Multiple Hits, Including Oxidative Stress, as Pathogenesis and Treatment Target in Non-Alcoholic Steatohepatitis (NASH)

Abstract
:1. Introduction
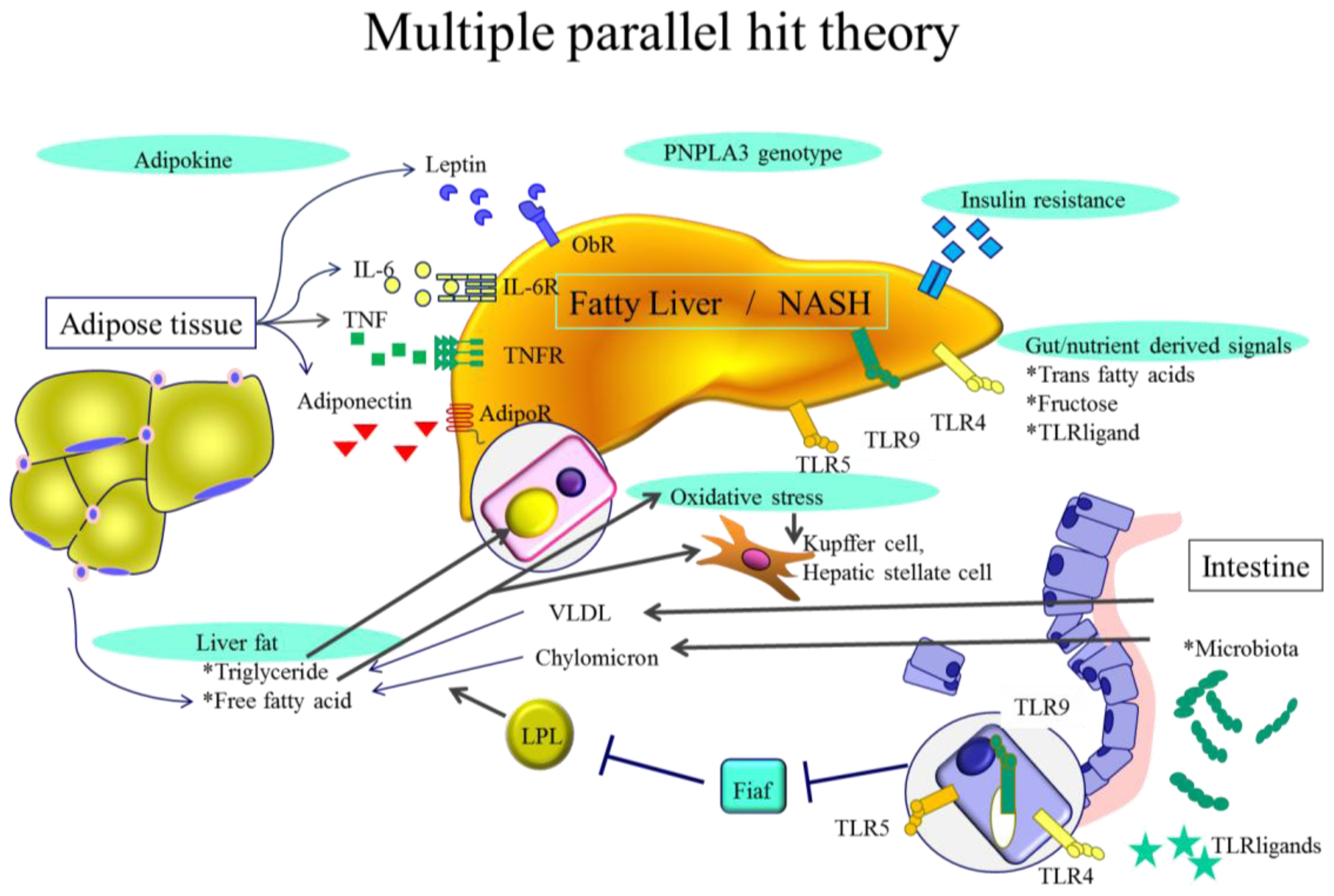
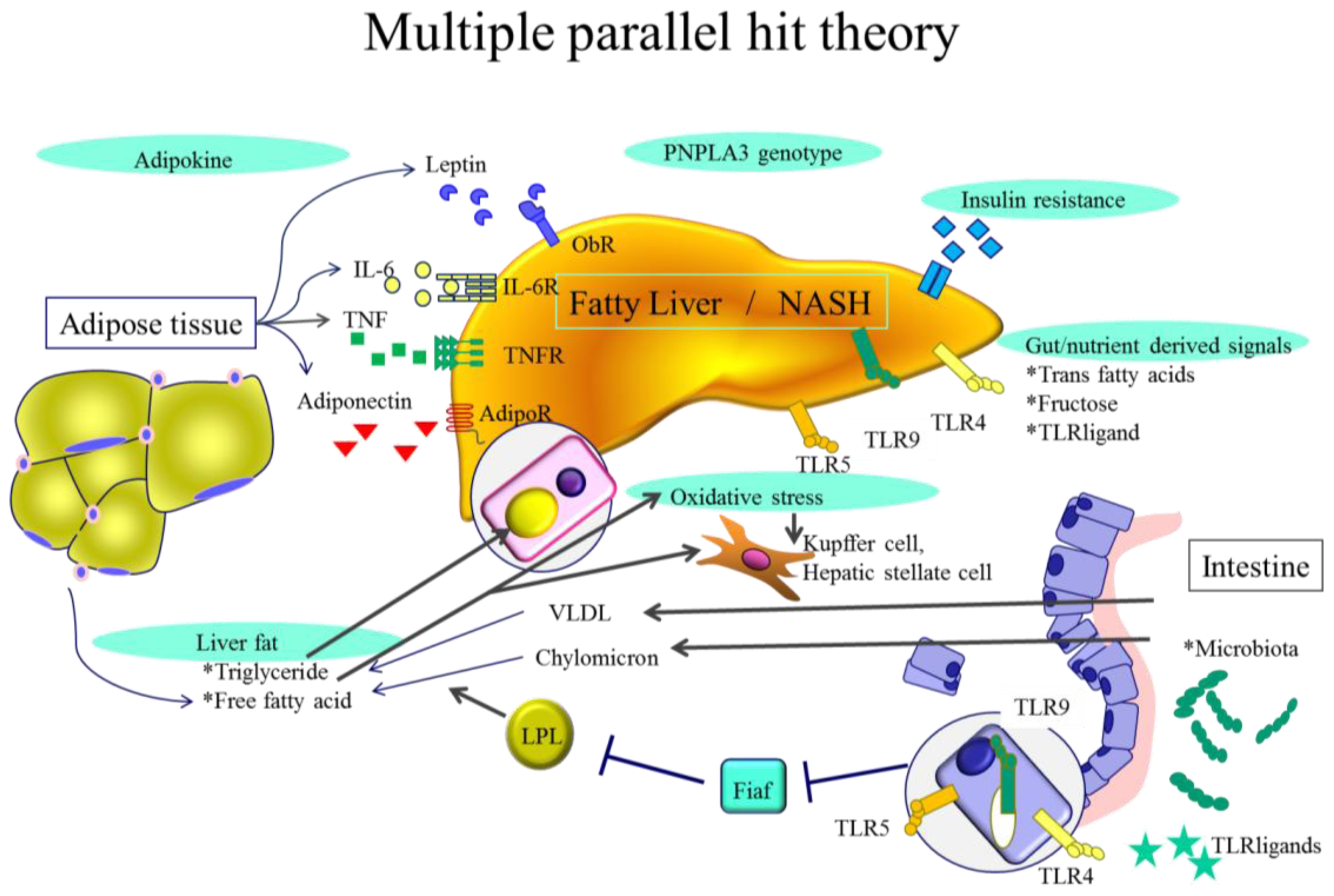
2. NASH Pathogenesis—General Characteristics
2.1. Genetic Background
2.2. Visceral Obesity
2.3. Insulin Resistance
2.4. Hepatic Steatosis
3. NASH Pathogenesis—Cellular Levels
3.1. Adipokines
3.2. Endotoxin and Gut Derived Signals
3.3. Toll-Like Receptors (TLRs)
3.4. Endoplasmic Reticulum (ER) Stress
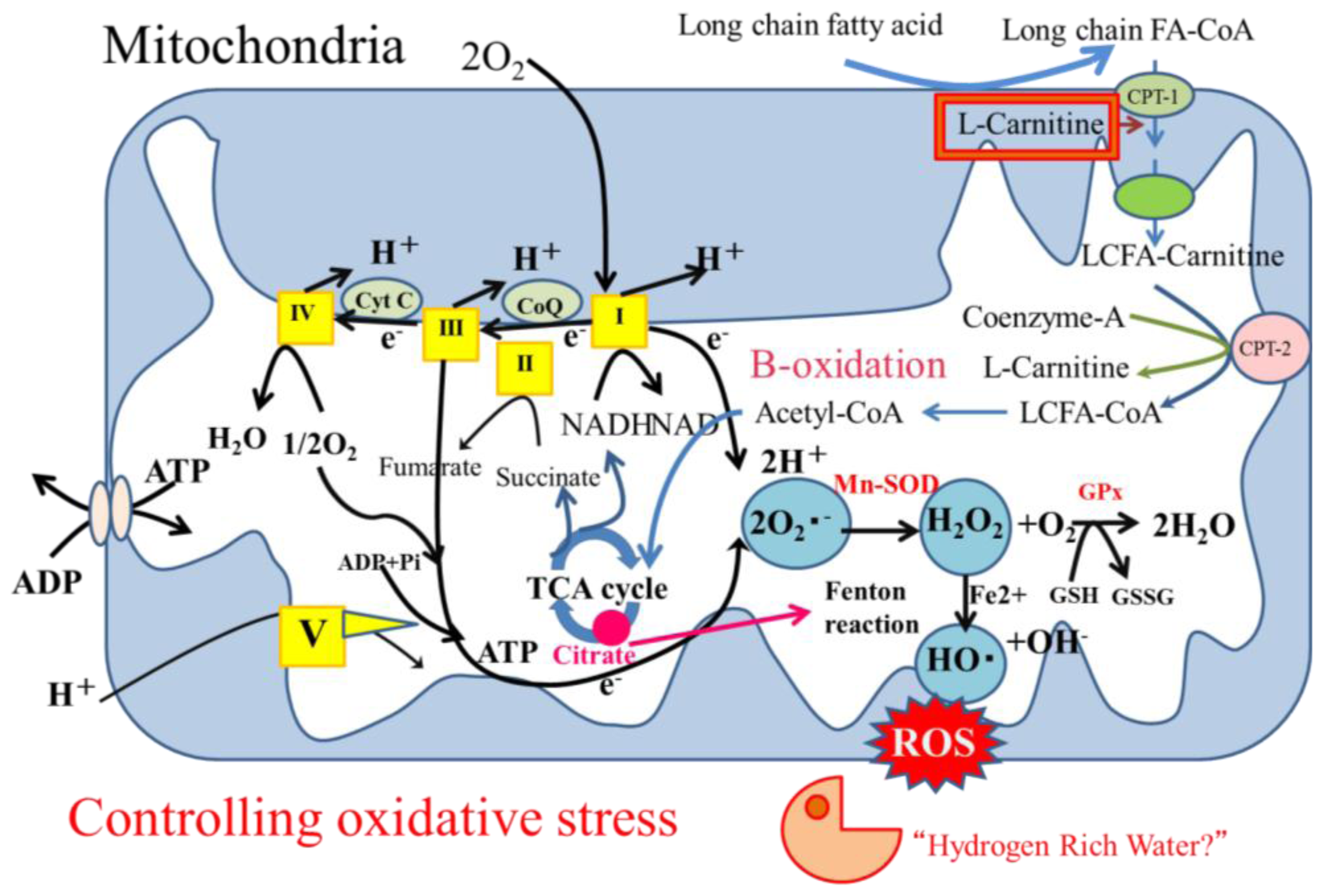
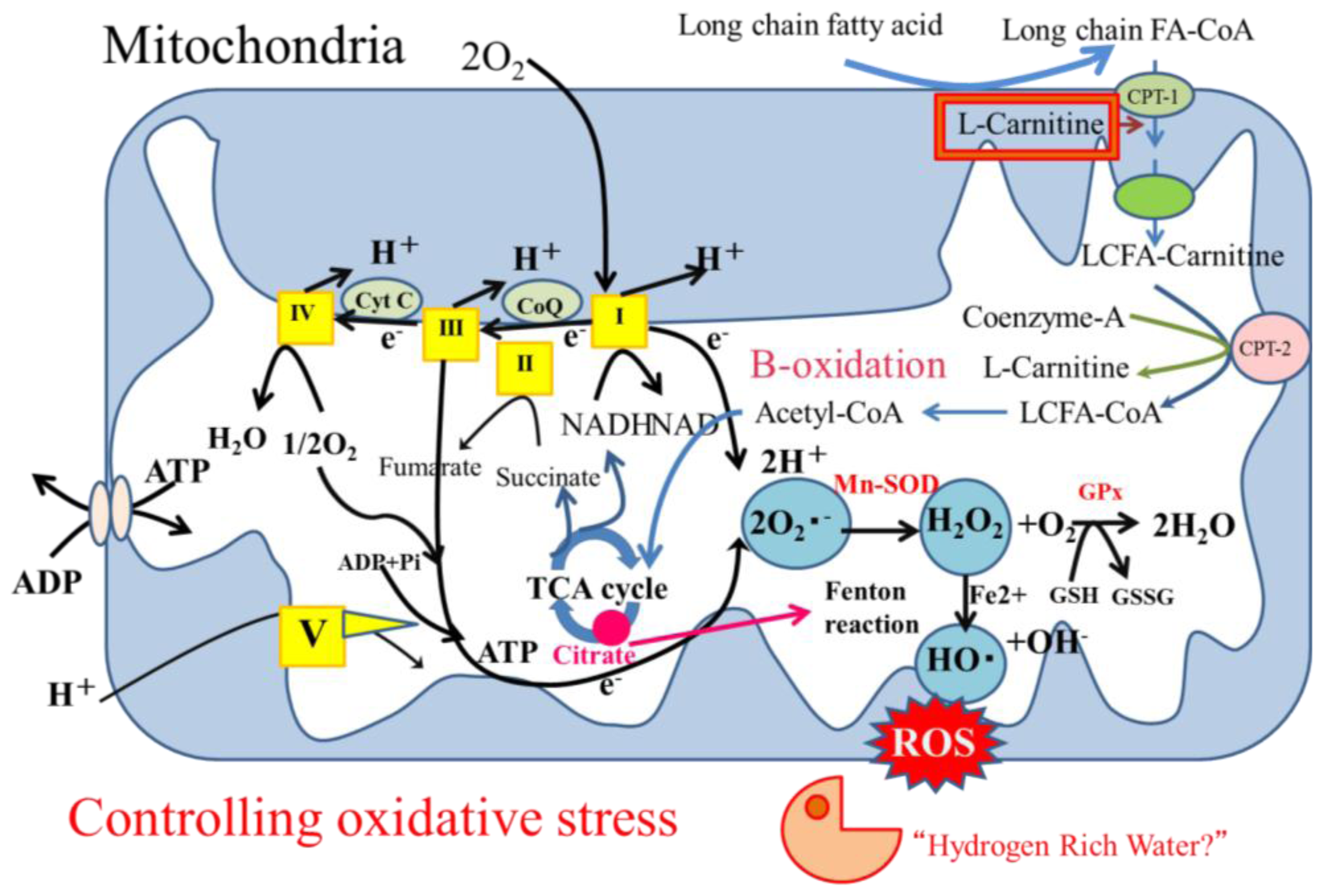
3.5. Oxidative Stress
3.5.1. Oxidative Stress in Hepatocytes
3.5.2. Oxidative Stress Affects outside Hepatocytes
4. Treatment for NASH
4.1. General Aspects
4.2. Hydrogen-Rich Water as a Candidate Treatment for NASH
4.2.1. Hydrogen as an Antioxidant Treatment
4.2.2. Hydrogen as an Antioxidant Treatment Candidate for NASH
5. Conclusions

{kind=link}
{kind=link}
| Targets | Hydrogen Administration | Reference Number |
|---|---|---|
| Disease models | ||
| Cerebral ischemia reperfusion injury | Gas | [18] |
| Liver ischemia reperfusion injury | Gas | [111] |
| Myocardial ischemia reperfusion injury | Gas | [112] |
| Lung ischemia reperfusion injury | Gas | [113] |
| Parkinson’s disease | Drinking water | [114] |
| Atherosclerosis | Drinking water | [115] |
| Obese diabetes | Drinking water | [116] |
| Type1 diabetes | Drinking water and intraperitoneal injection | [117] |
| Diabetic retinopathy | Intraperitoneal injection | [118] |
| Chemically-induced liver injury | Intraperitoneal injection | [119] |
| NASH | Drinking water | [70] |
| Cisplatin-induced renal injury | Gas and drinking water | [120] |
| Post renal transplant rejection | Drinking water | [19] |
| Intestinal transplant rejection | Graft storage in hydrogen bubbled preservative | [121] |
| Clinical trials | ||
| Glucose and lipid metabolism type 2 diabetes | Drinking water | [122] |
| Metabolic syndrome | Drinking water | [123] |
Conflicts of Interest
References
- Pacifico, L.; Anania, C.; Martino, F.; Poggiogalle, E.; Chiarelli, F.; Arca, M.; Chiesa, C. Management of metabolic syndrome in children and adolescents. Nutr. Metab. Cardiovasc. Dis 2011, 21, 455–466. [Google Scholar]
- Matteoni, C.A.; Younossi, Z.M.; Gramlich, T.; Boparai, N.; Liu, Y.C.; McCullough, A.J. Nonalcoholic fatty liver disease: A spectrum of clinical and pathological severity. Gastroenterology 1999, 116, 1413–1419. [Google Scholar]
- Brunt, E.M.; Kleiner, D.E.; Wilson, L.A.; Unalp, A.; Behling, C.E.; Lavine, J.E.; Neuschwander-Tetri, B.A. Portal chronic inflammation in nonalcoholic fatty liver disease (NAFLD): A histologic marker of advanced NAFLD-Clinicopathologic correlations from the nonalcoholic steatohepatitis clinical research network. Hepatology 2009, 49, 809–820. [Google Scholar]
- Yatsuji, S.; Hashimoto, E.; Tobari, M.; Taniai, M.; Tokushige, K.; Shiratori, K. Clinical features and outcomes of cirrhosis due to non-alcoholic steatohepatitis compared with cirrhosis caused by chronic hepatitis C. J. Gastroenterol. Hepatol 2009, 24, 248–254. [Google Scholar]
- Hatanaka, K.; Kudo, M.; Fukunaga, T.; Ueshima, K.; Chung, H.; Minami, Y.; Sakaguchi, Y.; Hagiwara, S.; Orino, A.; Osaki, Y. Clinical characteristics of NonBNonC-HCC: Comparison with HBV and HCV related HCC. Intervirology 2007, 50, 24–31. [Google Scholar]
- Fassio, E.; Alvarez, E.; Dominguez, N.; Landeira, G.; Longo, C. Natural history of nonalcoholic steatohepatitis: A longitudinal study of repeat liver biopsies. Hepatology 2004, 40, 820–826. [Google Scholar]
- Ono, M.; Saibara, T. Clinical features of nonalcoholic steatohepatitis in Japan: Evidence from the literature. J. Gastroenterol 2006, 41, 725–732. [Google Scholar]
- Gentile, C.L.; Pagliassotti, M.J. The role of fatty acids in the development and progression of nonalcoholic fatty liver disease. J. Nutr. Biochem 2008, 19, 567–576. [Google Scholar]
- Csak, T.; Ganz, M.; Pespisa, J.; Kodys, K.; Dolganiuc, A.; Szabo, G. Fatty acids and endotoxin activate inflammasome in hepatocytes which release danger signals to activate immune cells in steatohepatitis. Hepatology 2011, 54, 133–144. [Google Scholar]
