
Gene Set-Based Functionome Analysis of Pathogenesis in Epithelial Ovarian Serous Carcinoma and the Molecular Features in Different FIGO Stages

 ,
, 
Abstract
:
1. Introduction
2. Results
2.1. DNA Microarray Gene Expression Datasets and Gene Sets
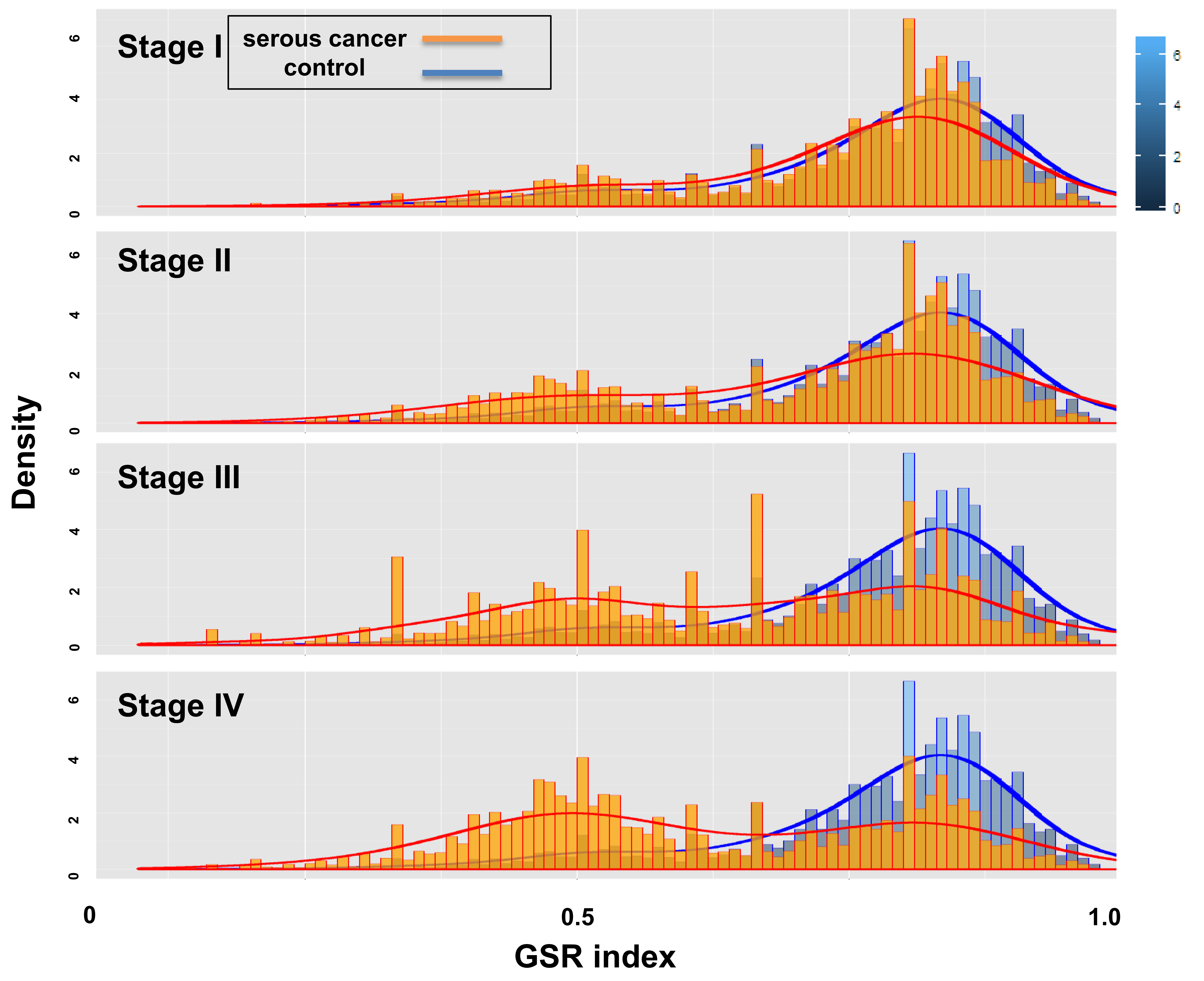
2.2. Means and Histograms of the Gene Set Regularity (GSR) Indices for the Four Stage Groups
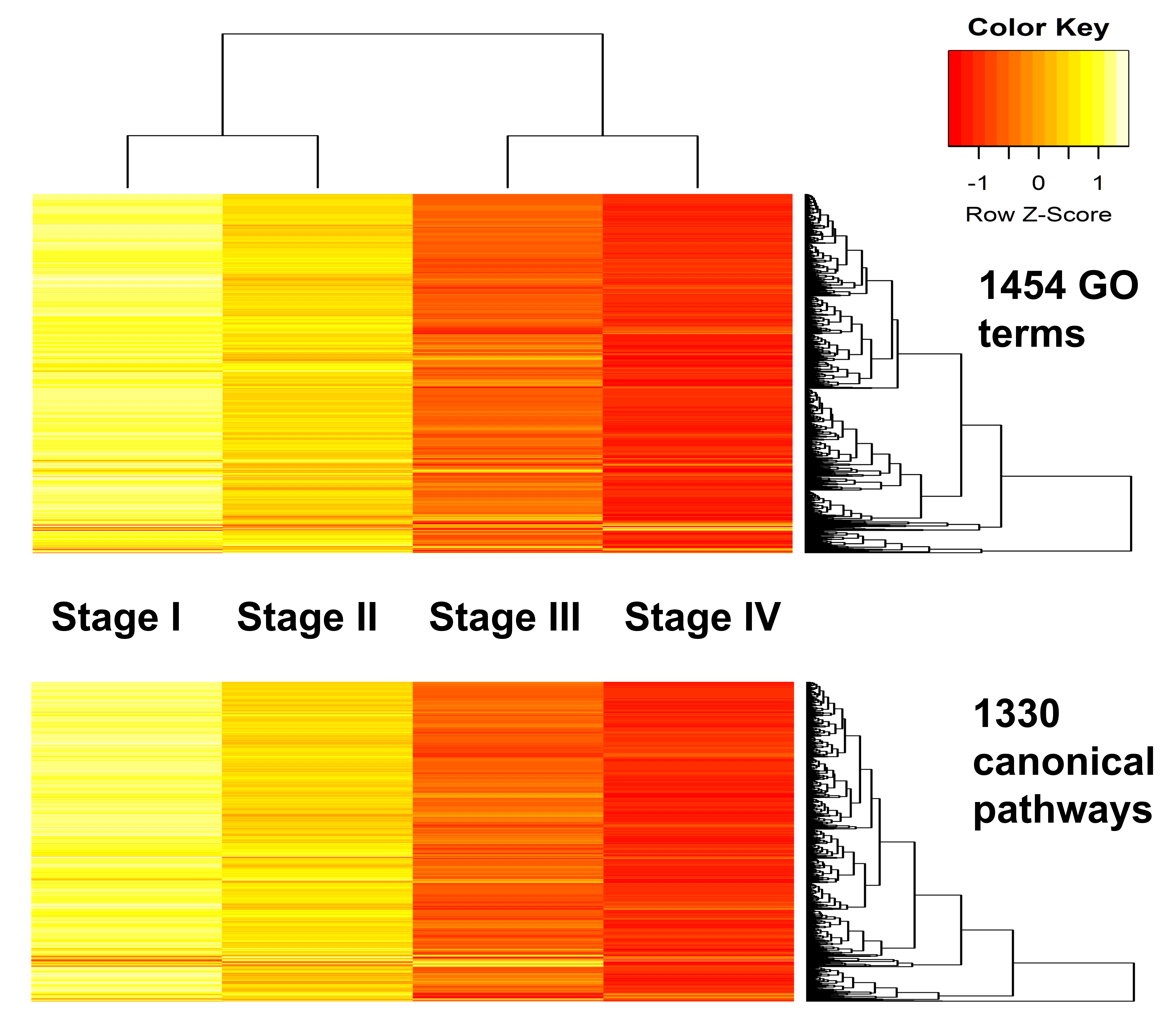
2.3. The Relationship of the Four Serous Carcinoma (SC) Stage Groups Revealed by Hierarchical Clustering
2.4. Function Regularity Patterns among the Four Stages Classified and Predicted by Machine Learning
2.5. The Most Significantly Deregulated Gene Ontology (GO) Terms and Canonical Pathways
2.6. The Commonly Deregulated GO Terms and Canonical Pathway Gene Sets among the Four Stages
2.7. The Elements of Serous Carcinoma Carcinogenesis Networks Discovered by Exploratory Factor Analysis (EFA)
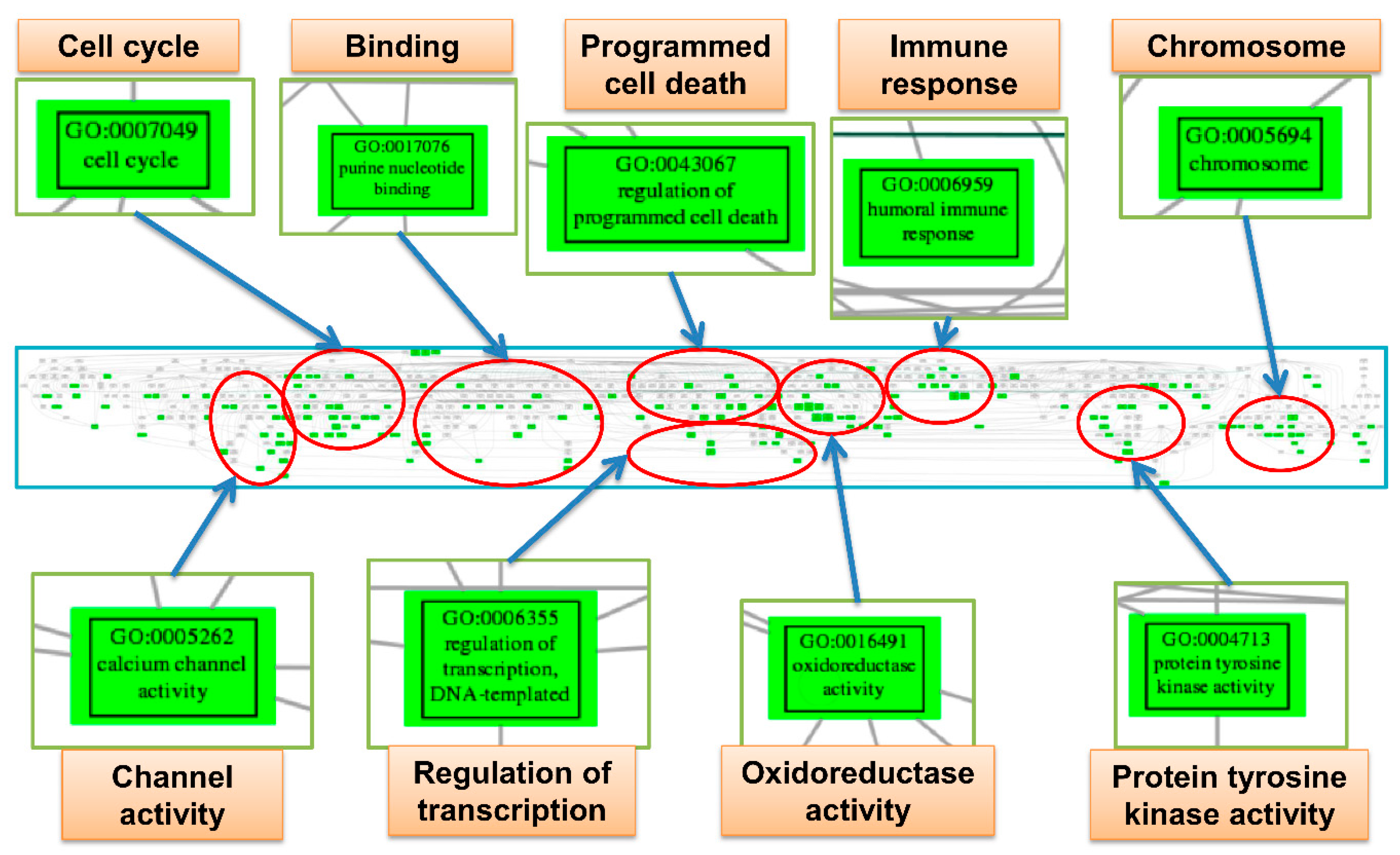
2.8. Trees of Deregulated Gene Ontology Terms for Serous Carcinoma
2.9. Interaction Network of SC Pathogenesis
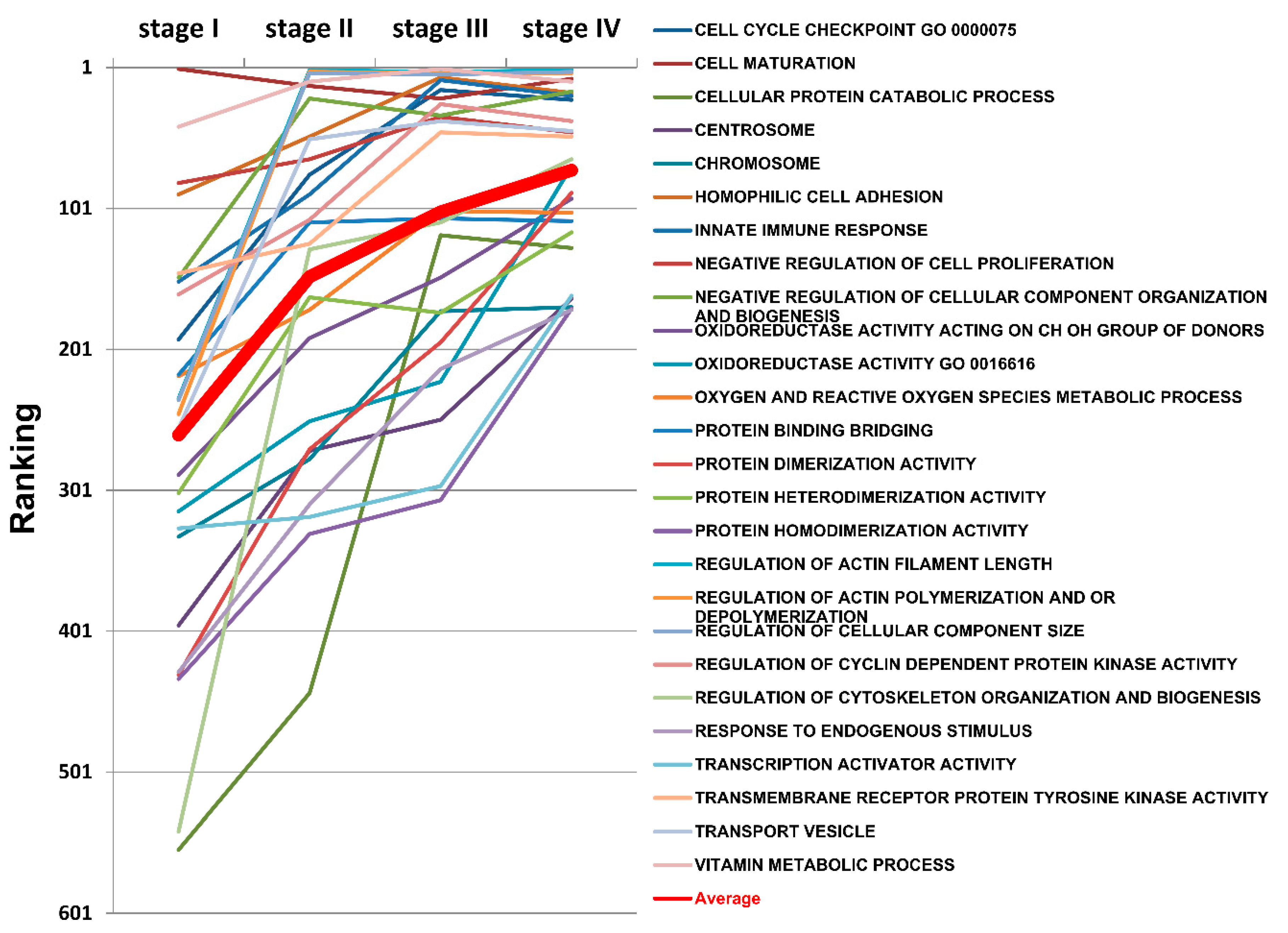
2.10. The Progressively Deregulated Functions in the Pathogenesis of SC from Stage I to IV
2.11. Differentially Expressed Genes in Ovarian Serous Carcinoma
3. Discussion
4. Materials and Methods
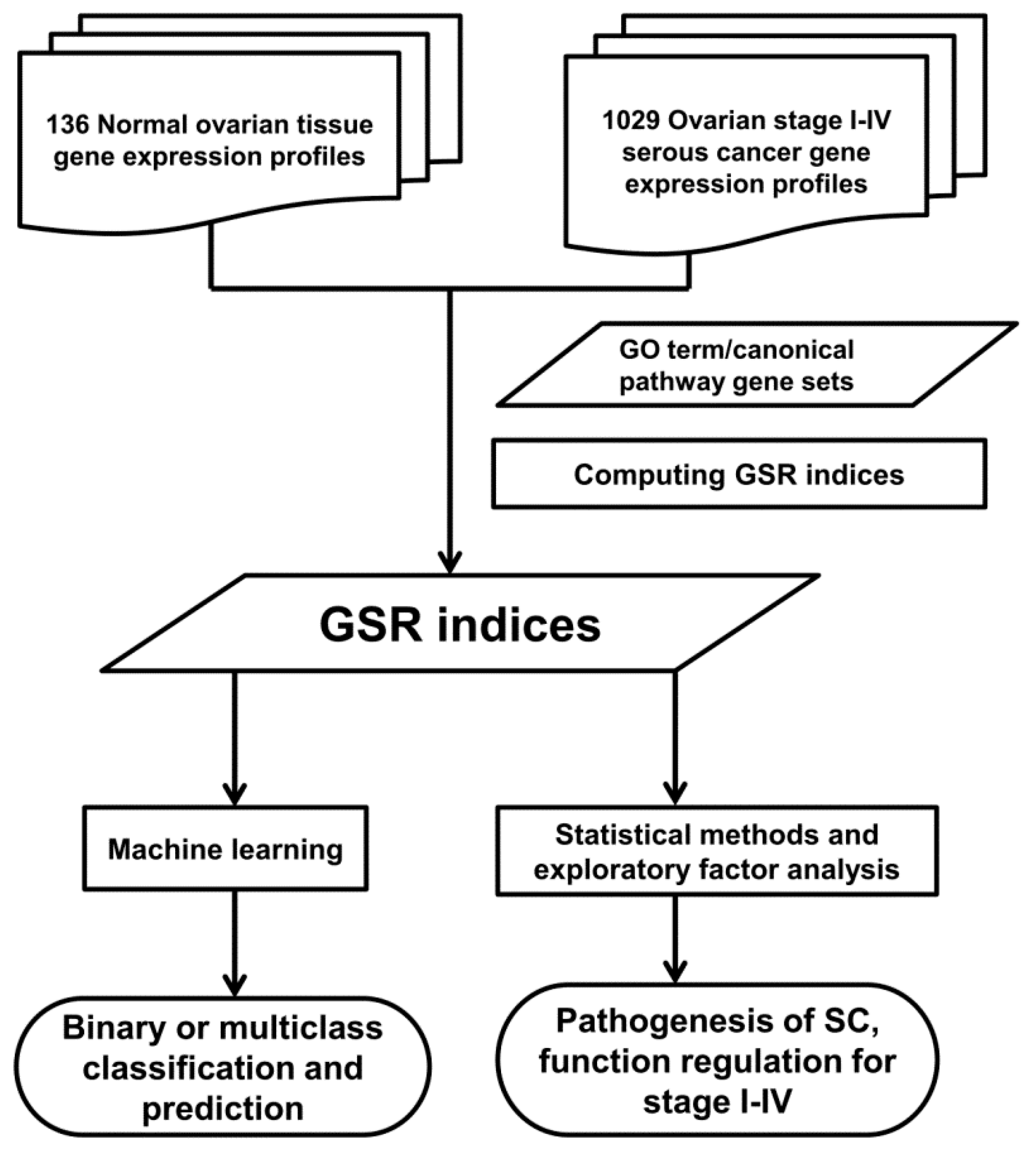
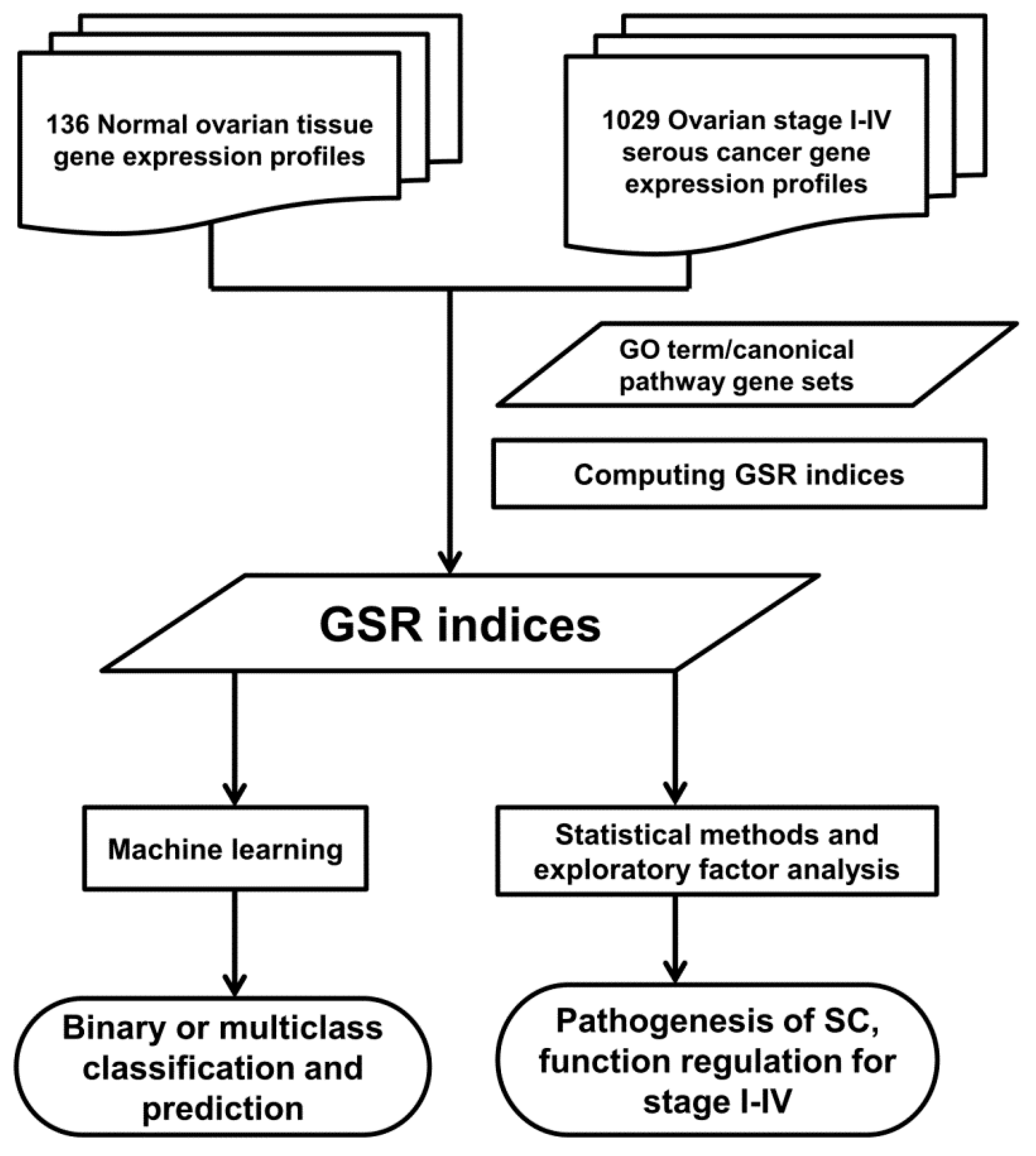
4.1. Workflow of Computing GSR Indices
4.2. Microarray Datasets, Gene Set Definition and Data Processing
4.3. Statistical Analysis
4.4. Classification and Prediction by Machine Learning
4.5. Hierarchical Clustering, Dendrogram and Heatmaps
4.6. Set Analysis
4.7. Exploratory Factor Analysis for the Deregulated GO Terms and Establishment of the GO Tree
4.8. Ranking Analysis
4.9. Construction of the Interaction Network
4.10. Detection of Differentially Expressed Genes in Ovarian Serous Carcinoma
5. Conclusions
Supplementary Materials
Author Contributions
Conflicts of Interest
Abbreviations
| SC | Serous carcinoma |
| FIGO | Federation of Gynecologists and Obstetrics |
| GO | Gene ontology |
| GSR | Gene set regularity |
| EFA | Exploratory factor analysis |
| EOC | Epithelial ovarian cancers |
| MSigDB | Molecular Signatures Database |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| PID | Pathway Interaction Database |
| GEO | Gene Expression Omnibus |
| SD | Standard deviation |
| SVM | Support vector machine |
| AUC | Area under curve |
| PI3K-AKT | Phosphoinositide 3-kinase-AKT |
| AKAP13 | A Kinase Anchor Protein 13 |
| AR TR | Androgen receptor transcription factor |
| EIF2 | Eukaryotic initiation factor-2 |
| mTOR | Mammalian target of rapamycin |
| FOXM1 | Forkhead box protein M1 |
| DIRAC | Differential Rank Conservation |
| PNG | Portable Network Graphics |
| ARACNE | Algorithm for the Reconstruction of Accurate Cellular Networks |
| GML | graph modeling language |
References
- Gilks, C.B.; Prat, J. Ovarian carcinoma pathology and genetics: Recent advances. Hum. Pathol. 2009, 40, 1213–1223. [Google Scholar] [CrossRef] [PubMed]
