Emodin Prevents Intrahepatic Fat Accumulation, Inflammation and Redox Status Imbalance During Diet-Induced Hepatosteatosis in Rats

 ,
,  , , and
, , and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Effects of Five Weeks HFD/HF Diet on Rats
2.2. Effects of Emodin on Body Weight, Liver Weight and Metabolic Parameters in HFD/HF Rats
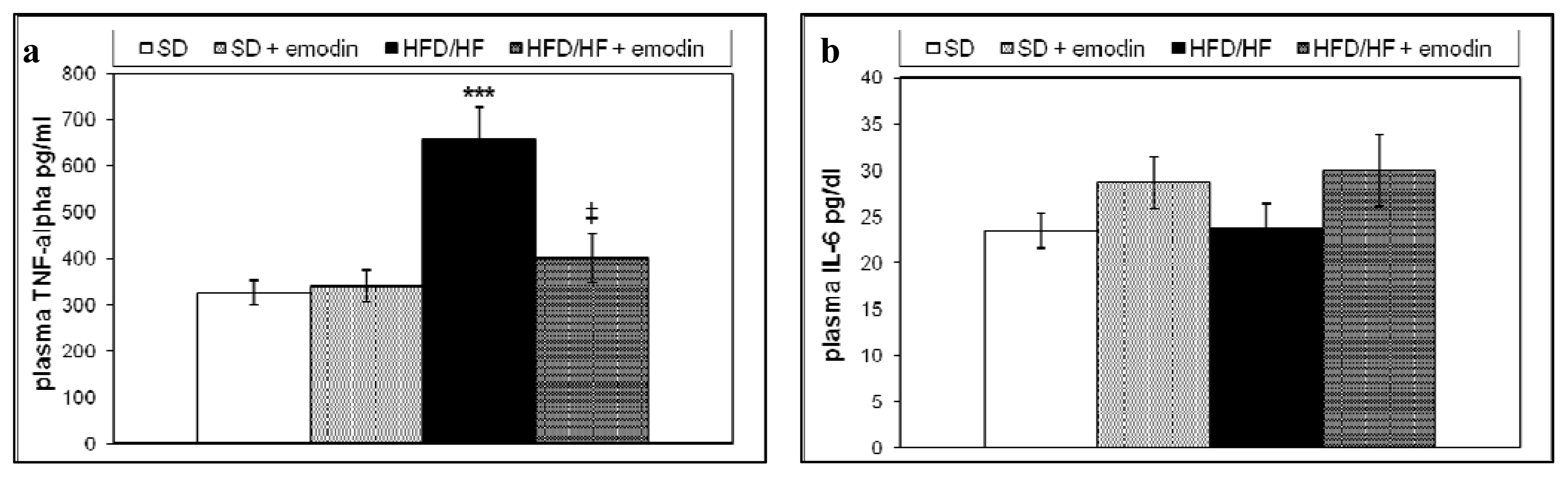
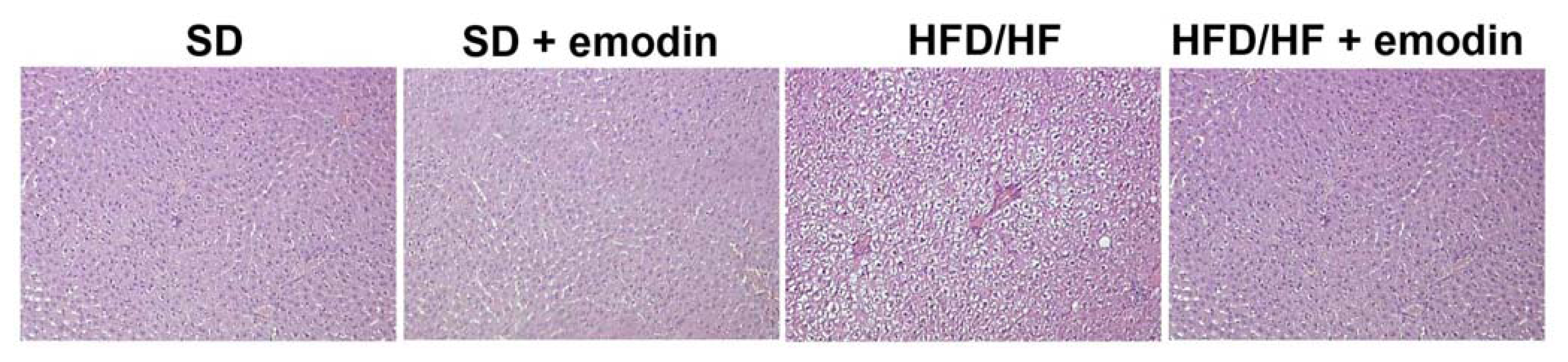
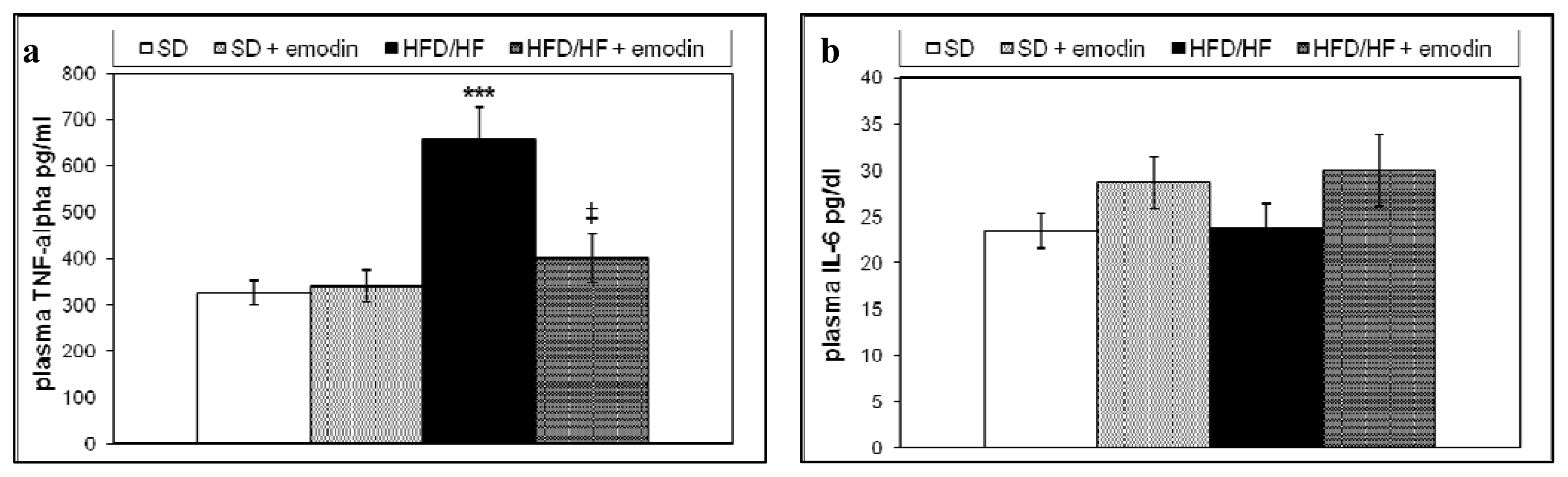
2.3. Hepatoprotective and Anti-Inflammatory Effects of Emodin in HFD/HF Rats
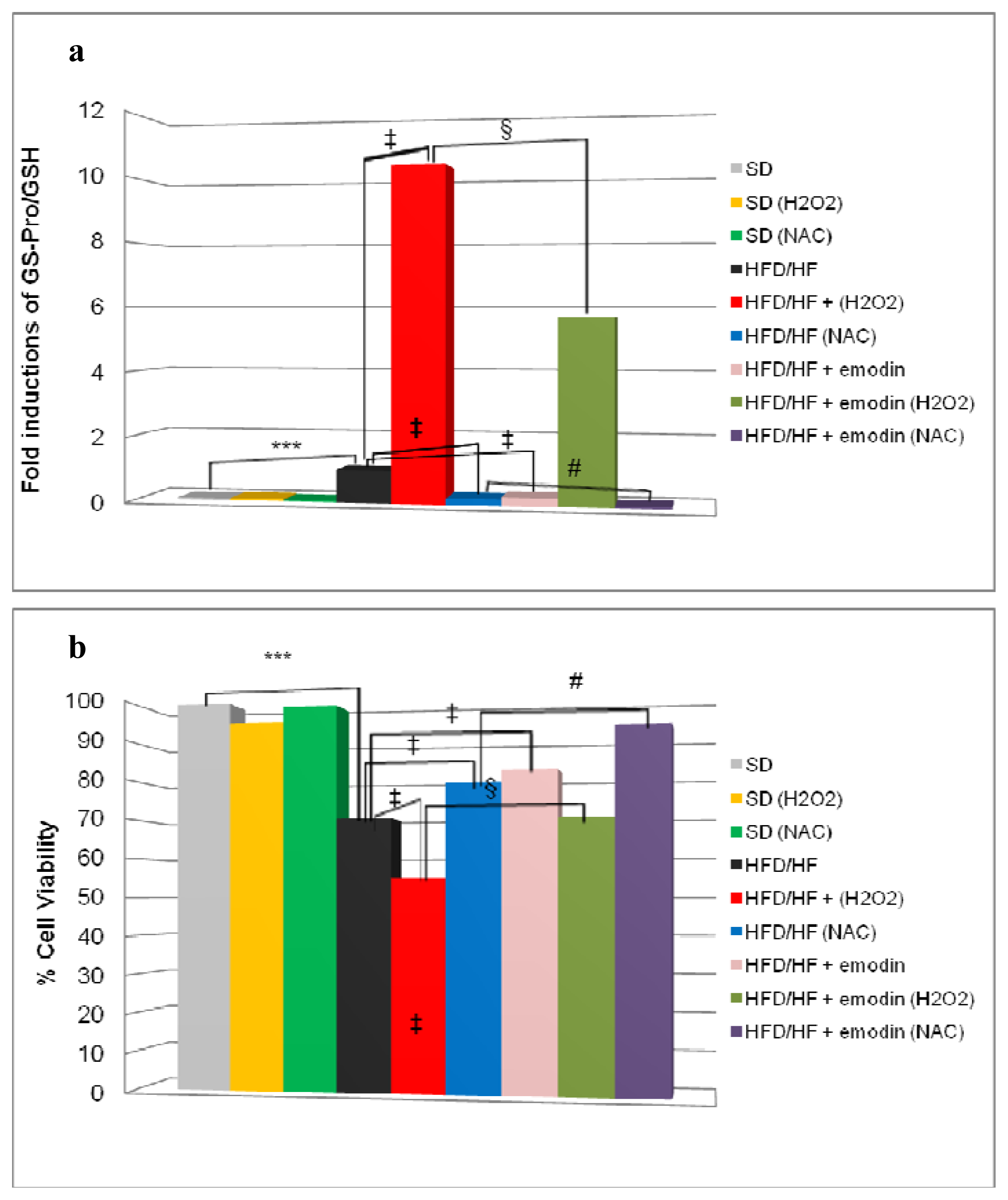
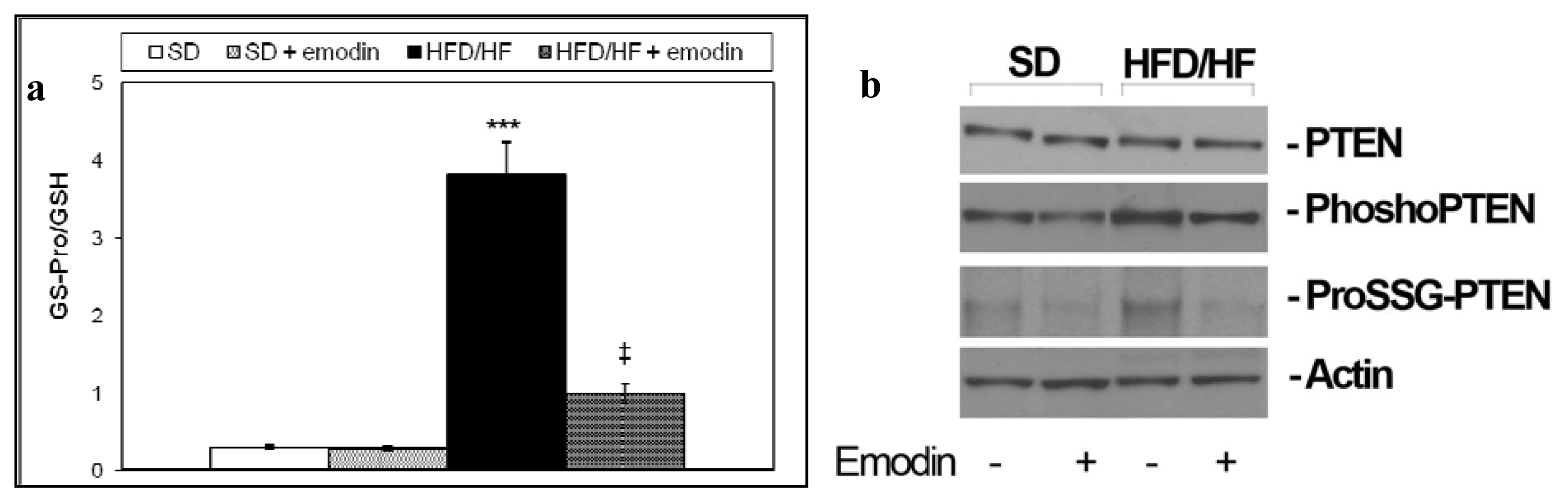
2.4. Emodin Promotes Recovery of Redox Status Imbalance in Primary Hepatocytes from HFD/HF Rats
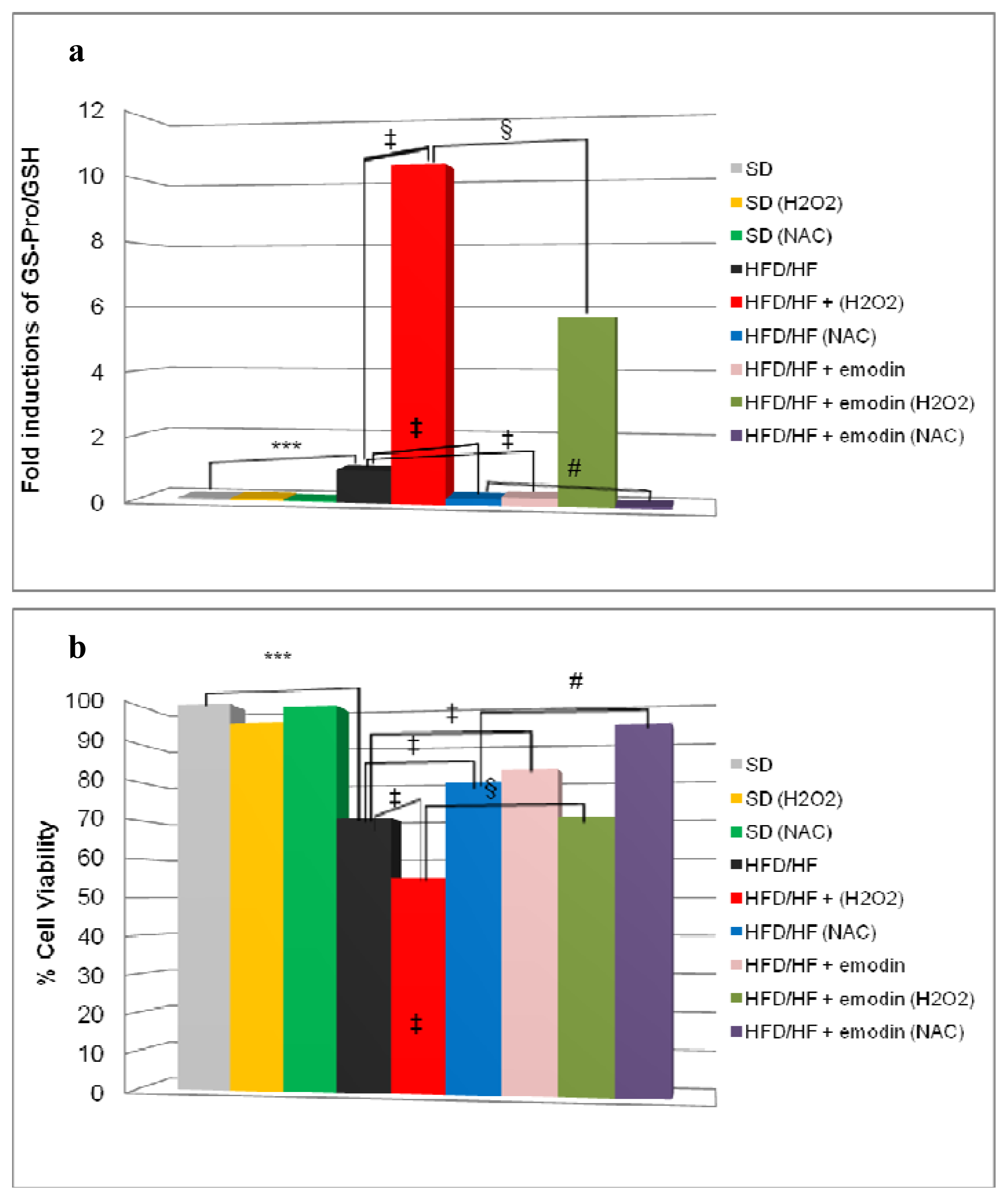
2.5. Emodin Protects HFD/HF Primary Hepatocytes Rats from Further Oxidative Stress Damage
3. Experimental Section
3.1. Animals and Primary Hepatocytes
3.2. Biochemical Determinations and Inflammatory Markers
3.3. Immunohistochemistry
3.4. High-Performance Liquid Chromatography of GSH
3.5. Immunoprecipitation and Western Blotting
3.6. Cell Viability
3.7. Statistical Analysis
4. Conclusions
- Conflicts of InterestNo conflict of interest exists.
References
- Mantena, S.K.; King, A.L.; Andringa, K.K.; Eccleston, H.B.; Bailey, S.M. Mitochondrial dysfunction and oxidative stress in the pathogenesis of alcohol- and obesity-induced fatty liver diseases. Free Radical Biol. Med 2008, 44, 1259–1272. [Google Scholar]