- Pessayre, D. Role of mitochondria in non-alcoholic fatty liver disease. J. Gastroenterol. Hepatol 2007, 22, S20–S27. [Google Scholar]
- Novo, E.; Busletta, C.; Bonzo, L.V.; Povero, D.; Paternostro, C.; Mareschi, K.; Ferrero, I.; David, E.; Bertolani, C.; Caligiuri, A.; et al. Intracellular reactive oxygen species are required for directional migration of resident and bone marrow-derived hepatic pro-fibrogenic cells. J. Hepatol 2011, 54, 964–974. [Google Scholar]
- Sanyal, A.J.; Chalasani, N.; Kowdley, K.V.; McCullough, A.; Diehl, A.M.; Bass, N.M.; Neuschwander-Tetri, B.A.; Lavine, J.E.; Tonascia, J.; Unalp, A.; et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N. Engl. J. Med 2010, 362, 1675–1685. [Google Scholar]
- Nan, Y.M.; Wu, W.J.; Fu, N.; Liang, B.L.; Wang, R.Q.; Li, L.X.; et al. Antioxidants vitamin E and 1-aminobenzotriazole prevent experimental non-alcoholic steatohepatitis in mice. Scand. J. Gastroenterol 2009, 44, 1121–1131. [Google Scholar]
- Bugianesi, E.; Gentilcore, E.; Manini, R.; Natale, S.; Vanni, E.; Villanova, N.; David, E.; Rizzetto, M.; Marchesini, G. A randomized controlled trial of metformin versus vitamin E or prescriptive diet in nonalcoholic fatty liver disease. Am. J. Gastroenterol 2005, 100, 1082–1090. [Google Scholar]
- Musso, G.; Gambino, R.; Cassader, M.; Pagano, G. A meta-analysis of randomized trials for the treatment of nonalcoholic fatty liver disease. Hepatology 2010, 52, 79–104. [Google Scholar]
- Steinhubl, S.R. Why have antioxidants failed in clinical trials? Am. J. Cardiol 2008, 101, S14–S19. [Google Scholar]
- Ohta, S. Molecular hydrogen is a novel antioxidant to efficiently reduce oxidative stress with potential for the improvement of mitochondrial diseases. Biochim. Biophys. Acta 2011, 1820, 586–594. [Google Scholar]
- Ohsawa, I.; Ishikawa, M.; Takahashi, K.; Watanabe, M.; Nishimaki, K.; Yamagata, K.; Katsura, K.; Katayama, Y.; Asoh, S.; Ohta, S. Hydrogen acts as a therapeutic antioxidant by selectively reducing cytotoxic oxygen radicals. Nat. Med 2007, 13, 688–694. [Google Scholar]
- Cardinal, J.S.; Zhan, J.; Wang, Y.; Sugimoto, R.; Tsung, A.; McCurry, K.R.; Billiar, T.R.; Nakao, A. Oral hydrogen water prevents chronic allograft nephropathy in rats. Kidney Int 2010, 77, 101–109. [Google Scholar]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar]
- Tiniakos, D.G.; Vos, M.B.; Brunt, E.M. Nonalcoholic fatty liver disease: Pathology and pathogenesis. Annu. Rev. Pathol 2010, 5, 145–171. [Google Scholar]
- Li, Z.; Yang, S.; Lin, H.; Huang, J.; Watkins, P.A.; Moser, A.B.; Desimone, C.; Song, X.Y.; Diehl, A.M. Probiotics and antibodies to TNF inhibit inflammatory activity and improve nonalcoholic fatty liver disease. Hepatology 2003, 37, 343–350. [Google Scholar]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet 2008, 40, 1461–1465. [Google Scholar]
- Valenti, L.; Alisi, A.; Galmozzi, E.; Bartuli, A.; Del Menico, B.; Alterio, A.; Dongiovanni, P.; Fargion, S.; Nobili, V. I148M patatin-like phospholipase domain-containing 3 gene variant and severity of pediatric nonalcoholic fatty liver disease. Hepatology 2010, 52, 1274–1280. [Google Scholar]
- Kawaguchi, T.; Sumida, Y.; Umemura, A.; Matsuo, K.; Takahashi, M.; Takamura, T.; Yasui, K.; Saibara, T.; Hashimoto, E.; Kawanaka, M.; et al. Genetic polymorphisms of the human PNPLA3 gene are strongly associated with severity of non-alcoholic fatty liver disease in Japanese. PloS One 2012, 7, e38322. [Google Scholar]
- Qiao, A.; Liang, J.; Ke, Y.; Li, C.; Cui, Y.; Shen, L.; Zhang, H.; Cui, A.; Liu, X.; Liu, C.; et al. Mouse patatin-like phospholipase domain-containing 3 influences systemic lipid and glucose homeostasis. Hepatology 2011, 54, 509–521. [Google Scholar]
- Tsuneto, A.; Hida, A.; Sera, N.; Imaizumi, M.; Ichimaru, S.; Nakashima, E.; Seto, S.; Maemura, K.; Akahoshi, M. Fatty liver incidence and predictive variables. Hypertens. Res 2010, 33, 638–643. [Google Scholar]
