1. Introduction
In-silico analysis of genetic variants aims to efficiently and accurately predict the presence and penetrance of phenotypic pathogenicity. Focusing on single nucleotide polymorphisms resulting in non-synonymous amino acid (AA) coding (nsSNPs), a number of existing tools provide assessments of predicted classification of variants based on various measures [
1–
3]. One approach involves multiple-species sequence alignments (MSAs) for analysis of phylogenetic diversity at the same locus as the nsSNP under consideration; although each differs in their analysis, all aforementioned measures implement this means of quantification.
A measure of diversity originally proposed by Grantham [
4] was later extended and implemented in Align-GVGD [
1] (an explanation of this implementation is outlined below, in Section 2). Greater diversity implies lesser evolutionary hold over the locus and thus a lower probability of deleterious outcome for novel variants. However this approach does not account for the number of species in the MSA and has been shown by Hicks
et al. [
5] to overestimate diversity while, at a critical MSA depth, classifying all nsSNPs as neutral. The aim of our research was to correct for this shortcoming. It is important to note that the specifically curated MSAs provided by the Align-GVGD team are not affected by this scenario; however, these only covered 11 genes at the time of our work.
In the absence of natural selection, random mutations between generations would, with time, render a genetic locus compared between species indistinguishable from a random sequence; the so-called entropy of the sequence increases with time. An analogous effect can be seen when two initially separated gases are allowed to mix; the ordered state of distinct groups of molecules progressively reaches an equilibrium state of indistinguishable mixture.
A rigorous treatment of this phenomenon is found in the second law of thermodynamics, the requirement of the thermodynamic entropy of a closed system to be non-decreasing.
James Clerk Maxwell’s famous thought experiment [
6] attempted to violate this law with the aid of a hypothetical demon redirecting molecules by opening and closing a frictionless door. Previously mixed gases would eventually separate much in the same way that homologous genes maintain a level of order; a low entropy distribution of gas molecules is analogous to tight evolutionary control. Natural selection operates as Charles Darwin’s demon; through survival or death, acting to open and close Maxwell’s hypothetical door.
Maxwell’s proposition was later countered by the close similarity between thermodynamic entropy and information entropy [
7], a measure of uncertainty in a data source. We recognised that highly controlled, low-information loci would display low Kolmogorov complexity [
8] and that an upper bound for such complexity could be determined through data compression. By utilising the compression ratio we could account for expected diversity for MSA depth and correct for the Grantham overestimation. High compressibility should correlate with deleterious nsSNP phenotypes.
2. Theoretical Background
Positive evolutionary selection of a non-synonymous AA change has been shown to—on a genome-wide scale—inversely correlate with the respective Grantham score (
GS), a quantified biochemical difference between individual AAs [
4]. Considering a pair of AAs
W (wild-type) and
V (variant), the
GS is the Euclidean distance between chemical properties of
W and
V; dimensions are normalised by mean AA distance (considering all 20 AAs) within the same dimension. Grantham’s original definition of
GS scaled the value by 50.723 such that the average pair-wise distance between AAs would be 100; we ignored this arbitrary adjustment as its linear nature has no effect on our analysis. The
GS accounts for differences in composition
Xc, polarity
Xp and molecular volume
Xv; with normalisation coefficients
α = 1.833,
β = 0.1018, and
γ = 0.000399:
An extension of the concept, the Grantham variance (
GV) is a measure of the diversity amongst a group of AAs [
1], in this case a column from the MSA. For the smallest rectangular prism encompassing all residues, the
GV is the length of the longest diagonal. For a set of AAs
A:
Thus GS is equivalent to GV for A consisting of only 2 AAs.
Align-GVGD includes a metric,
GD, which is either (i) the shortest Euclidean distance from
V to the rectangular prism surrounding the set
A; or (ii) zero if
V is contained within the said prism. Classification of novel nsSNPs is then achieved by reference to a plane in
GV-GD space that describes empirically determined probabilities of deleterious outcome. In the limit, the non-decreasing volume of the prism will encompass
V as MSA depth increases to include more distantly related species, hence the phenomenon described by Hicks
et al. [
5].
2.1. Correcting Overestimation of Diversity
Representing the set
A as an alphabetically sorted ASCII character string
S we defined
Sc as the compressed equivalent when utilising the DEFLATE algorithm [
9].
GV is adjusted by the ratio of the lengths of the plain and compressed strings with examples provided in
Table 1. As
GV does not necessarily overestimate diversity in a linear manner with respect to Kolmogorov complexity, we incorporated an additional factor
r to be found empirically.
2.2. Classification Metric
We combined these measures into a one-dimensional metric for quantitative comparison of nsSNPs. Such a metric would allow for analysis of clustering between sets of known deleterious and neutral variants and thus provide a means for classification of novel ones. An accurate metric should reflect intuition inferred from evolutionary theory and thus account for:
Wild-type and variant AA difference; increased GS implies greater change and thus a higher metric.
Evolutionary constraint of the specific AA position within the protein; increased GV′ of an appropriate MSA position reflects greater phylogenetic diversity, implying weaker negative selection and thus a lower metric.
- (a)
Complete constraint of a position (GV′ = 0) should leave the GS component unchanged whilst the limit as GV′ approaches infinity should be zero, i.e., absolute evolutionary freedom.
Thus we introduced an empirical, gene-specific constant k ≥ 1 and hypothesised that the Grantham Metric (GM) is an appropriate pathogenicity metric:
2.3. Empirical Constants
As Grantham’s original work compared the GS with the relative swapping frequency across the entire human genome, there was no consideration of gene-specific appropriateness of the coefficients α, β, and γ. We incorporated c, p and v to account for an individual gene’s sensitivity to specific properties.
2.4. Metric Clustering
In order to empirically assign values to k, r, c, p and v, a measure of fitness was required. As pathogenicity is a binary property, we required a measure of numerical clustering of two sets in one dimension. Increased clustering between the sample sets of deleterious and neutral metrics indicated improved distinction of the two groups by the model.
We utilised the cluster index proposed by Davies and Bouldin [
10] who were originally interested in the
similarity of clusters as against their
distinction as we are. The index is a function that satisfies two noteworthy criteria: (i) “if the distance between clusters increases while their dispersions remain constant, the [distinction] of the clusters [increases]”; and (ii) “if the distance between clusters remains constant while the dispersions increase, the [distinction] [decreases]” (original text altered to reflect our interest in
distinction). For sample sets
D (deleterious) and
N (neutral) and
x =
GM:
where distance is the absolute value of the difference of sample means χ̄ and the dispersion measure is the sample standard deviation s.
2.5. Model Training
Maximisation of the clustering index was achieved by computational means. We utilised a Particle Swarm Optimisation (PSO) algorithm [
11–
13] to achieve greatest clustering.
The swarm S contains n “particle” vectors, distributed randomly in ℝ5 at time t = 0, representing candidates for our 5 empirical constants. Each particle si ∈ S has an associated neighbourhood set Ni ⊂ S, constant over t and always including itself, i.e., si ∈ Ni. Note that the neighbourhood is not necessarily local with respect to the search space, thus allowing for swarm-wide dissemination of information regarding maxima.