- Benedet, J.L.; Bender, H.; Jones, H., III; Ngan, H.Y.; Pecorelli, S. FIGO staging classifications and clinical practice guidelines in the management of gynecologic cancers. FIGO Committee on Gynecologic Oncology. Int. J. Gynaecol. Obstet. 2000, 70, 209–262. [Google Scholar] [PubMed]
- Plaxe, S.C. Epidemiology of low-grade serous ovarian cancer. Am. J. Obstet. Gynecol. 2008, 198, 459.e8–459.e9. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Frede, J.; Fraser, S.P.; Oskay-Ozcelik, G.; Hong, Y.; Ioana Braicu, E.; Sehouli, J.; Gabra, H.; Djamgoz, M.B. Ovarian cancer: Ion channel and aquaporin expression as novel targets of clinical potential. Eur. J. Cancer. 2013, 49, 2331–2344. [Google Scholar] [CrossRef] [PubMed]
- Fehrenbacher, N.; Jaattela, M. Lysosomes as targets for cancer therapy. Cancer Res. 2005, 65, 2993–2995. [Google Scholar] [PubMed]
- Mabuchi, S.; Kuroda, H.; Takahashi, R.; Sasano, T. The PI3K/AKT/mTOR pathway as a therapeutic target in ovarian cancer. Gynecol. Oncol. 2015, 137, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Hofmann, J.; Lu, Y.; Mills, G.B.; Jaffe, R.B. Inhibition of phosphatidylinositol 3′-kinase increases efficacy of paclitaxel in in vitro and in vivo ovarian cancer models. Cancer Res. 2002, 62, 1087–1092. [Google Scholar] [PubMed]
- Furlong, F.; Fitzpatrick, P.; O’Toole, S.; Phelan, S.; McGrogan, B.; Maguire, A.; O’Grady, A.; Gallagher, M.; Prencipe, M.; McGoldrick, A.; et al. Low MAD2 expression levels associate with reduced progression-free survival in patients with high-grade serous epithelial ovarian cancer. J. Pathol. 2012, 226, 746–755. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.T.; Rubino, D.M.; Driggers, P.H.; Haddad, B.; Cisar, M.; Gray, K.; Segars, J.H. Expression of brx proto-oncogene in normal ovary and in epithelial ovarian neoplasms. Am. J. Obstet. Gynecol. 2000, 182, 286–295. [Google Scholar] [CrossRef]
- Liu, Y.; Yan, S.; Wondimu, A.; Bob, D.; Weiss, M.; Sliwinski, K.; Villar, J.; Notario, V.; Sutherland, M.; Colberg-Poley, A.M.; et al. Ganglioside synthase knockout in oncogene-transformed fibroblasts depletes gangliosides and impairs tumor growth. Oncogene 2010, 29, 3297–3306. [Google Scholar] [CrossRef] [PubMed]
- Prinetti, A.; Cao, T.; Illuzzi, G.; Prioni, S.; Aureli, M.; Gagliano, N.; Tredici, G.; Rodriguez-Menendez, V.; Chigorno, V.; Sonnino, S. A glycosphingolipid/caveolin-1 signaling complex inhibits motility of human ovarian carcinoma cells. J. Biol. Chem. 2011, 286, 40900–40910. [Google Scholar] [CrossRef] [PubMed]
- Miljan, E.A.; Bremer, E.G. Regulation of growth factor receptors by gangliosides. Sci. STKE 2002, 2002, re15. [Google Scholar] [CrossRef] [PubMed]
- Malpica, A.; Deavers, M.T.; Lu, K.; Bodurka, D.C.; Atkinson, E.N.; Gershenson, D.M.; Silva, E.G. Grading ovarian serous carcinoma using a two-tier system. Am. J. Surg. Pathol. 2004, 28, 496–504. [Google Scholar] [CrossRef] [PubMed]
- Kurman, R.J.; Shih, I.M. Pathogenesis of ovarian cancer: Lessons from morphology and molecular biology and their clinical implications. Int. J. Gynecol. Pathol. 2008, 27, 151–160. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar]
- Eddy, J.A.; Hood, L.; Price, N.D.; Geman, D. Identifying tightly regulated and variably expressed networks by Differential Rank Conservation (DIRAC). PLoS Comput. Biol. 2010, 27, e1000792. [Google Scholar] [CrossRef] [PubMed]