- Eguchi, Y.; Eguchi, T.; Mizuta, T.; Ide, Y.; Yasutake, T.; Iwakiri, R.; Hisatomi, A.; Ozaki, I.; Yamamoto, K.; Kitajima, Y.; et al. Visceral fat accumulation and insulin resistance are important factors in nonalcoholic fatty liver disease. J. Gastroenterol 2006, 41, 462–469. [Google Scholar]
- Nobili, V.; Svegliati-Baroni, G.; Alisi, A.; Miele, L.; Valenti, L.; Vajro, P. A 360-degree overview of paediatric NAFLD: Recent insights. J. Hepatol 2013, 58, 1218–1229. [Google Scholar]
- Musso, G.; Cassader, M.; de Michieli, F.; Rosina, F.; Orlandi, F.; Gambino, R. Nonalcoholic steatohepatitis versus steatosis: Adipose tissue insulin resistance and dysfunctional response to fat ingestion predict liver injury and altered glucose and lipoprotein metabolism. Hepatology 2012, 56, 933–942. [Google Scholar]
- Yamaguchi, K.; Yang, L.; McCall, S.; Huang, J.; Yu, X.X.; Pandey, S.K.; Bhanot, S.; Monia, B.P.; Li, Y.X.; Diehl, A.M. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology 2007, 45, 1366–1374. [Google Scholar]
- Yu, X.X.; Murray, S.F.; Pandey, S.K.; Booten, S.L.; Bao, D.; Song, X.Z.; Kelly, S.; Chen, S.; McKay, R.; Monia, B.P.; et al. Antisense oligonucleotide reduction of DGAT2 expression improves hepatic steatosis and hyperlipidemia in obese mice. Hepatology 2005, 42, 362–371. [Google Scholar]
- Kantartzis, K.; Machicao, F.; Machann, J.; Schick, F.; Fritsche, A.; Haring, H.U.; Stefan, N. The DGAT2 gene is a candidate for the dissociation between fatty liver and insulin resistance in humans. Clin. Sci 2009, 116, 531–537. [Google Scholar]
- Corbetta, S.; Bulfamante, G.; Cortelazzi, D.; Barresi, V.; Cetin, I.; Mantovani, G.; Bondioni, S.; Beck-Peccoz, P.; Spada, A. Adiponectin expression in human fetal tissues during mid- and late gestation. J. Clin. Endocrinol. Metab 2005, 90, 2397–2402. [Google Scholar]
- Polyzos, S.A.; Toulis, K.A.; Goulis, D.G.; Zavos, C.; Kountouras, J. Serum total adiponectin in nonalcoholic fatty liver disease: A systematic review and meta-analysis. Metabolism 2011, 60, 313–326. [Google Scholar]
- Carbone, F.; la Rocca, C.; Matarese, G. Immunological functions of leptin and adiponectin. Biochimie 2012, 94, 2082–2088. [Google Scholar]
- Shimada, M.; Kawahara, H.; Ozaki, K.; Fukura, M.; Yano, H.; Tsuchishima, M.; Bondioni, S.; Beck-Peccoz, P.; Spada, A. Usefulness of a combined evaluation of the serum adiponectin level, HOMA-IR, and serum type IV collagen 7S level to predict the early stage of nonalcoholic steatohepatitis. Am. J. Gastroenterol 2007, 102, 1931–1938. [Google Scholar]
- Younossi, Z.M.; Jarrar, M.; Nugent, C.; Randhawa, M.; Afendy, M.; Stepanova, M.; Bondioni, S.; Beck-Peccoz, P.; Spada, A. A novel diagnostic biomarker panel for obesity-related nonalcoholic steatohepatitis (NASH). Obes. Surg 2008, 18, 1430–1437. [Google Scholar]
- Argentou, M.; Tiniakos, D.G.; Karanikolas, M.; Melachrinou, M.; Makri, M.G.; Kittas, C.; Kalfarentzos, F. Adipokine serum levels are related to liver histology in severely obese patients undergoing bariatric surgery. Obes. Surg 2009, 19, 1313–1323. [Google Scholar]
- Lemoine, M.; Ratziu, V.; Kim, M.; Maachi, M.; Wendum, D.; Paye, F.; Bastard, J.P.; Poupon, R.; Housset, C.; Capeau, J.; et al. Serum adipokine levels predictive of liver injury in non-alcoholic fatty liver disease. Liver Int 2009, 29, 1431–1438. [Google Scholar]
- Kaser, S.; Moschen, A.; Cayon, A.; Kaser, A.; Crespo, J.; Pons-Romero, F.; Ebenbichler, C.F.; Patsch, J.R.; Tilg, H. Adiponectin and its receptors in non-alcoholic steatohepatitis. Gut 2005, 54, 117–121. [Google Scholar]
- Matsunami, T.; Sato, Y.; Ariga, S.; Sato, T.; Shimomura, T.; Kashimura, H.; Hasegawa, Y.; Yukawa, M. Regulation of synthesis and oxidation of fatty acids by adiponectin receptors (AdipoR1/R2) and insulin receptor substrate isoforms (IRS-1/-2) of the liver in a nonalcoholic steatohepatitis animal model. Metabolism 2011, 60, 805–814. [Google Scholar]
- Nannipieri, M.; Cecchetti, F.; Anselmino, M.; Mancini, E.; Marchetti, G.; Bonotti, A.; Baldi, S.; Solito, B.; Giannetti, M.; Pinchera, A.; et al. Pattern of expression of adiponectin receptors in human liver and its relation to nonalcoholic steatohepatitis. Obes. Surg 2009, 19, 467–474. [Google Scholar]