- Clément, S.; Negro, F. Hepatitis C virus: The viral way to fatty liver. J. Hepatol 2007, 46, 985–987. [Google Scholar]
- Wree, A.; Kahraman, A.; Gerken, G.; Canbay, A. Obesity affects the liver—the link between adipocytes and hepatocytes. Digestion 2011, 83, 124–133. [Google Scholar]
- Khashab, M.A.; Liangpunsakul, S.; Chalasani, N. Nonalcoholic fatty liver disease as a component of the metabolic syndrome. Curr. Gastroenterol. Rep 2008, 10, 73–80. [Google Scholar]
- Alisi, A.; Feldstein, A.E.; Villani, A.; Raponi, M.; Nobili, V. nonalcoholic fatty liver disease: A multidisciplinary approach. Nat. Rev. Gastroenterol. Hepatol 2012, in press. [Google Scholar]
- Brunt, E.M. Pathology of nonalcoholic fatty liver disease. Nat. Rev. Gastroenterol. Hepatol 2010, 7, 195–203. [Google Scholar]
- Feldstein, A.E. Novel insights into the pathophysiology of nonalcoholic fatty liver disease. Semin. Liver Dis 2010, 30, 391–401. [Google Scholar]
- Moore, J.B. Non-alcoholic fatty liver disease: The hepatic consequence of obesity and the metabolic syndrome. Proc. Nutr. Soc 2010, 69, 211–220. [Google Scholar]
- Day, C.P.; James, O.F. Steatohepatitis: a tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar]
- Bugianesi, E.; Moscatiello, S.; Ciaravella, M.F.; Marchesini, G. Insulin resistance in nonalcoholic fatty liver disease. Curr. Pharm. Des 2010, 16, 1941–1951. [Google Scholar]