- Alexandros, K.; Alex, S.; Kurt, H.; Achim, Z. kernlab—An S4 Package for Kernel Methods in R. J. Stat. Softw. 2004, 11, 1–20. [Google Scholar]
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.C.; Muller, M. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 2011, 12, 77. [Google Scholar] [CrossRef] [PubMed]
- Schroder, M.S.; Gusenleitner, D.; Quackenbush, J.; Culhane, A.C.; Haibe-Kains, B. RamiGO: An R/Bioconductor package providing an AmiGO visualize interface. Bioinformatics 2013, 29, 666–668. [Google Scholar] [CrossRef] [PubMed]
- AmiGO 2. Available online: http://amigo2.berkeleybop.org/amigo (accessed on 15 January 2016).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Stage | Case | Control | Total | Case Mean (SD) | Control Mean (SD) | p Value * |
|---|---|---|---|---|---|---|
| I | 34 | 136 | 170 | 0.7425 (0.1511) | 0.7752 (0.1370) | <0.05 |
| II | 39 | 136 | 175 | 0.7088 (0.1745) | 0.7752 (0.1369) | <0.05 |
| III | 689 | 136 | 825 | 0.6483 (0.2007) | 0.7738 (0.1548) | <0.05 |
| IV | 131 | 136 | 267 | 0.6197 (0.1922) | 0.7737 (0.1413) | <0.05 |
| Classification | Stage | Sensitivity (SD) | Specificity (SD) | Accuracy (SD) | AUC |
|---|---|---|---|---|---|
| Binary | I | 0.9488 (0.0857) | 1.0000 (0.0000) | 0.9882 (0.0205) | 0.9692 |
| II | 0.9655 (0.0568) | 1.0000 (0.0000) | 0.9914 (0.0138) | 0.9807 | |
| III | 0.9920 (0.0069) | 0.9769 (0.0363) | 0.9890 (0.0079) | 0.9835 | |
| IV | 0.9929 (0.0149) | 0.9961 (0.0121) | 0.9943 (0.0091) | 0.9942 | |
| Multiclass | I–IV | NA | NA | 0.9038 (0.0054) | NA |
| Ranking | Stage I | Stage II | Stage III | Stage IV |
|---|---|---|---|---|
| 1 | Calcium channel activity | lysosomal membrane | protein tyrosine kinase activity | lysosomal membrane |
| 2 | Cell maturation | vacuolar membrane | vitamin metabolic process | vacuolar membrane |
| 3 | Oxygen binding | regulation of actin filament length | oxidoreductase activity acting on the aldehyde or OXO group of donors | regulation of actin filament length |
| 4 | Secretin-like receptor activity | regulation of actin polymerization and/or depolymerization | regulation of actin filament length | regulation of cellular component size |
| 5 | Lysosomal membrane | regulation of cellular component size | regulation of actin polymerization and/or depolymerization | regulation of actin polymerization and/or depolymerization |
| 6 | Vacuolar membrane | amino acid derivative metabolic process | regulation of cellular component size | vacuolar part |
| 7 | Developmental maturation | response to hormone stimulus | spindle pole | cell division |
| 8 | Taste receptor activity | vacuolar part | homophilic cell adhesion | cytokinesis |
| 9 | Hematopoietin interferon class D200 domain Cytokine receptor binding | neuropeptide signaling pathway | single-stranded DNA binding | cell maturation |
| 10 | Cofactor transporter activity | G-protein coupled receptor binding | innate immune response | amino acid derivative metabolic process |
| 11 | Auxiliary transport protein activity | vitamin metabolic process | spindle | vitamin metabolic process |
| 12 | Hormone activity | steroid hormone receptor binding | damaged DNA binding | response to hormone stimulus |