- Ma, H.; Gomez, V.; Lu, L.; Yang, X.; Wu, X.; Xiao, S.Y. Expression of adiponectin and its receptors in livers of morbidly obese patients with non-alcoholic fatty liver disease. J. Gastroenterol. Hepatol 2009, 24, 233–237. [Google Scholar]
- Miele, L.; Valenza, V.; la Torre, G.; Montalto, M.; Cammarota, G.; Ricci, R.; Masciana, R.; Forgione, A.; Gabrieli, M.L.; Perotti, G.; et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology 2009, 49, 1877–1887. [Google Scholar]
- Brun, P.; Castagliuolo, I.; di Leo, V.; Buda, A.; Pinzani, M.; Palu, G.; Martines, D. Increased intestinal permeability in obese mice: New evidence in the pathogenesis of nonalcoholic steatohepatitis. Am. J. Physiol. Gastrointest. Liver Physiol 2007, 292, G518–G525. [Google Scholar]
- Cani, P.D.; Bibiloni, R.; Knauf, C.; Waget, A.; Neyrinck, A.M.; Delzenne, N.M.; Burcelin, R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 2008, 57, 1470–1481. [Google Scholar]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar]
- Henao-Mejia, J.; Elinav, E.; Jin, C.; Hao, L.; Mehal, W.Z.; Strowig, T.; Thaiss, C.A.; Kau, A.L.; Eisenbarth, S.C.; Jurczak, M.J.; et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 2012, 482, 179–185. [Google Scholar]
- Tamaki, N.; Takaki, A.; Tomofuji, T.; Endo, Y.; Kasuyama, K.; Ekuni, D.; Yasunaka, T.; Yamamoto, K.; Morita, M. Stage of hepatocellular carcinoma is associated with periodontitis. J. Clin. Periodontol 2011, 38, 1015–1020. [Google Scholar]
- Yoneda, M.; Naka, S.; Nakano, K.; Wada, K.; Endo, H.; Mawatari, H.; Imajo, K.; Nomura, R.; Hokamura, K.; Ono, M.; et al. Involvement of a periodontal pathogen, Porphyromonas gingivalis on the pathogenesis of non-alcoholic fatty liver disease. BMC Gastroenterol 2012, 12. [Google Scholar] [CrossRef]
- Endo, H.; Niioka, M.; Kobayashi, N.; Tanaka, M.; Watanabe, T. Butyrate-producing probiotics reduce nonalcoholic fatty liver disease progression in rats: New insight into the probiotics for the gut-liver axis. PloS One 2013, 8, e63388. [Google Scholar]
- Xu, R.Y.; Wan, Y.P.; Fang, Q.Y.; Lu, W.; Cai, W. Supplementation with probiotics modifies gut flora and attenuates liver fat accumulation in rat nonalcoholic fatty liver disease model. J. Clin. Biochem. Nutr 2012, 50, 72–77. [Google Scholar]
- Yoshimoto, S.; Loo, T.M.; Atarashi, K.; Kanda, H.; Sato, S.; Oyadomari, S.; Iwakura, Y.; Oshima, K.; Morita, H.; Hattori, M.; et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 2013, 499, 97–101. [Google Scholar]
- Tomita, K.; Teratani, T.; Suzuki, T.; Shimizu, M.; Sato, H.; Narimatsu, K.; Okada, Y.; Kurihara, C.; Irie, R.; Yokoyama, H.; et al. Free cholesterol accumulation in hepatic stellate cells: Mechanism of liver fibrosis aggravation in nonalcoholic steatohepatitis in mice. Hepatology 2013. [Google Scholar] [CrossRef]
- Leroux, A.; Ferrere, G.; Godie, V.; Cailleux, F.; Renoud, M.L.; Gaudin, F.; Naveau, S.; Prevot, S.; Makhzami, S.; Perlemuter, G.; et al. Toxic lipids stored by Kupffer cells correlates with their pro-inflammatory phenotype at an early stage of steatohepatitis. J. Hepatol 2012, 57, 141–149. [Google Scholar]
- Miura, K.; Yang, L; van Rooijen, N.; Brenner, D.A.; Ohnishi, H.; Seki, E. Toll-like receptor 2 and palmitic acid cooperatively contribute to the development of nonalcoholic steatohepatitis through inflammasome activation in mice. Hepatology 2013, 57, 577–589. [Google Scholar]
- Rivera, C.A.; Gaskin, L.; Allman, M.; Pang, J.; Brady, K.; Adegboyega, P.; Pruitt, K. Toll-like receptor-2 deficiency enhances non-alcoholic steatohepatitis. BMC Gastroenterol 2010, 10. [Google Scholar] [CrossRef]
- Vijay-Kumar, M.; Sanders, C.J.; Taylor, R.T.; Kumar, A.; Aitken, J.D.; Sitaraman, S.V.; Neish, A.S.; Uematsu, S.; Akira, S.; Williams, I.R.; et al. Deletion of TLR5 results in spontaneous colitis in mice. J. Clin. Invest 2007, 117, 3909–3921. [Google Scholar]
- Vijay-Kumar, M.; Aitken, J.D.; Carvalho, F.A.; Cullender, T.C.; Mwangi, S.; Srinivasan, S.; Sitaraman, S.V.; Knight, R.; Ley, R.E.; Gewirtz, A.T. Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science 2010, 328, 228–231. [Google Scholar]