- Feldstein, A.E.; Charatcharoenwitthaya, P.; Treeprasertsuk, S.; Benson, J.T.; Enders, F.B.; Angulo, P. The natural history of non-alcoholic fatty liver disease in children: A follow-up study for up to 20 years. Gut 2009, 58, 1538–1544. [Google Scholar]
- Cohen, J.C.; Horton, J.D.; Hobbs, H.H. Human fatty liver disease: Old questions and new insights. Science 2011, 332, 1519–1523. [Google Scholar]
- Albano, E.; Mottaran, E.; Occhino, G.; Reale, E.; Vidali, M. Review article: Role of oxidative stress in the progression of non-alcoholic steatosis. Aliment. Pharmacol. Ther 2005, 22, 71–73. [Google Scholar]
- Tarantino, G.; Savastano, S.; Colao, A. Hepatic steatosis, low-grade chronic inflammation and hormone/growth factor/adipokine imbalance. World J. Gastroenterol 2010, 16, 4773–4783. [Google Scholar]
- Alisi, A.; Carsetti, R.; Nobili, V. Pathogen- or damage-associated molecular patterns during nonalcoholic fatty liver disease development. Hepatology 2011, 54, 1500–1502. [Google Scholar]
- Alisi, A.; Nobili, V. Nonalcoholic fatty liver disease: Targeted therapy in children—what is the right way? Nat. Rev. Gastroenterol. Hepatol 2011, 8, 425–426. [Google Scholar]
- Lavine, J.E.; Schwimmer, J.B.; Van Natta, M.L.; Molleston, J.P.; Murray, K.F.; Rosenthal, P.; Abrams, S.H.; Scheimann, A.O.; Sanyal, A.J.; Chalasani, N.; et al. Nonalcoholic Steatohepatitis Clinical Research Network. Effect of vitamin E or metformin for treatment of nonalcoholic fatty liver disease in children and adolescents: the TONIC randomized controlled trial. JAMA 2011, 305, 1659–1668. [Google Scholar]
