar] [CrossRef] [PubMed]
- Demily, C.; Assouline, M.; Boddaert, N.; Barcia, G.; Besmond, C.; Poisson, A.; Sanlaville, D.; Munnich, A. Genetic approaches in autism spectrum disorders. Neuropsychiatr. Enfance Adolesc. 2016, 64, 395–401. [Google Scholar] [CrossRef]
- Lai, M.C.; Lombardo, M.V.; Baron-Cohen, S. Autism. Lancet 2014, 383, 896–910. [Google Scholar] [CrossRef]
- Tordjman, S.; Anderson, G.M.; Bellissant, E.; Botbol, M.; Charbuy, H.; Camus, F.; Graignic, R.; Kermarrec, S.; Fougerou, C.; Cohen, D.; et al. Day and nighttime excretion of 6-sulphatoxymelatonin in adolescents and young adults with autistic disorder. Psychoneuroendocrinology 2012, 37, 1990–1997. [Google Scholar] [CrossRef] [PubMed]
- Haag, G.; Tordjman, S.; Duprat, A.; Urwand, S.; Jardin, F.; Clement, M.C.; Cukierman, A.; Druon, C.; Maufras du Chatellier, A.; et al. Presentation of a diagnostic grid of the progressive stages of infantile autism as observed in treatment. Int. J. Psychoanal. 2005, 86, 335–352. [Google Scholar] [CrossRef] [PubMed]
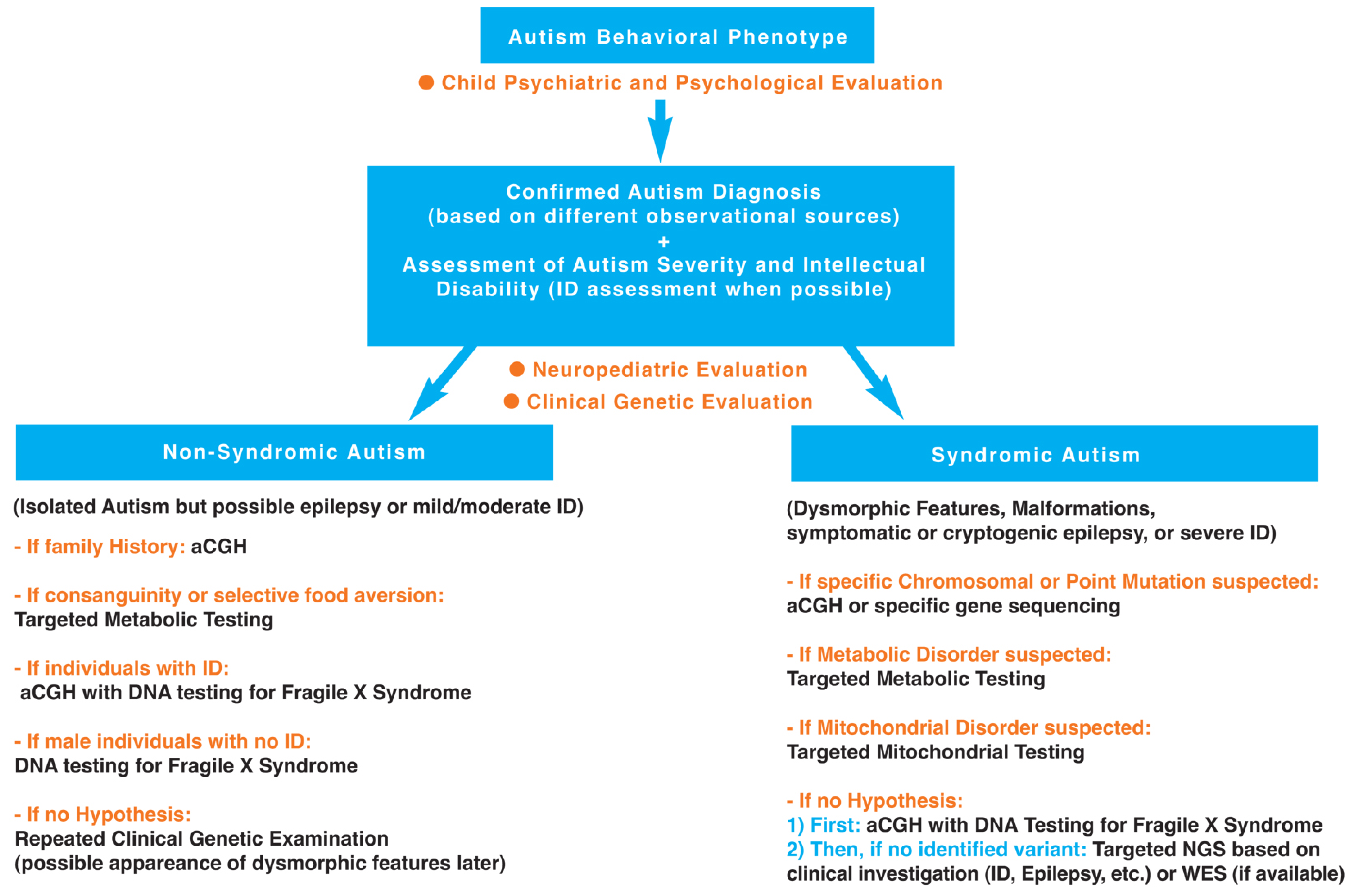
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).