- Letran, S.E.; Lee, S.J.; Atif, S.M.; Flores-Langarica, A.; Uematsu, S.; Akira, S.; Cunningham, A.F.; McSorley, S.J. TLR5-deficient mice lack basal inflammatory and metabolic defects but exhibit impaired CD4 T cell responses to a flagellated pathogen. J. Immunol 2011, 186, 5406–5412. [Google Scholar]
- Musso, G.; Gambino, R.; Cassader, M. Cholesterol metabolism and the pathogenesis of non-alcoholic steatohepatitis. Prog. Lipid Res 2013, 52, 175–191. [Google Scholar]
- Fu, S.; Yang, L.; Li, P.; Hofmann, O.; Dicker, L.; Hide, W.; Lin, X.; Watkins, S.M.; Ivanov, A.R.; Hotamisligil, G.S. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature 2011, 473, 528–531. [Google Scholar] [Green Version]
- Park, S.W.; Zhou, Y.; Lee, J.; Lu, A.; Sun, C.; Chung, J.; Ueki, K.; Ozcan, U. The regulatory subunits of PI3K, p85α and p85β, interact with XBP-1 and increase its nuclear translocation. Nat. Med 2010, 16, 429–437. [Google Scholar]
- Park, S.W.; Zhou, Y.; Lee, J.; Ozcan, U. Sarco(endo)plasmic reticulum Ca2+-ATPase 2b is a major regulator of endoplasmic reticulum stress and glucose homeostasis in obesity. Proc. Natl. Acad. Sci. USA 2010, 107, 19320–19325. [Google Scholar]
- Canova, N.K.; Kmonickova, E.; Martinek, J.; Zidek, Z.; Farghali, H. Thapsigargin, a selective inhibitor of sarco-endoplasmic reticulum Ca2+-ATPases, modulates nitric oxide production and cell death of primary rat hepatocytes in culture. Cell Biol. Toxicol 2007, 23, 337–354. [Google Scholar]
- Barker, J.R.; Koestler, B.J.; Carpenter, V.K.; Burdette, D.L.; Waters, C.M.; Vance, R.E.; Valdivia, R.H. STING-dependent recognition of cyclic di-AMP mediates type I interferon responses during Chlamydia trachomatis infection. MBio 2013, 4. [Google Scholar] [CrossRef]
- De Minicis, S.; Candelaresi, C.; Agostinelli, L.; Taffetani, S.; Saccomanno, S.; Rychlicki, C.; Trozzi, L.; Marzioni, M.; Benedetti, A.; Svegliati-Baroni, G. Endoplasmic Reticulum stress induces hepatic stellate cell apoptosis and contributes to fibrosis resolution. Liver Int 2012, 32, 1574–1584. [Google Scholar]
- Kawai, D.; Takaki, A.; Nakatsuka, A.; Wada, J.; Tamaki, N.; Yasunaka, T.; Koike, K.; Tsuzaki, R.; Matsumoto, K.; Miyake, Y.; et al. Hydrogen-rich water prevents progression of nonalcoholic steatohepatitis and accompanying hepatocarcinogenesis in mice. Hepatology 2012, 56, 912–921. [Google Scholar]
- Bugianesi, E. Non-alcoholic steatohepatitis and cancer. Clin. Liver Dis 2007, 11, 191–207. [Google Scholar]
- Mittler, R.; Vanderauwera, S.; Suzuki, N.; Miller, G.; Tognetti, V.B.; Vandepoele, K.; Gollery, M.; Shulaev, V.; van Breusegem, F. ROS signaling: The new wave? Trends Plant Sci 2011, 16, 300–309. [Google Scholar]
- Nelson, J.E.; Wilson, L.; Brunt, E.M.; Yeh, M.M.; Kleiner, D.E.; Unalp-Arida, A.; Kowdley, K.V. Relationship between the pattern of hepatic iron deposition and histological severity in nonalcoholic fatty liver disease. Hepatology 2011, 53, 448–457. [Google Scholar]
- Van de Wier, B.; Balk, J.M.; Haenen, G.R.; Giamouridis, D.; Bakker, J.A.; Bast, B.C.; den Hartog, G.J.; Koek, G.H.; Bast, A. Elevated citrate levels in non-alcoholic fatty liver disease: The potential of citrate to promote radical production. FEBS Lett 2013, 587, 2461–2466. [Google Scholar]
- Rolo, A.P.; Teodoro, J.S.; Palmeira, C.M. Role of oxidative stress in the pathogenesis of nonalcoholic steatohepatitis. Free Radic. Biol. Med 2012, 52, 59–69. [Google Scholar]
- Fritz, R.; Bol, J.; Hebling, U.; Angermuller, S.; Volkl, A.; Fahimi, H.D.; Mueller, S. Compartment-dependent management of H2O2 by peroxisomes. Free Radic. Biol. Med 2007, 42, 1119–1129. [Google Scholar]
- Zangar, R.C.; Davydov, D.R.; Verma, S. Mechanisms that regulate production of reactive oxygen species by cytochrome P450. Toxicol. Appl. Pharmacol 2004, 199, 316–331. [Google Scholar]
- Rachek, L.I.; Yuzefovych, L.V.; Ledoux, S.P.; Julie, N.L.; Wilson, G.L. Troglitazone, but not rosiglitazone, damages mitochondrial DNA and induces mitochondrial dysfunction and cell death in human hepatocytes. Toxicol. Appl. Pharmacol 2009, 240, 348–354. [Google Scholar]