- Pradeep, K.; Mohan, C.V.; Gobianand, K.; Karthikeyan, S. Silymarin modulates the oxidant-antioxidant imbalance during diethylnitrosamine induced oxidative stress in rats. Eur. J. Pharmacol 2007, 560, 110–116. [Google Scholar]
- Comar, K.M.; Kirby, D.F. Herbal remedies in gastroenterology. J. Clin. Gastroenterol 2005, 39, 457–468. [Google Scholar]
- Di Sario, A.; Bendia, E.; Taffetani, S.; Omenetti, A.; Candelaresi, C.; Marzioni, M.; De Minicis, S.; Benedetti, A. Hepatoprotective and antifibrotic effect of a new silybin-phosphatidylcholine-Vitamin E complex in rats. Dig. Liver Dis 2005, 37, 869–876. [Google Scholar]
- Loguercio, C.; Federico, A.; Trappoliere, M.; Tuccillo, C.; de Sio, I.; Di Leva, A.; Niosi, M.; D’Auria, M.V.; Papasso, R.; Del Vecchio Blanco, C. The effect of a silybin-vitamin e-phospholipid complex on nonalcoholic fatty liver disease: a pilot study. Dig. Dis. Sci 2007, 52, 2387–2395. [Google Scholar]
- Shapiro, H.; Bruck, R. Therapeutic potential of curcumin in non-alcoholic steatohepatitis. Nutr. Res. Rev 2005, 18, 212–221. [Google Scholar]
- Jang, E.M.; Choi, M.S.; Jung, U.J.; Kim, M.J.; Kim, H.J.; Jeon, S.M.; Shin, S.K.; Seong, C.N.; Lee, M.K. Beneficial effects of curcumin on hyperlipidemia and insulin resistance in high-fat-fed hamsters. Metabolism 2008, 57, 1576–1583. [Google Scholar]