| 13 | Organic anion transmembrane transporter activity | aromatic compound metabolic process | Rho protein signal transduction | calcium channel activity |
| 14 | Response to hormone stimulus | cell maturation | microtubule cytoskeleton | coenzyme binding |
| 15 | Potassium channel regulator activity | chaperone binding | structure specific DNA binding | developmental maturation |
| Ranking | Stage I | Stage II | Stage III | Stage IV |
|---|---|---|---|---|
| 1 | Reactome CD28-dependent PI3K AKT signaling | Biocarta AKAP13 pathway | PID AR TF pathway | KEGG glycosphingolipid biosynthesis ganglio series |
| 2 | Biocarta AKAP13 pathway | Reactome CD28-dependent PI3K AKT signaling | KEGG glycosphingolipid biosynthesis ganglio series | Biocarta AKAP13 pathway |
| 3 | KEGG ascorbate and aldarate metabolism | Reactome PI3K events in ERBB4 signaling | Biocarta CK1 pathway | PID AR TF pathway |
| 4 | KEGG glycosphingolipid biosynthesis ganglio series | KEGG ascorbate and aldarate metabolism | Reactome COPI-mediated transport | Reactome CD28-dependent PI3K AKT signaling |
| 5 | Reactome signaling by NOTCH3 | Reactome GPVI-mediated activation cascade | Reactome G0 and early G1 | Reactome GPVI-mediated activation cascade |
| 6 | Reactome packaging of telomere ends | Biocarta MTA3 pathway | Reactome sphingolipid de novo biosynthesis | Reactome hormone-sensitive lipase HSL-mediated triacylglycerol hydrolysis |
| 7 | Reactome meiotic synapsis | Reactome GAB1 signalosome | KEGG cell cycle | Reactome termination of O glycan biosynthesis |
| 8 | KEGG retinol metabolism | Reactome PI3K AKT activation | Reactome DARPP 32 events | KEGG aldosterone-regulated sodium reabsorption |
| 9 | Reactome apoptotic cleavage of cell adhesion proteins | Reactome post-chaperonin tubulin folding pathway | Reactome meiotic synapsis | Reactome PI3K events in ERBB4 signaling |
| 10 | Reactome cytosolic sulfonation of small molecules | KEGG glyoxylate and dicarboxylate metabolism | PID AR pathway | KEGG inositol phosphate metabolism |
| 11 | Reactome digestion of dietary carbohydrate | Reactome GABA synthesis release reuptake and degradation | Reactome neurotransmitter release cycle | Reactome G0 and early G1 |
| 12 | Reactome peptide ligand binding receptors | Reactome packaging of telomere ends | PID AJDISS 2 pathway | KEGG acute myeloid leukemia |
| 13 | Reactome synthesis of PIPS at the plasma membrane | KEGG glycosphingolipid biosynthesis ganglio series | Reactome signaling by Rho GTPases | KEGG tryptophan metabolism |
| 14 | Reactome xenobiotics | PID TGFBR pathway | KEGG progesterone-mediated oocyte maturation | Reactome downregulation of ERBB2 ERBB3 signaling |
| 15 | SA TRKA receptor | Reactome adenylate cyclase inhibitory pathway | Reactome trans Golgi network vesicle budding | Reactome meiotic synapsis |
| Gene Symbol | Alias | Related GO Terms or Pathways | p Value |
|---|---|---|---|
| AOX1 | Aldehyde Oxidase 1 | Catalytic activity (GO:0003824) | 3.51 × 10−133 |
| Aldehyde oxidase activity (GO:0004031) | |||
| Small molecule metabolic process (G0:0044281) | |||