- Ricci, C.; Pastukh, V.; Leonard, J.; Turrens, J.; Wilson, G.; Schaffer, D.; Schaffer, S.W. Mitochondrial DNA damage triggers mitochondrial-superoxide generation and apoptosis. Am. J. Physiol. Cell Physiol 2008, 294, C413–C422. [Google Scholar]
- Gredilla, R.; Bohr, V.A.; Stevnsner, T. Mitochondrial DNA repair and association with aging—An update. Exp. Gerontol 2010, 45, 478–488. [Google Scholar]
- Leclere, R.; Torregrosa-Munumer, R.; Kireev, R.; Garcia, C.; Vara, E.; Tresguerres, J.A.; Gredilla, R. Effect of estrogens on base excision repair in brain and liver mitochondria of aged female rats. Biogerontology 2013, 14, 383–394. [Google Scholar]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J 2009, 417, 1–13. [Google Scholar]
- Scarpulla, R.C. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim. Biophys. Acta 2011, 1813, 1269–1278. [Google Scholar]
- Aharoni-Simon, M.; Hann-Obercyger, M.; Pen, S.; Madar, Z.; Tirosh, O. Fatty liver is associated with impaired activity of PPARγ-coactivator 1alpha (PGC1α) and mitochondrial biogenesis in mice. Lab. Invest 2011, 91, 1018–1028. [Google Scholar]
- Chiappini, F.; Barrier, A.; Saffroy, R.; Domart, M.C.; Dagues, N.; Azoulay, D.; Sebagh, M.; Franc, B.; Chevalier, S.; Debuire, B.; et al. Exploration of global gene expression in human liver steatosis by high-density oligonucleotide microarray. Lab. Invest 2006, 86, 154–165. [Google Scholar]
- Hui, J.M.; Hodge, A.; Farrell, G.C.; Kench, J.G.; Kriketos, A.; George, J. Beyond insulin resistance in NASH: TNF-α or adiponectin? Hepatology 2004, 40, 46–54. [Google Scholar]
- Hernandez-Gea, V.; Hilscher, M.; Rozenfeld, R.; Lim, M.P.; Nieto, N.; Werner, S.; Devi, L.A.; Friedman, S.L. Endoplasmic reticulum stress induces fibrogenic activity in hepatic stellate cells through autophagy. J. Hepatol 2013, 59, 98–104. [Google Scholar]
- Blaner, W.S.; O’Byrne, S.M.; Wongsiriroj, N.; Kluwe, J.; D’Ambrosio, D.M.; Jiang, H.; Schwabe, R.F.; Hillman, E.M.; Piantedosi, R.; Libien, J. Hepatic stellate cell lipid droplets: A specialized lipid droplet for retinoid storage. Biochim. Biophys. Acta 2009, 1791, 467–473. [Google Scholar]
- Hernandez-Gea, V.; Ghiassi-Nejad, Z.; Rozenfeld, R.; Gordon, R.; Fiel, M.I.; Yue, Z.; Czaja, M.J.; Friedman, S.L. Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology 2012, 142, 938–946. [Google Scholar]
- Oddy, W.H.; Herbison, C.E.; Jacoby, P.; Ambrosini, G.L.; O’Sullivan, T.A.; Ayonrinde, O.T.; Olynyk, J.K.; Black, L.J.; Beilin, L.J.; Mori, T.A.; et al. The Western dietary pattern is prospectively associated with nonalcoholic fatty liver disease in adolescence. Am. J. Gastroenterol 2013, 108, 778–785. [Google Scholar]
- Goodpaster, B.H.; Delany, J.P.; Otto, A.D.; Kuller, L.; Vockley, J.; South-Paul, J.E.; Thomas, S.B.; Brown, J.; McTigue, K.; Hames, K.C.; et al. Effects of diet and physical activity interventions on weight loss and cardiometabolic risk factors in severely obese adults: A randomized trial. JAMA 2010, 304, 1795–1802. [Google Scholar]
- Neuschwander-Tetri, B.A.; Ford, D.A.; Acharya, S.; Gilkey, G.; Basaranoglu, M.; Tetri, L.H.; Brunt, E.M. Dietary trans-fatty acid induced NASH is normalized following loss of trans-fatty acids from hepatic lipid pools. Lipids 2012, 47, 941–950. [Google Scholar]
- Soetens, B.; Braet, C.; Moens, E. Thought suppression in obese and non-obese restrained eaters: Piece of cake or forbidden fruit? Eur. Eat. Disord. Rev 2008, 16, 67–76. [Google Scholar]
- Musso, G.; Cassader, M.; Rosina, F.; Gambino, R. Impact of current treatments on liver disease, glucose metabolism and cardiovascular risk in non-alcoholic fatty liver disease (NAFLD): A systematic review and meta-analysis of randomised trials. Diabetologia 2012, 55, 885–904. [Google Scholar]
- Svegliati-Baroni, G.; Saccomanno, S.; Rychlicki, C.; Agostinelli, L.; de Minicis, S.; Candelaresi, C.; Faraci, G.; Pacetti, D.; Vivarelli, M.; Nicolini, D.; et al. Glucagon-like peptide-1 receptor activation stimulates hepatic lipid oxidation and restores hepatic signalling alteration induced by a high-fat diet in nonalcoholic steatohepatitis. Liver Int 2011, 31, 1285–1297. [Google Scholar] [Green Version]