- Tang, Y.; Zheng, S.; Chen, A. Curcumin eliminates leptin’s effects on hepatic stellate cell activation via interrupting leptin signaling. Endocrinology 2009, 150, 3011–3020. [Google Scholar]
- Dong, H.; Lu, F.E.; Gao, Z.Q.; Xu, L.J.; Wang, K.F.; Zou, X. Effetcs of emodin on treating murine nonalcohlic fatty liver induced by high caloric laboratory chaw. World J. Gastroenterol 2005, 11, 1339–1344. [Google Scholar]
- Samuel, V.T. Fructose induced lipogenesis: From sugar to fat to insulin resistance. Trends Endocrinol. Metab 2011, 22, 60–65. [Google Scholar]
- Lim, J.S.; Mietus-Snyder, M.; Valente, A.; Schwarz, J.M.; Lustig, R.H. The role of fructose in the pathogenesis of NAFLD and the metabolic syndrome. Nat. Rev. Gastroenterol. Hepatol 2010, 7, 251–264. [Google Scholar]
- Spruss, A.; Bergheim, I. Dietary fructose and intestinal barrier: Potential risk factor in the pathogenesis of nonalcoholic fatty liver disease. J. Nutr. Biochem 2009, 20, 657–662. [Google Scholar]
- Alisi, A.; Manco, M.; Pezzullo, M.; Nobili, V. Fructose at the center of necroinflammation and fibrosis in nonalcoholic steatohepatitis. Hepatology 2011, 53, 372–373. [Google Scholar]
- Kohli, R.; Kirby, M.; Xanthakos, S.A.; Softic, S.; Feldstein, A.E.; Saxena, V.; Tang, P.H.; Miles, L.; Miles, M.V.; Balistreri, W.F.; et al. High-fructose, medium chain trans fat diet induces liver fibrosis and elevates plasma coenzyme Q9 in a novel murine model of obesity and nonalcoholic steatohepatitis. Hepatology 2010, 52, 934–944. [Google Scholar]