Let St represent the updated values of the particles at time t. Prior to iteration of the optimisation at time T the historically optimal candidate vectors are determined both globally (sj ∈ S) and locally (sj ∈ Ni; 0 < i < n):
Iteration of each vector si involves a stochastic traversal of the search space with an attraction g and l towards
and
respectively; with the distance to historically optimal points:
and where
are random vectors with elements uniformly distributed [−1, 1]:
2.6. Classification of New Variants
2.6.1. Binary
Binary classification as deleterious or neutral was done by selecting the designation (allocation to set D or N) that maximised the cluster index R. Conversely, a decrease in R would have signified increased similarity between the two sets, a sign that the model was separating them less accurately.
It can be shown that the cut-off for designation is:
i.e., for variant AA V:
and conversely
2.6.2. Continuous
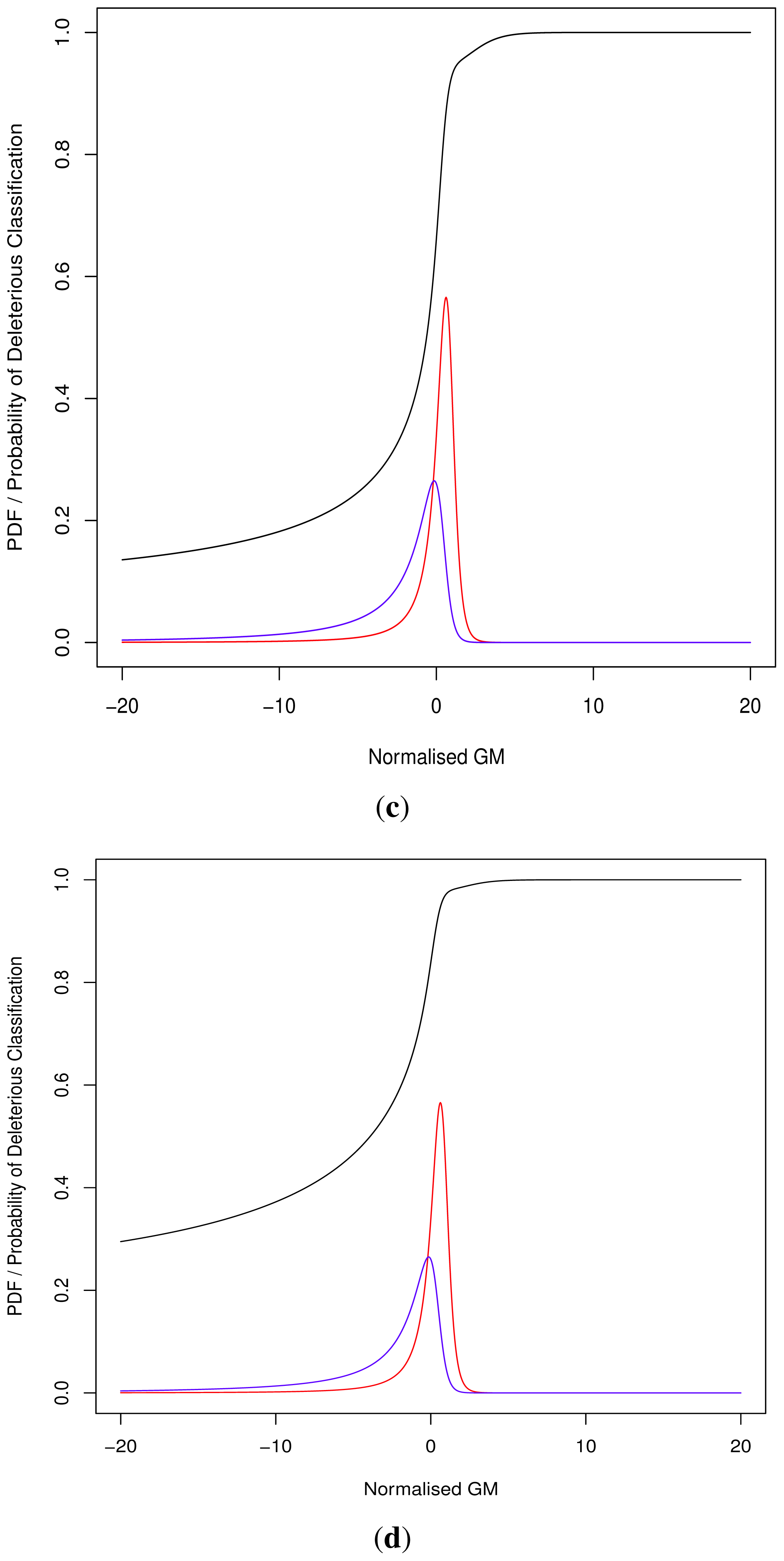
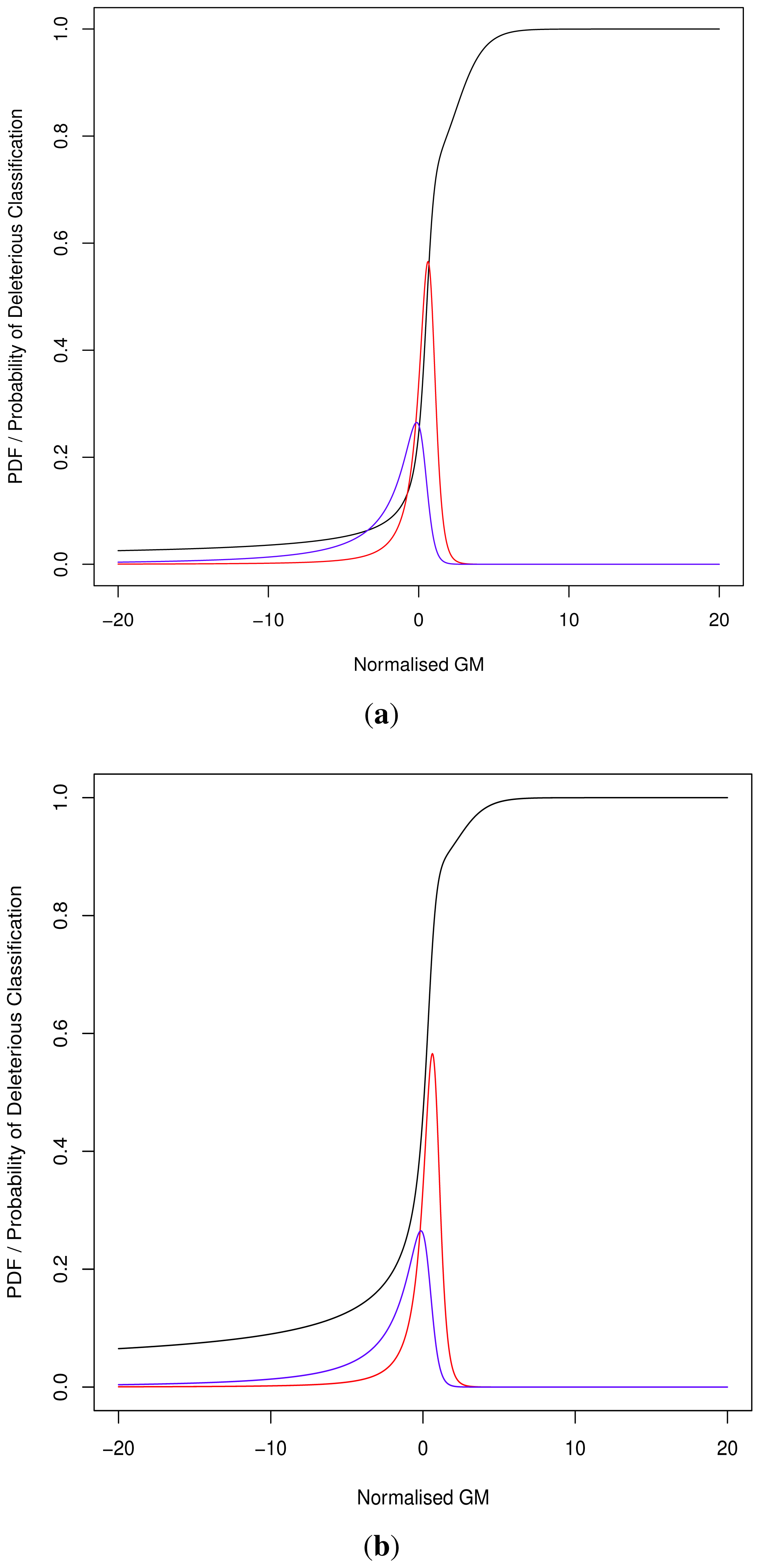
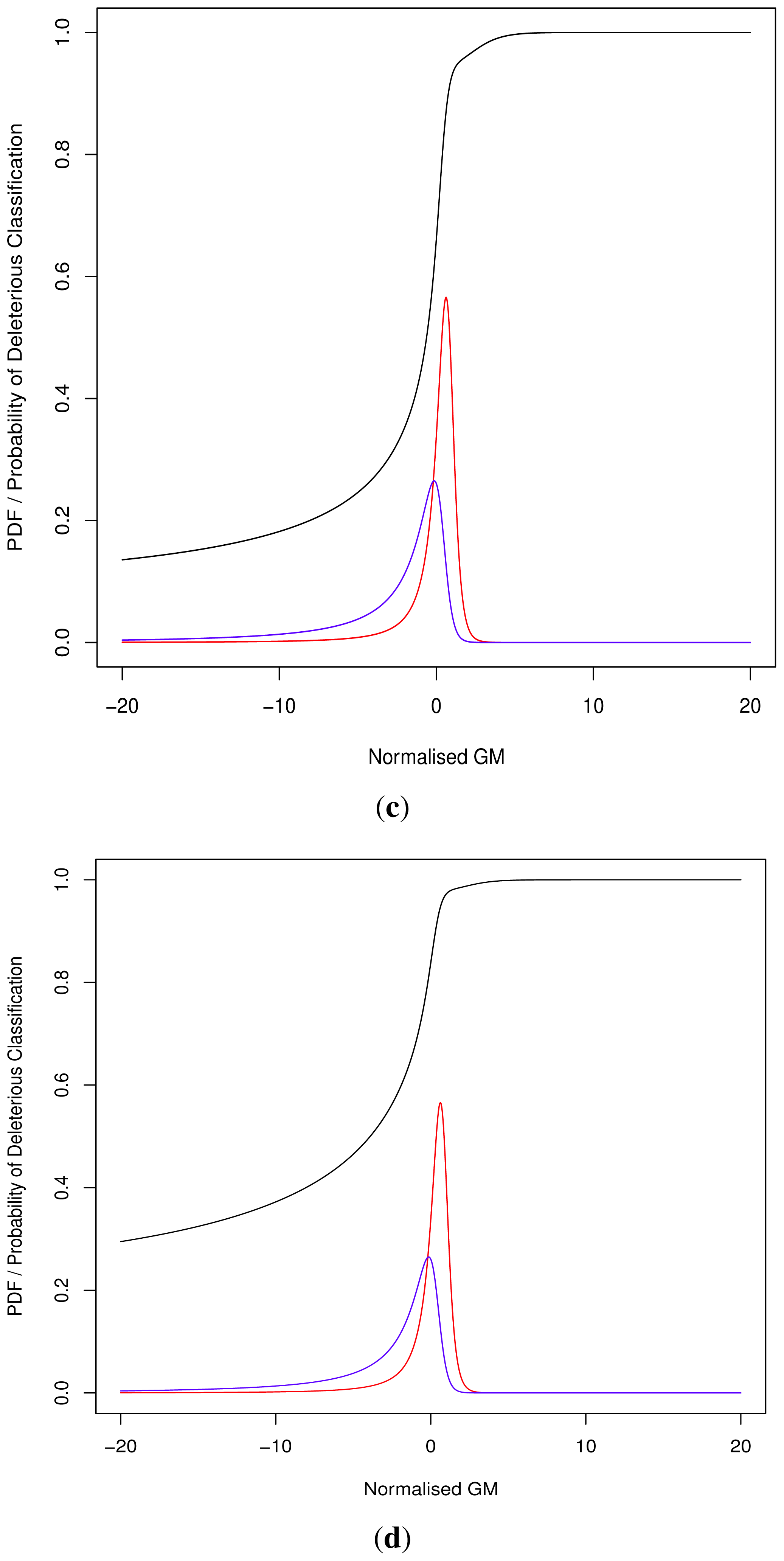
As the cut-off C differs we cannot directly compare pathogenicity metrics between genes. Thus we hypothesised a normalised metric
for modelling with continuous distributions fD(x) and fN(x) for the deleterious and neutral sets respectively.
The models fD(x) and fN(x) can then be used for Bayesian updating of the probability of deleterious classification Pr(V ∈ D). Given that point probability on a continuous distribution is undefined we consider an infinitesimally narrow region of width ε and then limit this to 0. For sufficiently small ε > 0, and M being the event that
, first note that the sets D and N are mutually exclusive and exhaustive, i.e.,:
3. Results
The HumVar [
3] training data was utilised for testing of the suitability of our algorithm. This is a large dataset of known deleterious and neutral (
MAF > 1%) variants, grouped by gene. Pre-computed MSAs accompanying the data were utilised for a leave-one-out cross-validation; for each gene we iterated through all known variants, individually excluding them and training a model based on the remaining variants of the same gene. The variant in question was then classified under the binary method and the designation was compared with the known classification.
Only genes with at least 3 variants per set were included. This, after exclusion of the variant being classified, allowed for at least 2 remaining variants in order to calculate a meaningful standard deviation for the clustering metric.
The same variants were classified using the Align-GVGD [
1] web interface and the SIFT [
2] submitting for alignment script that allows specification of MSA. The HumVar-associated MSAs were used and each variant was classified once with SIFT and multiple times with Align-GVGD with varying MSA depth. Comparison with PolyPhen [
3] could not be performed with this dataset as it was used to train the classifier and would thus favourably skew results. SIFT returned a classification of NOT SCORED for 242 (4.29%) variants.