- Cuthbertson, D.J.; Irwin, A.; Gardner, C.J.; Daousi, C.; Purewal, T.; Furlong, N.; Goenka, N.; Thomas, E.L.; Adams, V.L.; Pushpakom, S.P.; et al. Improved glycaemia correlates with liver fat reduction in obese, type 2 diabetes, patients given glucagon-like peptide-1 (GLP-1) receptor agonists. PloS One 2012, 7, e50117. [Google Scholar]
- Kern, M.; Kloting, N.; Niessen, H.G.; Thomas, L.; Stiller, D.; Mark, M.; Klein, T.; Bluher, M. Linagliptin improves insulin sensitivity and hepatic steatosis in diet-induced obesity. PLoS One 2012, 7, e38744. [Google Scholar]
- Hoofnagle, J.H.; van Natta, M.L.; Kleiner, D.E.; Clark, J.M.; Kowdley, K.V.; Loomba, R.; Neuschwander-Tetri, B.A.; Sanyal, A.J.; Tonascia, J. Vitamin E and changes in serum alanine aminotransferase levels in patients with non-alcoholic steatohepatitis. Aliment. Pharmacol. Ther 2013, 38, 134–143. [Google Scholar]
- Malaguarnera, M.; Gargante, M.P.; Russo, C.; Antic, T.; Vacante, M.; Avitabile, T.; li Volti, G.; Galvano, F. l-carnitine supplementation to diet: A new tool in treatment of nonalcoholic steatohepatitis—A randomized and controlled clinical trial. Am. J. Gastroenterol 2010, 105, 1338–1345. [Google Scholar]
- Zein, C.O.; Lopez, R.; Fu, X.; Kirwan, J.P.; Yerian, L.M.; McCullough, A.J.; Hazen, S.L.; Feldstein, A.E. Pentoxifylline decreases oxidized lipid products in nonalcoholic steatohepatitis: New evidence on the potential therapeutic mechanism. Hepatology 2012, 56, 1291–1299. [Google Scholar]
- Zein, C.O.; Yerian, L.M.; Gogate, P.; Lopez, R.; Kirwan, J.P.; Feldstein, A.E.; McCullough, A.J. Pentoxifylline improves nonalcoholic steatohepatitis: A randomized placebo-controlled trial. Hepatology 2011, 54, 1610–1619. [Google Scholar]
- Nelson, A.; Torres, D.M.; Morgan, A.E.; Fincke, C.; Harrison, S.A. A pilot study using simvastatin in the treatment of nonalcoholic steatohepatitis: A randomized placebo-controlled trial. J. Clin. Gastroenterol 2009, 43, 990–994. [Google Scholar]
- Deushi, M.; Nomura, M.; Kawakami, A.; Haraguchi, M.; Ito, M.; Okazaki, M.; Ishii, H.; Yoshida, M. Ezetimibe improves liver steatosis and insulin resistance in obese rat model of metabolic syndrome. FEBS Lett 2007, 581, 5664–5670. [Google Scholar]
- Yoneda, M.; Fujita, K.; Nozaki, Y.; Endo, H.; Takahashi, H.; Hosono, K.; Suzuki, K.; Mawatari, H.; Kirikoshi, H.; Inamori, M.; et al. Efficacy of ezetimibe for the treatment of non-alcoholic steatohepatitis: An open-label, pilot study. Hepatol. Res 2010, 40, 613–621. [Google Scholar]
- Leuschner, U.F.; Lindenthal, B.; Herrmann, G.; Arnold, J.C.; Rossle, M.; Cordes, H.J.; Zeuzem, S.; Hein, J.; Berg, T. High-dose ursodeoxycholic acid therapy for nonalcoholic steatohepatitis: A double-blind, randomized, placebo-controlled trial. Hepatology 2010, 52, 472–479. [Google Scholar]
- Pietu, F.; Guillaud, O.; Walter, T.; Vallin, M.; Hervieu, V.; Scoazec, J.Y.; Dumortier, J. Ursodeoxycholic acid with vitamin E in patients with nonalcoholic steatohepatitis: Long-term results. Clin. Res. Hepatol. Gastroenterol 2012, 36, 146–155. [Google Scholar]
- Ford, A.C.; Peyrin-Biroulet, L. Opportunistic Infections With Anti-Tumor Necrosis Factor-α Therapy in Inflammatory Bowel Disease: Meta-Analysis of Randomized Controlled Trials. Am. J. Gastroenterol 2013, 108, 1268–1276. [Google Scholar]
- Bast, A.; Haenen, G.R. Ten misconceptions about antioxidants. Trends Pharmacol. Sci 2013, 34, 430–436. [Google Scholar]
- Watson, J. Oxidants, antioxidants and the current incurability of metastatic cancers. Open Biol 2013, 3. [Google Scholar] [CrossRef]
- Yae, T.; Tsuchihashi, K.; Ishimoto, T.; Motohara, T.; Yoshikawa, M.; Yoshida, G.J.; Wada, T.; Masuko, T.; Mogushi, K.; Tanaka, H.; et al. Alternative splicing of CD44 mRNA by ESRP1 enhances lung colonization of metastatic cancer cell. Nat. Commun 2012, 3. [Google Scholar] [CrossRef]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov 2009, 8, 579–591. [Google Scholar]
- Hackam, D.G. Review: Antioxidant supplements for primary and secondary prevention do not decrease mortality. ACP J. Club. 2007, 147, p. 4. Available online: http://acpjc.acponline.org/Content/147/1/Issue/ACPJC-2007-147-1-004.htm (accessed on 11 October 2013).