- Alisi, A.; Da Sacco, L.; Bruscalupi, G.; Piemonte, F.; Panera, N.; De Vito, R.; Leoni, S.; Bottazzo, G.F.; Masotti, A.; Nobili, V. Mirnome analysis reveals novel molecular determinants in the pathogenesis of diet-induced nonalcoholic fatty liver disease. Lab. Invest 2011, 91, 283–293. [Google Scholar]
- Tiniakos, D.G. Nonalcoholic fatty liver disease/nonalcoholic steatohepatitis: Histological diagnostic criteria and scoring systems. Eur. J. Gastroenterol. Hepatol 2010, 22, 643–650. [Google Scholar]
- Hebbard, L.; George, J. Animal models of nonalcoholic fatty liver disease. Nat. Rev. Gastroenterol. Hepatol 2011, 8, 35–44. [Google Scholar]
- Lin, C.C.; Chang, C.H.; Yang, J.J.; Namba, T.; Hattori, M. Hepatoprotective effects of emodin from Ventilago leiocarpa. J. Ethnopharmacol 1996, 52, 107–111. [Google Scholar]
- Dong, M.X.; Jia, Y.; Zhang, Y.B.; Li, C.C.; Geng, Y.T.; Zhou, L.; Li, X.Y.; Liu, J.C.; Niu, Y.C. Emodin protects rat liver from CCl(4)-induced fibrogenesis via inhibition of hepatic stellate cells activation. World J. Gastroenterol 2009, 15, 4753–4762. [Google Scholar]
- Zhao, Y.L.; Wang, J.B.; Zhou, G.D.; Shan, L.M.; Xiao, X.H. Investigations of free anthraquinones from rhubarb against α-naphthylisothiocyanate-induced cholestatic liver injury in rats. Basic Clin. Pharmacol. Toxicol 2009, 104, 463–469. [Google Scholar]