3.1. Assessment of Classification
3.1.1. Binary
A total of 5645 variants across 212 genes were tested and a Matthews correlation coefficient (MCC) [
14] of 0.38 was achieved at the theoretically optimal cut-off
GM =
C with a maximal MCC of 0.41 at
. Compared with random allocation with equal probability of assignment to each set, this equates to
χ2(1) statistics of 833.23 (
p < 10
−16) and 968.49 (
p < 10
−16) respectively. This remained unchanged when the values
c,
p, and
v were locked at 1 (
i.e., using Grantham’s original dimension scales). Allowing
r to vary for each classification diminished performance and an optimal constant value of 2.47 was found empirically. As such, all further testing was performed in this manner, leaving
k as the only free variable.
The nature of the value of r reflects the relation between the DEFLATE algorithm and evolutionary history. One could not necessarily expect this to be a non-arbitrary value such as an integer nor any common constant and, additionally, it would change should a different compression algorithm be utilised.
We achieved sensitivity of 64.56% and specificity of 83.42% with the confusion matrix shown in
Table 2. Exclusion of the DEFLATE adjustment (
r = 0) reduces sensitivity to 58.11% and specificity to 83.24%; the greater effect on sensitivity is to be expected as this is, as described below, in keeping with Align-GVGD’s decreasing performance with increasing MSA depth.
3.1.2. Continuous
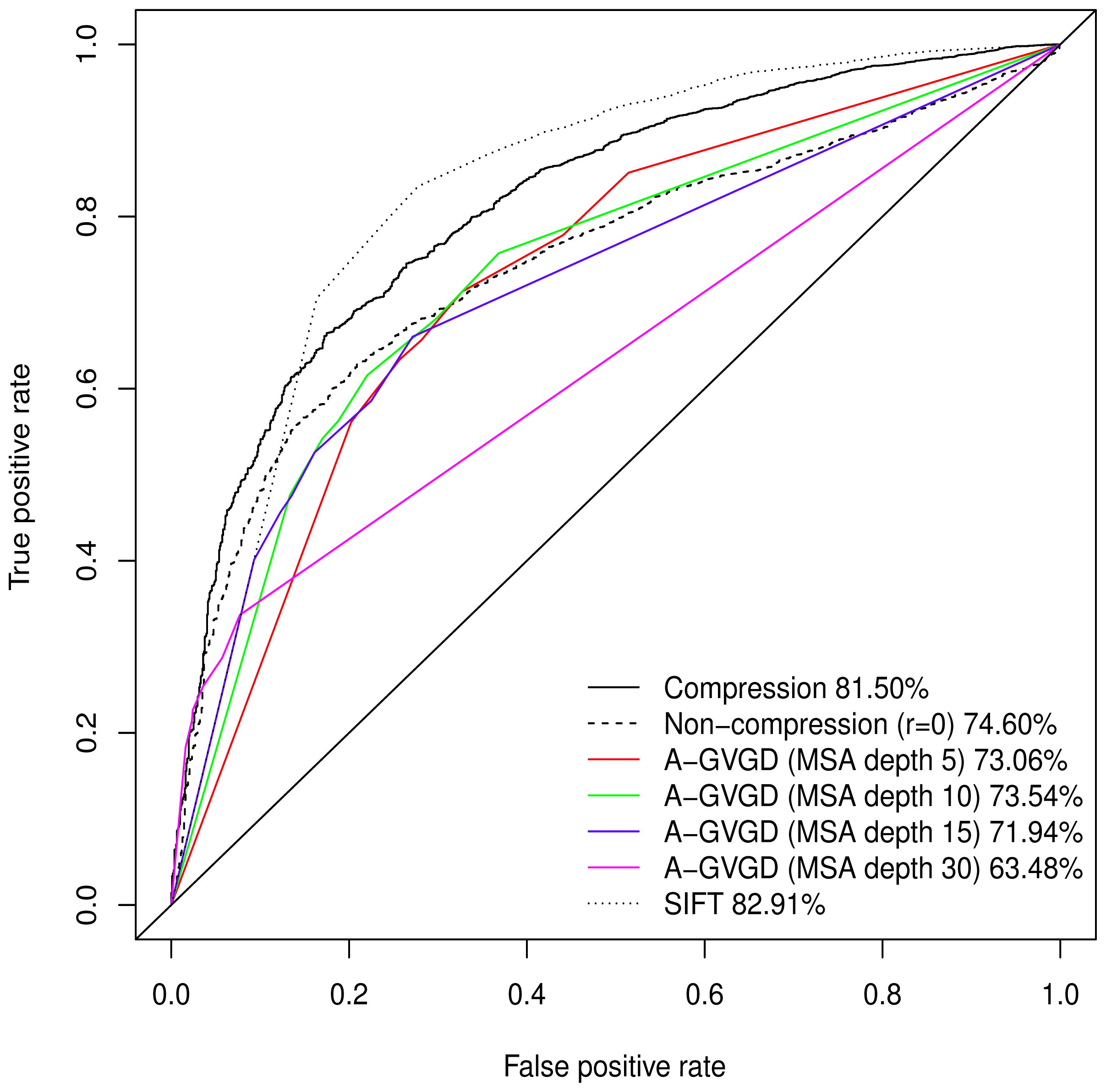
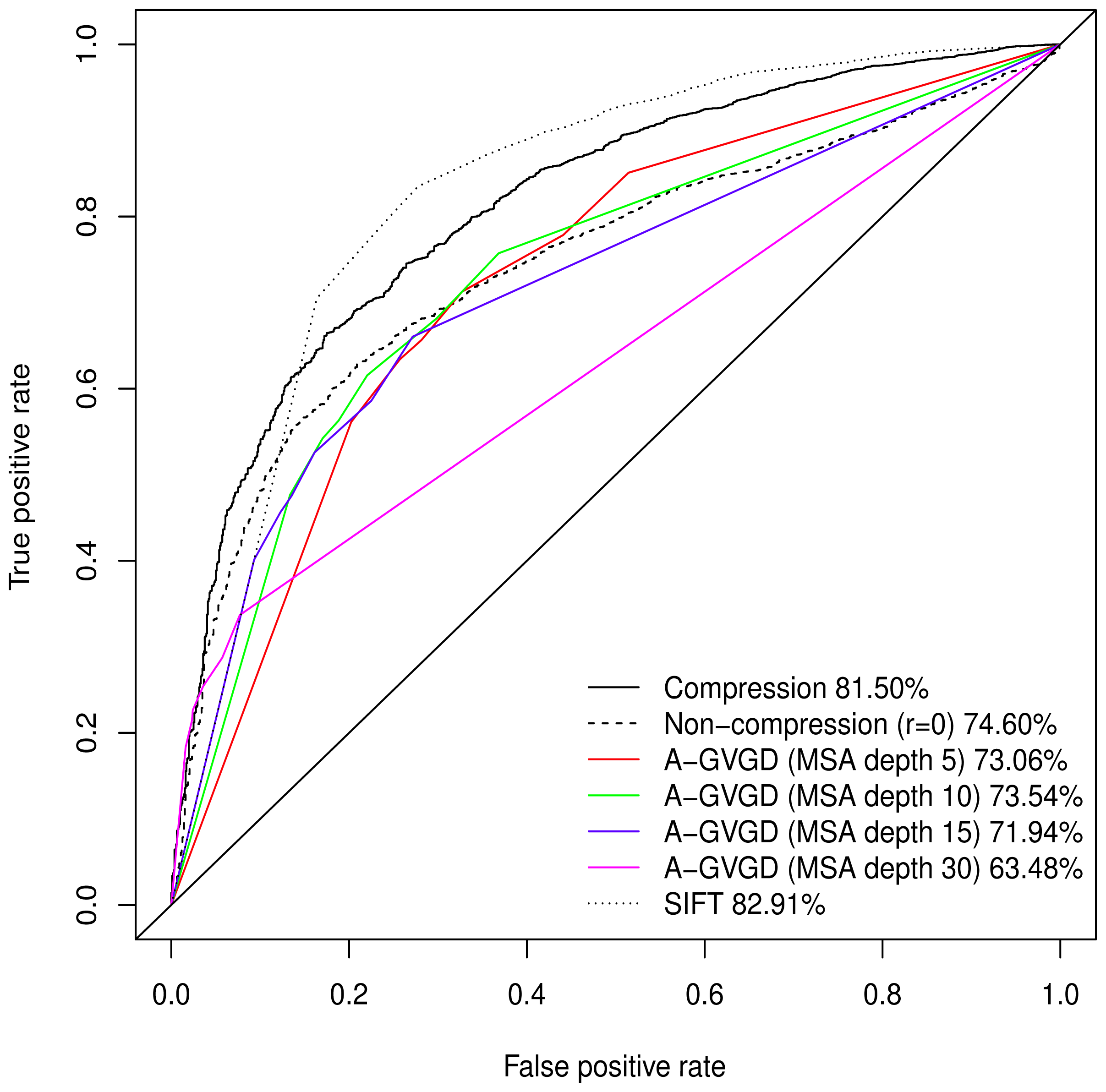
Normalised values were used to produce a Receiver Operating Characteristic (ROC) plot with an area under the curve (AUC) of 81.50%. Align-GVGD had a peak AUC of 73.54% (MSA depth of 10), decreasing with greater MSA depth, as originally shown by Hicks
et al. [
5] (
Figure 1); the pre-computed MSAs accompanying HumVar are ordered by similarity to the human protein, thus the shallower MSAs used in testing include closely related species rather than the more distantly related ones to which Align-GVGD is sensitive. The peak Align-GVGD performance was poorer than our model even without DEFLATE adjustment, which had an AUC of 74.60%. SIFT classification resulted in an AUC of 82.91%, which was marginally better than our model although, as noted above, not all variants were assigned a classification.
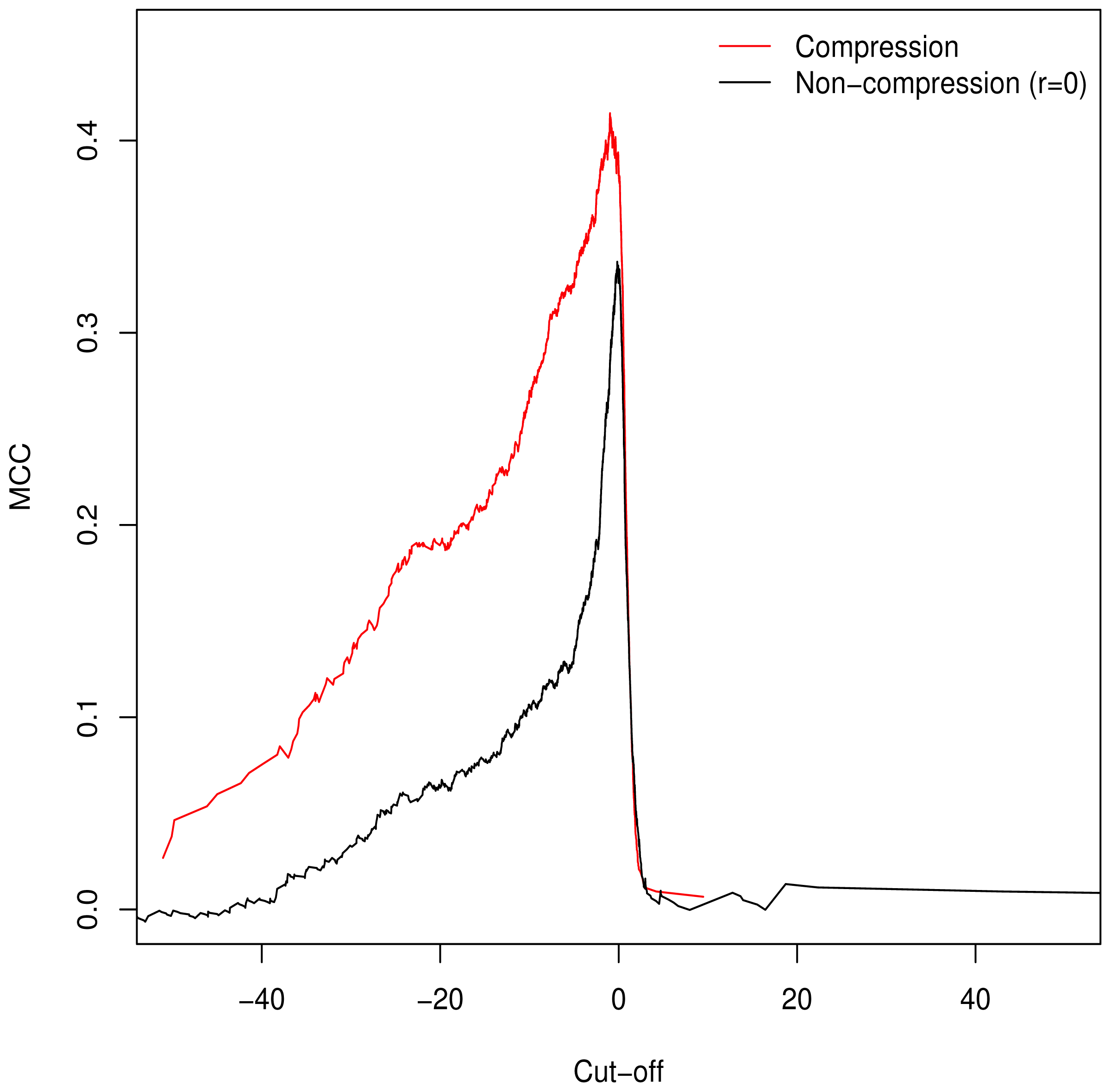
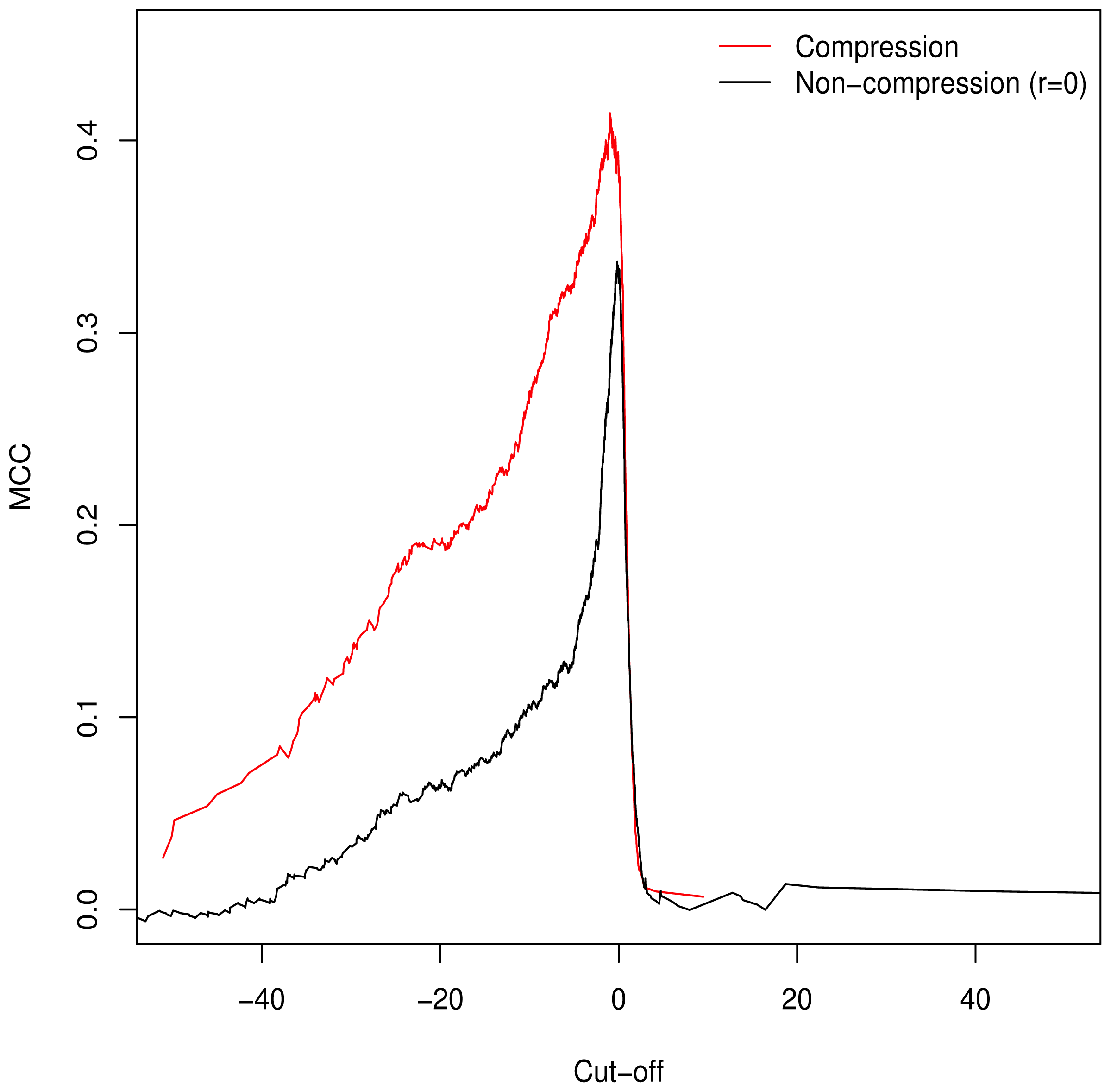
3.2. Validation of Clustering Hypothesis