- Ohta, S. Recent progress toward hydrogen medicine: Potential of molecular hydrogen for preventive and therapeutic applications. In Curr. Pharm. Des; 2011; Volume 17, pp. 2241–2252. [Google Scholar]
- Winterbourn, C.C. Biological reactivity and biomarkers of the neutrophil oxidant, hypochlorous acid. Toxicology 2002, 181–182, 223–227. [Google Scholar]
- Liao, J.K. Linking endothelial dysfunction with endothelial cell activation. J. Clin. Invest 2013, 123, 540–541. [Google Scholar]
- Fontanari, P.; Badier, M.; Guillot, C.; Tomei, C.; Burnet, H.; Gardette, B.; Jammes, Y. Changes in maximal performance of inspiratory and skeletal muscles during and after the 7.1-MPa Hydra 10 record human dive. Eur. J. Appl. Physiol 2000, 81, 325–328. [Google Scholar]
- Fukuda, K.I.; Asoh, S.; Ishikawa, M.; Yamamoto, Y.; Ohsawa, I.; Ohta, S. Inhalation of hydrogen gas suppresses hepatic injury caused by ischemia/reperfusion through reducing oxidative stress. Biochem. Bioph. Res. Co 2007, 361, 670–674. [Google Scholar]
- Hayashida, K.; Sano, M.; Ohsawa, I.; Shinmura, K.; Tamaki, K.; Kimura, K.; Endo, J.; Katayama, T.; Kawamura, A.; Kohsaka, S.; et al. Inhalation of hydrogen gas reduces infarct size in the rat model of myocardial ischemia-reperfusion injury. Biochem. Biophys. Res. Commun 2008, 373, 30–35. [Google Scholar]
- Kawamura, T.; Huang, C.S.; Tochigi, N.; Lee, S.; Shigemura, N.; Billiar, T.R.; Okumura, M.; Nakao, A.; Toyoda, Y. Inhaled hydrogen gas therapy for prevention of lung transplant-induced ischemia/reperfusion injury in rats. Transplantation 2010, 90, 1344–1351. [Google Scholar]
- Fu, Y.; Ito, M.; Fujita, Y.; Ichihara, M.; Masuda, A.; Suzuki, Y.; Maesawa, S.; Kajita, Y.; Hirayama, M.; Ohsawa, I.; et al. Molecular hydrogen is protective against 6-hydroxydopamine-induced nigrostriatal degeneration in a rat model of Parkinson’s disease. Neurosci. Lett 2009, 453, 81–85. [Google Scholar]
- Ohsawa, I.; Nishimaki, K.; Yamagata, K.; Ishikawa, M.; Ohta, S. Consumption of hydrogen water prevents atherosclerosis in apolipoprotein E knockout mice. Biochem. Biophys. Res. Commun 2008, 377, 1195–1198. [Google Scholar]
- Kamimura, N.; Nishimaki, K.; Ohsawa, I.; Ohta, S. Molecular hydrogen improves obesity and diabetes by inducing hepatic FGF21 and stimulating energy metabolism in db/db mice. Obesity 2011, 19, 1396–1403. [Google Scholar]
- Amitani, H.; Asakawa, A.; Cheng, K.; Amitani, M.; Kaimoto, K.; Nakano, M.; Ushikai, M.; Li, Y.; Tsai, M.; Li, J.B.; et al. Hydrogen improves glycemic control in type1 diabetic animal model by promoting glucose uptake into skeletal muscle. PloS One 2013, 8, e53913. [Google Scholar]
- Feng, Y.; Wang, R.; Xu, J.; Sun, J.; Xu, T.; Gu, Q.; Wu, X. Hydrogen-rich saline prevents early neurovascular dysfunction resulting from inhibition of oxidative stress in STZ-diabetic rats. Curr. Eye Res 2013, 38, 396–404. [Google Scholar]
- Sun, H.; Chen, L.; Zhou, W.; Hu, L.; Li, L.; Tu, Q.; Chang, Y.; Liu, Q.; Sun, X.; Wu, M.; et al. The protective role of hydrogen-rich saline in experimental liver injury in mice. J. Hepatol 2011, 54, 471–480. [Google Scholar]
- Nakashima-Kamimura, N.; Mori, T.; Ohsawa, I.; Asoh, S.; Ohta, S. Molecular hydrogen alleviates nephrotoxicity induced by an anti-cancer drug cisplatin without compromising anti-tumor activity in mice. Cancer Chemother. Pharmacol 2009, 64, 753–761. [Google Scholar]
- Buchholz, B.M.; Kaczorowski, D.J.; Sugimoto, R.; Yang, R.; Wang, Y.; Billiar, T.R.; McCurry, K.R.; Bauer, A.J.; Nakao, A. Hydrogen inhalation ameliorates oxidative stress in transplantation induced intestinal graft injury. Am. J. Transplant 2008, 8, 2015–2024. [Google Scholar]
- Kajiyama, S.; Hasegawa, G.; Asano, M.; Hosoda, H.; Fukui, M.; Nakamura, N.; Kitawaki, J.; Imai, S.; Nakano, K.; Ohta, M.; et al. Supplementation of hydrogen-rich water improves lipid and glucose metabolism in patients with type 2 diabetes or impaired glucose tolerance. Nutr. Res 2008, 28, 137–143. [Google Scholar]
- Nakao, A.; Toyoda, Y.; Sharma, P.; Evans, M.; Guthrie, N. Effectiveness of hydrogen rich water on antioxidant status of subjects with potential metabolic syndrome—An open label pilot study. J. Clin. Biochem. Nutr 2010, 46, 140–149. [Google Scholar]
- Huang, C.S.; Kawamura, T.; Lee, S.; Tochigi, N.; Shigemura, N.; Buchholz, B.M.; Kloke, J.D.; Billiar, T.R.; Toyoda, Y.; Nakao, A. Hydrogen inhalation ameliorates ventilator-induced lung injury. Crit. Care 2010, 14, R234:1–R234:15. [Google Scholar]
- Fujii, M.; Shibazaki, Y.; Wakamatsu, K.; Honda, Y.; Kawauchi, Y.; Suzuki, K.; Arumugam, S.; Watanabe, K.; Ichida, T.; Asakura, H.; et al. A murine model for non-alcoholic steatohepatitis showing evidence of association between diabetes and hepatocellular carcinoma. Med. Mol. Morphol 2013, 46, 141–152. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Takaki, A.; Kawai, D.; Yamamoto, K. Multiple Hits, Including Oxidative Stress, as Pathogenesis and Treatment Target in Non-Alcoholic Steatohepatitis (NASH). Int. J. Mol. Sci. 2013, 14, 20704-20728. https://doi.org/10.3390/ijms141020704
Takaki A, Kawai D, Yamamoto K. Multiple Hits, Including Oxidative Stress, as Pathogenesis and Treatment Target in Non-Alcoholic Steatohepatitis (NASH). International Journal of Molecular Sciences. 2013; 14(10):20704-20728. https://doi.org/10.3390/ijms141020704
Chicago/Turabian StyleTakaki, Akinobu, Daisuke Kawai, and Kazuhide Yamamoto. 2013. "Multiple Hits, Including Oxidative Stress, as Pathogenesis and Treatment Target in Non-Alcoholic Steatohepatitis (NASH)" International Journal of Molecular Sciences 14, no. 10: 20704-20728. https://doi.org/10.3390/ijms141020704
APA StyleTakaki, A., Kawai, D., & Yamamoto, K. (2013). Multiple Hits, Including Oxidative Stress, as Pathogenesis and Treatment Target in Non-Alcoholic Steatohepatitis (NASH). International Journal of Molecular Sciences, 14(10), 20704-20728. https://doi.org/10.3390/ijms141020704