- Srinivas, G.; Anto, R.J.; Srinivas, P.; Vidhyalakshmi, S.; Senan, V.P.; Karunagaran, D. Emodin induces apoptosis of human cervical cancer cells through poly(ADP-ribose) polymerase cleavage and activation of caspase-9. Eur. J. Pharmacol 2003, 473, 117–125. [Google Scholar]
- Ding, Y.; Zhao, L.; Mei, H.; Zhang, S.L.; Huang, Z.H.; Duan, Y.Y.; Ye, P. Exploration of Emodin to treat α-naphthylisothiocyanate-induced cholestatic hepatitis via anti-inflammatory pathway. Eur. J. Pharmacol 2008, 590, 377–386. [Google Scholar]
- Hsu, C.M.; Hsu, Y.A.; Tsai, Y.; Shieh, F.K.; Huang, S.H.; Wan, L.; Tsai, F.J. Emodin inhibits the growth of hepatoma cells: finding the common anti-cancer pathway using Huh7, Hep3B, and HepG2 cells. Biochem. Biophys. Res. Commun 2010, 392, 473–478. [Google Scholar]
- Zhan, Y.; Li, D.; Wei, H.; Wang, Z.; Huang, X.; Xu, Q.; Lu, H. Emodin on hepatic fibrosis in rats. Chin. Med. J 2000, 113, 599–601. [Google Scholar]
- Tilg, H. The role of cytokines in non-alcoholic fatty liver disease. Dig. Dis 2010, 28, 179–185. [Google Scholar]
- Tetri, L.H.; Basaranoglu, M.; Brunt, E.M.; Yerian, L.M.; Neuschwander-Tetri, B.A. Severe NAFLD with hepatic necroinflammatory changes in mice fed trans fats and a high-fructose corn syrup equivalent. Am. J. Physiol. Gastrointest. Liver Physiol 2008, 295, G987–G995. [Google Scholar]
- Bertola, A.; Bonnafous, S.; Anty, R.; Patouraux, S.; Saint-Paul, M.C.; Iannelli, A.; Gugenheim, J.; Barr, J.; Mato, J.M.; Le Marchand-Brustel, Y.; et al. Hepatic expression patterns of inflammatory and immune response genes associated with obesity and NASH in morbidly obese patients. PLoS One 2010, 5, e13577. [Google Scholar]