| EIF3F | Eukaryotic Translation Initiation Factor 3, Subunit F | Translation initiation factor activity (GO:0003743) | 2.00 × 10−132 |
| Protein binding (GO:0005515) | |||
| TRanslation (GO:0006412) | |||
| Eukaryotic translation initiation (Reactome) | |||
| Activation of the mRNA upon binding of the cap-binding complex and eIFs and subsequent binding to 43S (Reactome) | |||
| DFNA5 | Deafness, Autosomal Dominant 5 | Apoptotic process (GO:0006915) | 1.26 × 10−128 |
| Negative regulation of cell proliferation (GO:0008285) | |||
| Positive regulation of intrinsic apoptotic signaling Pathway (GO:2001244) | |||
| PTGIS | Prostaglandin I2 (Prostacyclin) Synthase | Monooxygenase activity (GO:0004497) | 6.85 × 10−125 |
| Protein binding (GO:0005515) | |||
| Oxidoreductase activity acting on paired donors with Incorporation or reduction of molecular oxygen (GO:0016705) | |||
| TSPAN5 | Tetraspanin 5 | Positive regulation of Notch signaling pathway (GO:0045747) | 7.08 × 10−124 |
| Protein maturation (GO:0051604) | |||
| BAMBI | BMP and Activin Membrane-Bound Inhibitor | Positive regulation of cell proliferation (GO:0008284) | 2.13 × 10−108 |
| Transforming growth factor β receptor signaling pathway (GO:0007179) | |||
| TGF-β receptor signaling (PID) | |||
| SPOCK1 | Sparc/Osteonectin, Cwcv and Kazal-Like Domains Proteoglycan (Testican) 1 | Serine-type endopeptidase inhibitor activity (GO:0004867) | 2.13 × 10−108 |
| Cysteine-type endopeptidase inhibitor activity (GO:0004869) | |||
| Calcium ion binding (GO:0005509) | |||
| Protein binding (GO:0005515) | |||
| Metalloendopeptidase inhibitor activity (GO:0008191) | |||
| GFPT2 | Glutamine-Fructose-6-Phosphate Transaminase 2 | Glutamine-fructose-6-phosphate transaminase (isomerizing) activity (GO:0004360) | 8.91 × 10−107 |
| Carbohydrate binding (GO:0030246) | |||
| Amino sugar and nucleotide sugar metabolism (KEGG) | |||
| C21orf62 | Chromosome 21 Open Reading Frame 62 | Unclear | 1.35 × 10−106 |
| FLRT2 | Fibronectin Leucine Rich Transmembrane Protein 2 | Receptor signaling protein activity (GO:0005057) | 5.29 × 10−104 |
| Protein binding (GO:0005515) | |||
| Fibroblast growth factor receptor signaling pathway (GO:0008543) | |||
| Cell adhesion (GO:0007155) |
| Gene Symbol | Alias | Related GO Terms or Pathways | p Value |
|---|---|---|---|
| C14orf2 | Chromosome 14 Open Reading Frame 2 | unclear | 8.15 × 10−78 |
| COX6B1 | Cytochrome C Oxidase Subunit VIb Polypeptide 1 | transcriptional regulation by TP53(Reactome) | 2.59 × 10−66 |
| gene expression (Reactome) | |||
| transcription initiation from RNA polymerase II | |||
| promoter ( GO:0006367) | |||
| gene expression (GO:0010467) | |||
| TRIAP1 | TP53 Regulated Inhibitor of Apoptosis 1 | p53 binding (GO:0002039) | 3.44 × 10−65 |
| DNA damage response signal transduction by p53 class mediator resulting in cell cycle arrest (GO:0006977) | |||
| DNA damage response signal transduction by p53 class mediator (GO:0030330) | |||
| negative regulation of apoptotic process (GO:0043066) | |||
| RBX1 | Ring-Box 1, E3 Ubiquitin Protein Ligase | contributes to ubiquitin-protein transferase activity (GO:0004842) | 9.37 × 10−63 |
| DNA repair (GO:0006281) | |||
| MAPK cascade (GO:0000165) | |||