Unlike the AUC, the MCC is defined at a specific cut-off point for binary classification; additionally it is of use in assessing classification accuracy as it controls for differences in the sizes of the positive and negative sets [
15,
16] (a naive classification model can simply assign all predictions to the larger set, guaranteeing ≥50% accuracy). As such, we used it to determine the empirically optimal cut-off value. The MCC was calculated for the full range of possible cut-off values;
Figure 2 demonstrates peaking around
, which is in keeping with our theorised cut-off of
GM =
C as described in
Equation (6).
4. Discussion
4.1. Choice of Binary or Continuous Classification
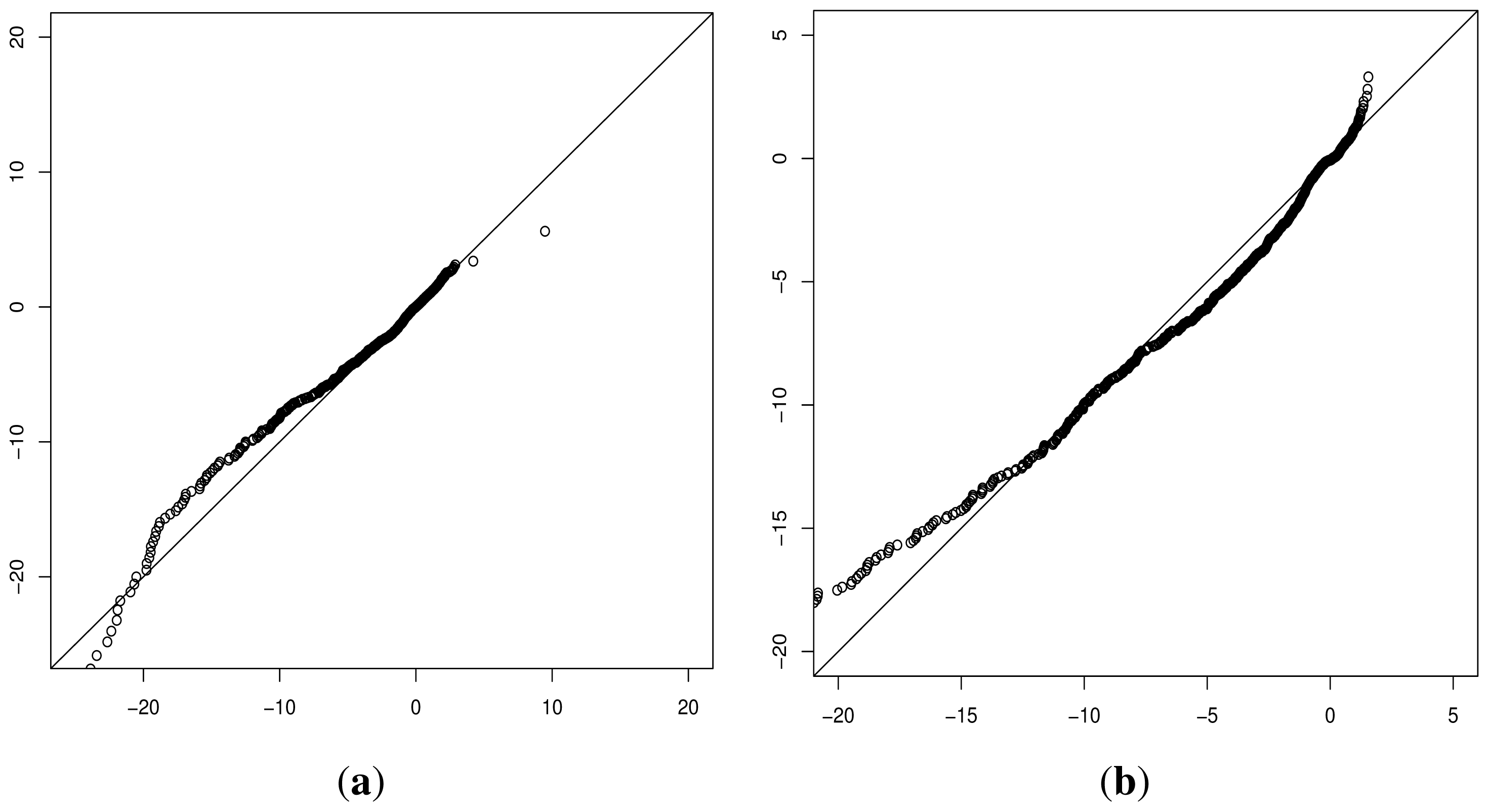
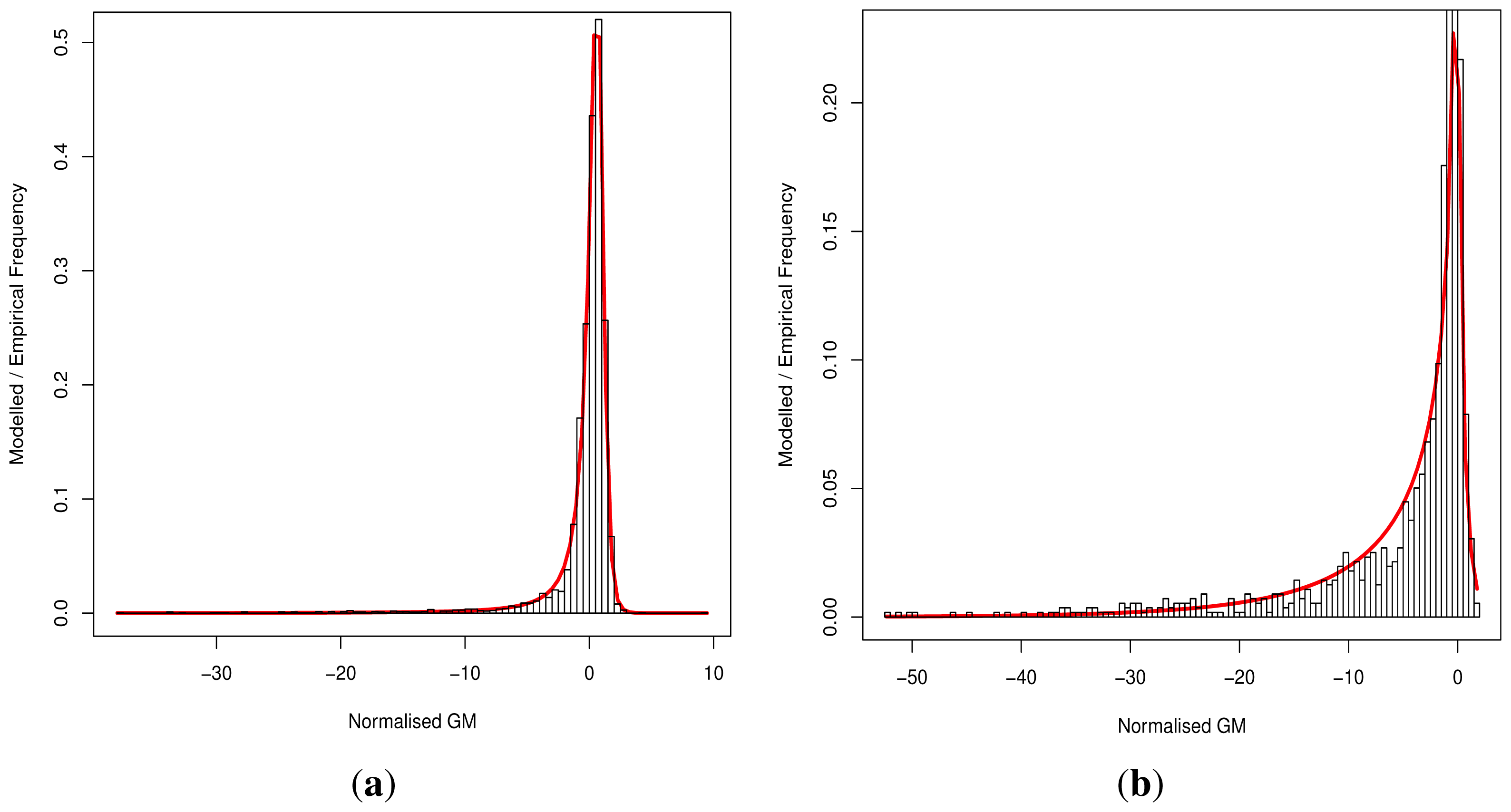
During the development of our approach, we were questioned as to the need for both methods of classification. Ideally a perfect binary classifier would be produced with 100% certain results. In the absence of such ideal circumstances, we rely on probabilistic measures of certainty such as positive and negative predictive values. Despite this, researchers may require a more nuanced comparison of multiple variants, perhaps in the triage of further laboratory investigations where those with a greater classification metric (despite the same binary classification) may receive preferential attention.