| signaling by ERBB2 (Reactome) | |||
| RAF/MAP kinase cascade (Reactome) | |||
| CGRRF1 | Cell Growth Regulator with Ring Finger Domain 1 | response to stress (GO:0006950) | 1.25 × 10−61 |
| cell cycle arrest (GO:0007050) | |||
| negative regulation of cell proliferation (GO:0008285) | |||
| LSM6 | LSM6 Homolog, U6 Small Nuclear RNA and MRNA Degradation Associated | cytoplasmic mRNA processing body (GO:0000932) | 6.16 × 10−60 |
| spliceosomal complex (GO:0005681) | |||
| U6 snRNP (GO:0005688) | |||
| nucleolus (GO:0005730) | |||
| small nucleolar ribonucleoprotein complex (GO:0005732) | |||
| deadenylation-dependent mRNA decay (Reactome) | |||
| COX5A | Cytochrome C Oxidase Subunit Va | cytochrome-c oxidase activity (GO:0004129) | 1.71 × 10−59 |
| transcriptional regulation by TP53 (Reactome) | |||
| mitochondrial electron transport; cytochrome c to oxygen (GO:0006123) | |||
| transcription initiation from RNA polymerase II promoter (GO:0006367) | |||
| gene expression (GO:0010467) | |||
| TIMM8B | Translocase of Inner Mitochondrial Membrane 8 Homolog B (Yeast) | protein targeting to mitochondrion (GO:0006626) | 1.54 × 10−58 |
| protein transport (GO:0015031) | |||
| cellular protein metabolic process (GO:0044267) | |||
| chaperone-mediated protein transport (GO:0072321) | |||
| SNX6 | Sorting Nexin 6 | type I transforming growth factor beta receptor binding (GO:0034713) | 1.62 × 10−58 |
| phosphatidylinositol binding (GO:0035091) | |||
| protein homodimerization activity (GO:0042803) | |||
| TGF-β receptor signaling pathway (Reactome) | |||
| negative regulation of epidermal growth factor-activated receptor activity (GO:0007175) | |||
| negative regulation of transforming growth factor β receptor signaling pathway (GO:0030512) | |||
| IER3IP1 | Immediate Early Response 3 Interacting Protein 1 | regulation of fibroblast apoptotic process (GO:2000269) | 1.88 × 10−58 |
| endoplasmic reticulum (GO:0005783) |
| Gene Symbol | Alias | Related GO Terms or Pathways |
|---|---|---|
| UFC1 | Ubiquitin-Fold Modifier Conjugating Enzyme 1 | protein binding (GO:0005515) |
| response to endoplasmic reticulum stress (GO:0034976) protein ufmylation (GO:0071569) | ||
| SOX12 | SRY (Sex Determining Region Y)-Box 12 | transcription regulatory region sequence-specific DNA binding (GO:0000976) |
| transcriptional activator activity, RNA polymerase II core promoter proximal region sequence-specific binding (GO:0001077) | ||
| RNA polymerase II transcription coactivator activity (GO:0001105) | ||
| DNA binding (GO:0003677) | ||
| molecular mechanisms of cancer (QIAGEN) | ||
| APOC3 | Apolipoprotein C-III | phospholipid binding (GO:0005543) |
| cholesterol binding (GO:0015485) | ||
| enzyme regulator activity (GO:0030234) | ||
| lipase inhibitor activity (GO:0055102) | ||
| signal transduction (Reactome) | ||
| G-protein coupled receptor signaling pathway (GO:0007186) | ||
| RAB11FIP2 | RAB11 Family Interacting Protein 2 (Class I) | Rab GTPase binding (GO:0017137) |
| protein kinase binding (GO:0019901) | ||
| protein homodimerization activity (GO:0042803) | ||
| PCOLCE2 | Procollagen C-Endopeptidase Enhancer 2 | protein binding (GO:0005515) |
| collagen binding (GO:0005518) | ||