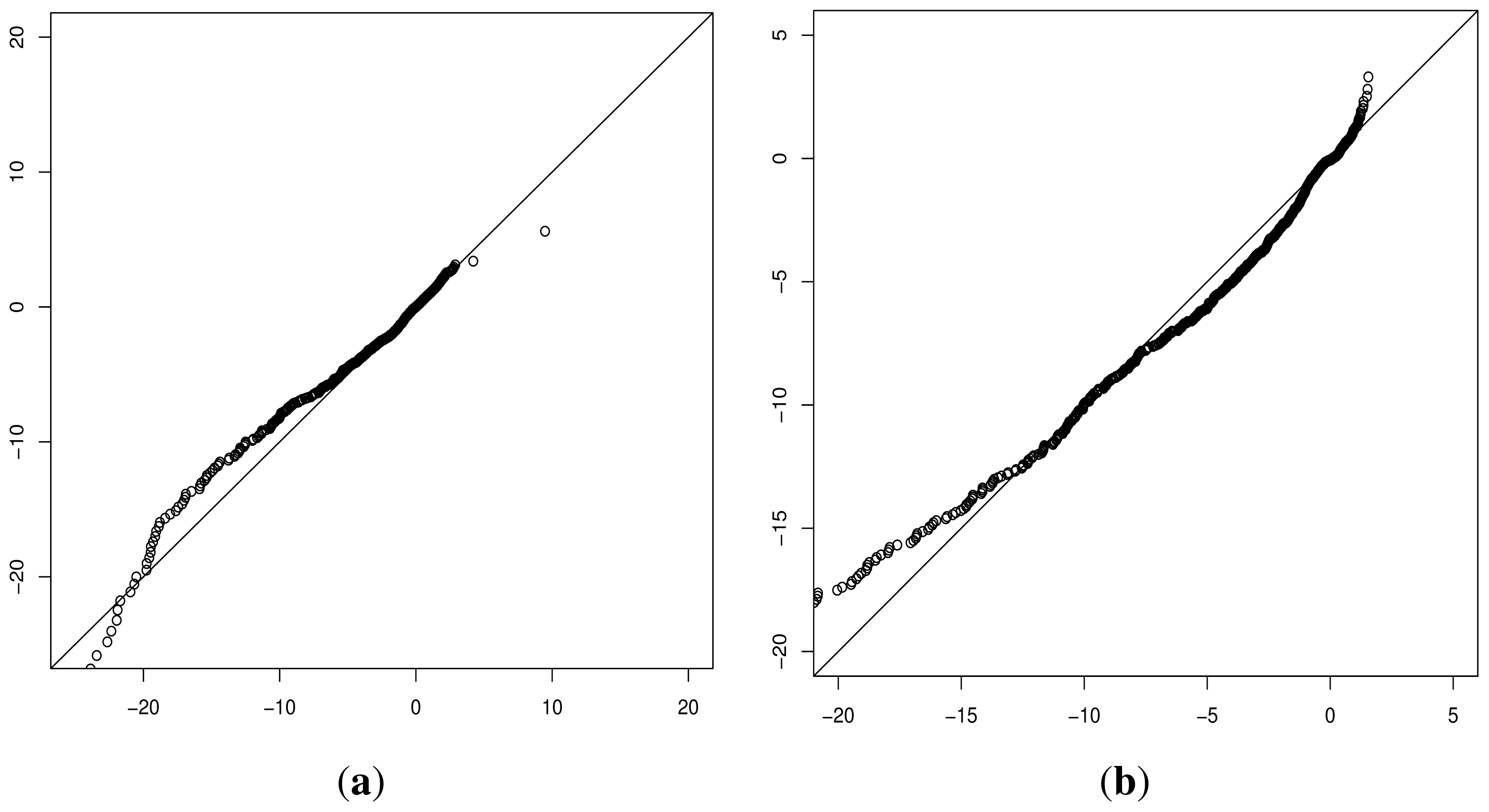
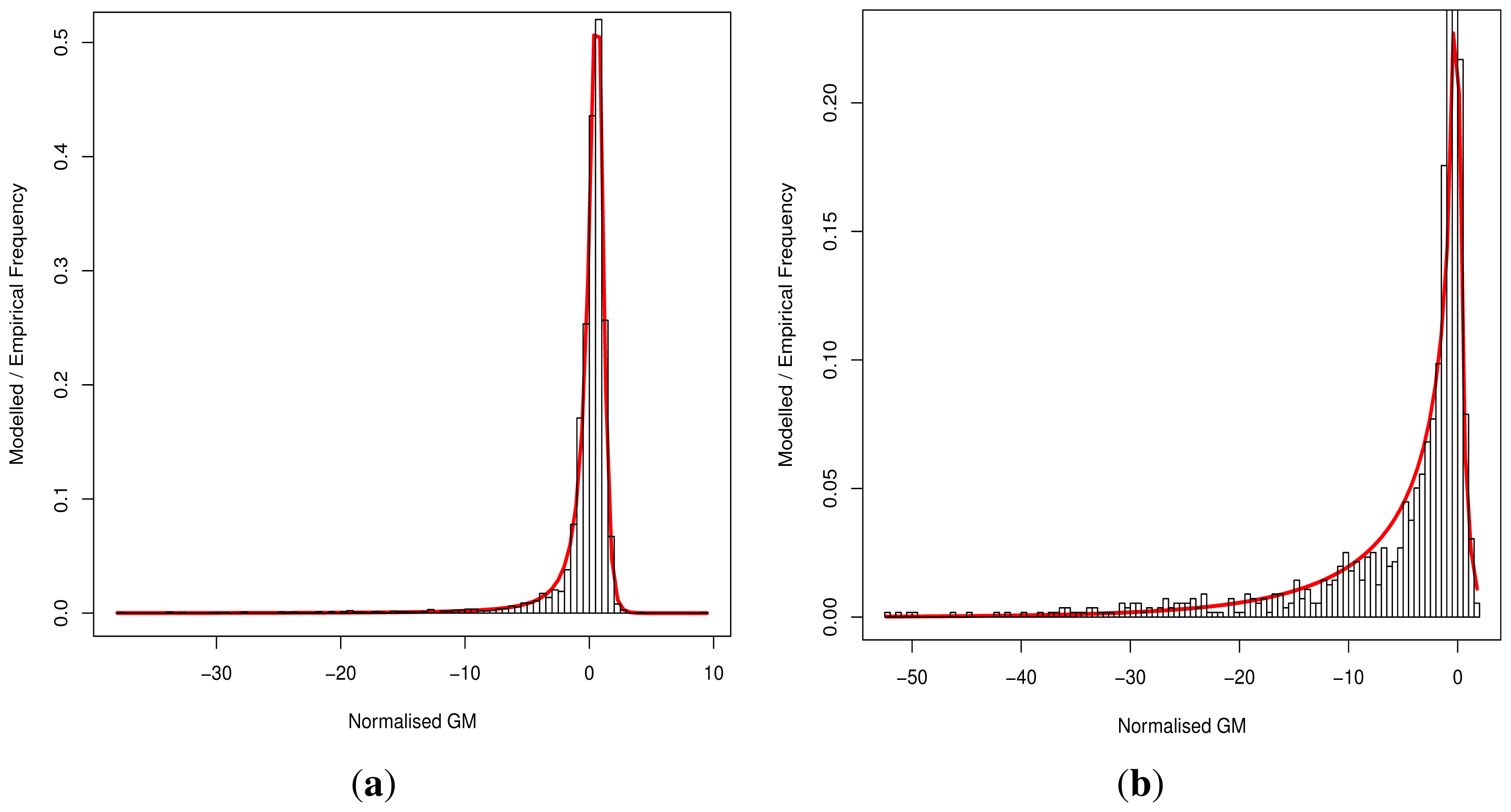
Although progress was made towards determining optimal distributions for
fD(
x) and
fN(
x) as required by
Equation (8), goodness-of-fit tests showed tail deviation such that we believe that they are currently unsuitable for accurate reflection of probability. Despite this, the normalised value
can still be used for inter-gene comparison of the probability of deleterious outcome; this will provide ordinal relations with magnitudes correlating positively with actual probability. Details of modelling progress are outlined in the Supplementary Information wherein we have demonstrated promising outcomes with a statistically feasible choice of models that result in posterior probability monotonically increasing with respect to
.
It has been suggested that in this case only the continuous approach is required; however, the binary method is a logical prerequisite as
Equation (7) relies on
Equation (6).
4.2. Application and Limitations
The scope of evolutionary constraint must be taken into consideration when utilising our model. Genes associated with pathology presenting after reproductive age are intuitively less well suited whilst those associated with congenital abnormalities should be more so. Furthermore, lowered genotypic penetrance will affect results.
Additionally, we recognise that our approach is limited to genes with adequate numbers of known nsSNPs (at least 2 for each of deleterious and neutral). Although our proposed method is not universally applicable, it demonstrates a significant utilisation of additional data where such information is available.
4.3. Choice of Classifier
Researchers have many options available for in-silico classification of novel nsSNPs. The suitability of each is dependent on individual use cases; as already noted, both our approach and SIFT do not assign classifications to all SNPs whilst Align-GVGD is limited by careful MSA curation. In situations amenable to GM-analysis we have demonstrated an objective improvement over Align-GVGD and suggest substitution. Additionally, the Bayesian updating detailed in Section 4.1 is of great benefit in that it allows a simple means of incorporating our methodology into an existing classification pipeline.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}