| heparin binding (GO:0008201) | ||
| peptidase activator activity (GO:0016504) | ||
| collagen formation (Reactome) | ||
| positive regulation of peptidase activity (GO:0010952) | ||
| STAT2 | Signal Transducer and Activator Of Transcription 2, 113 kDa | DNA binding (GO:0003677) |
| transcription factor activity, sequence-specific DNA binding (GO:0003700) | ||
| signal transducer activity (GO:0004871) | ||
| Jak-STAT signaling pathway (KEGG) | ||
| transcription, DNA-templated (GO:0006351) | ||
| regulation of transcription, DNA-templated (GO:0006355) | ||
| regulation of transcription from RNA polymerase II promoter (GO:0006357) | ||
| AR | Androgen Receptor | RNA polymerase II core promoter proximal region sequence-specific DNA binding (GO:0000978) |
| RNA polymerase II transcription factor binding (GO:0001085) | ||
| DNA binding (GO:0003677) | ||
| chromatin binding (GO:0003682) | ||
| signaling by Rho GTPases (Reactome) | ||
| regulation of transcription, DNA-templated (GO:0006355) | ||
| INSIG2 | Insulin Induced Gene 2 | transcription factor binding (GO:0008134) |
| regulation of cholesterol biosynthesis by SREBP (Reactome) | ||
| cholesterol biosynthetic process (GO:0006695) | ||
| response to sterol depletion (GO:0006991) | ||
| cholesterol metabolic process (GO:0008203) | ||
| negative regulation of steroid biosynthetic process (GO:0010894) | ||
| POLR2G | Polymerase (RNA) II (DNA Directed) Polypeptide G | nucleic acid binding (GO:0003676) |
| single-stranded DNA binding (GO:0003697) | ||
| single-stranded RNA binding (GO:0003727) | ||
| translation initiation factor binding (GO:0031369) | ||
| mRNA splicing, via spliceosome (GO:0000398) | ||
| DNA repair (GO:0006281) | ||
| CHODL | Chondrolectin | carbohydrate binding (GO:0030246) |
| regulation of neuron projection development (GO:0010975) | ||
| perinuclear region of cytoplasm (GO:0048471) | ||
| COL4A1 | Collagen, Type IV, Alpha 1 | extracellular matrix structural constituent (GO:0005201) |
| protein binding (GO:0005515) | ||
| extracellular matrix constituent conferring elasticity (GO:0030023) | ||
| platelet-derived growth factor binding (GO:0048407) | ||
| focal adhesion (KEGG) | ||
| patterning of blood vessels (GO:0001569) | ||
| receptor-mediated endocytosis (GO:0006898) | ||
| RAB9A | RAB9A, Member RAS Oncogene Family | GTPase activity (GO:0003924) |
| GTP binding (GO:0005525) | ||
| GDP binding (GO:0019003) | ||
| signal transduction (GO:0007165) | ||
| small GTPase-mediated signal transduction (GO:0007264) | ||
| EN1 | Engrailed Homeobox 1 | RNA polymerase II core promoter proximal region sequence-specific DNA binding (GO:0000978) |
| transcriptional repressor activity, RNA polymerase II core promoter proximal region sequence-specific binding (GO:0001078) | ||
| DNA binding (GO:0003677) | ||
| sequence-specific DNA binding (GO:0043565) | ||
| ATP1B1 | ATPase, Na+/K+ Transporting, Beta 1 Polypeptide | ATPase activator activity (GO:0001671) |
| response to hypoxia (GO:0001666) | ||
| potassium ion transport (GO:0006813) | ||
| sodium ion transport (GO:0006814) | ||
| cellular calcium ion homeostasis (GO:0006874) | ||
| GNAT1 | Guanine Nucleotide Binding Protein (G Protein), Alpha Transducing Activity Polypeptide 1 | acyl binding (GO:0000035) |
| G-protein coupled receptor binding (GO:0001664) | ||
| GTPase activity (GO:0003924) | ||
| signal transducer activity (GO:0004871) | ||
| activation of the phototransduction cascade (Reactome) | ||
| G-protein coupled receptor signaling pathway (GO:0007186) | ||
| PDCD6IP | Programmed Cell Death 6 Interacting Protein | SH3 domain binding (GO:0017124) |
| proteinase activated receptor binding (GO:0031871) | ||
| protein homodimerization activity (GO:0042803) | ||
| protein dimerization activity (GO:0046983) | ||
| cell separation after cytokinesis (GO:0000920) | ||
| apoptotic process (GO:0006915) | ||
| regulation of centrosome duplication (GO:0010824) | ||
| PDHB | Pyruvate Dehydrogenase (Lipoamide) Beta | catalytic activity (GO:0003824) |
| pyruvate dehydrogenase activity (GO:0004738) | ||
| glucose metabolic process (GO:0006006) | ||
| acetyl-CoA biosynthetic process from pyruvate (GO:0006086) | ||
| pyruvate metabolic process (GO:0006090) | ||
| tricarboxylic acid cycle (GO:0006099) | ||
| GCNT3 | Glucosaminyl (N-Acetyl) Transferase 3, Mucin Type | acetylglucosaminyltransferase activity (GO:0005975) carbohydrate metabolic process (GO:0008375) |
| protein O-linked glycosylation (GO:0006493) | ||
| post-translational protein modification (GO:0043687) | ||
| FXYD3 | FXYD Domain Containing Ion Transport Regulator 3 | ion channel activity (GO:0005216) |
| chloride channel activity (GO:0005254) | ||
| sodium channel regulator activity (GO:0017080) | ||
| ATPase binding (GO:0051117) | ||
| CHGA | Chromogranin A | protein binding (GO:0005515) |
| Peptide hormone biosynthesis (Reactome) | ||
| Androgen biosynthesis (Reactome) | ||
| Signaling by GPCR (Reactome) |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, C.-M.; Chuang, C.-M.; Wang, M.-L.; Yang, M.-J.; Chang, C.-C.; Yen, M.-S.; Chiou, S.-H. Gene Set-Based Functionome Analysis of Pathogenesis in Epithelial Ovarian Serous Carcinoma and the Molecular Features in Different FIGO Stages. Int. J. Mol. Sci. 2016, 17, 886. https://doi.org/10.3390/ijms17060886
Chang C-M, Chuang C-M, Wang M-L, Yang M-J, Chang C-C, Yen M-S, Chiou S-H. Gene Set-Based Functionome Analysis of Pathogenesis in Epithelial Ovarian Serous Carcinoma and the Molecular Features in Different FIGO Stages. International Journal of Molecular Sciences. 2016; 17(6):886. https://doi.org/10.3390/ijms17060886
Chicago/Turabian StyleChang, Chia-Ming, Chi-Mu Chuang, Mong-Lien Wang, Ming-Jie Yang, Cheng-Chang Chang, Ming-Shyen Yen, and Shih-Hwa Chiou. 2016. "Gene Set-Based Functionome Analysis of Pathogenesis in Epithelial Ovarian Serous Carcinoma and the Molecular Features in Different FIGO Stages" International Journal of Molecular Sciences 17, no. 6: 886. https://doi.org/10.3390/ijms17060886
APA StyleChang, C.-M., Chuang, C.-M., Wang, M.-L., Yang, M.-J., Chang, C.-C., Yen, M.-S., & Chiou, S.-H. (2016). Gene Set-Based Functionome Analysis of Pathogenesis in Epithelial Ovarian Serous Carcinoma and the Molecular Features in Different FIGO Stages. International Journal of Molecular Sciences, 17(6), 886. https://doi.org/10.3390/ijms17060886