- Ferreira, A.V.; Mario, E.G.; Porto, L.C.; Andrade, S.P.; Botion, L.M. High-carbohydrate diet selectively induces tumor necrosis factor-α production in mice liver. Inflammation 2011, 34, 139–145. [Google Scholar]
- Han, D.; Hanawa, N.; Saberi, B.; Kaplowitz, N. Mechanisms of liver injury. III. Role of glutathione redox status in liver injury. Am. J. Physiol. Gastrointest. Liver Physiol 2006, 291, G1–G7. [Google Scholar]
- Alisi, A.; Piemonte, F.; Pastore, A.; Panera, N.; Passatelli, C.; Tozzi, G.; Petrini, S.; Pietrobattista, A.; Bottazzo, G.F.; Nobili, V. Glutathionylation of p65NF-kappaB correlates with proliferating/apoptotic hepatoma cells exposed to pro- and anti-oxidants. Int. J. Mol. Med 2009, 24, 319–326. [Google Scholar]
- Yu, C.X.; Li, S.; Whorton, A.R. Redox regulation of PTEN by S-nitrosothiols. Mol. Pharmacol 2005, 68, 847–854. [Google Scholar]
- Alisi, A.; Bruscalupi, G.; Pastore, A.; Petrini, S.; Panera, N.; Massimi, M.; Tozzi, G.; Leoni, S.; Piemonte, F.; Nobili, V. Redox homeostasis and posttranslational modifications/activity of phosphatase and tensin homolog in hepatocytes from rats with diet-induced hepatosteatosis. J. Nutr. Biochem 2012, 23, 169–178. [Google Scholar]
- Peyrou, M.; Bourgoin, L.; Foti, M. PTEN in non-alcoholic fatty liver disease/non-alcoholic steatohepatitis and cancer. Dig. Dis 2010, 1, 236–246. [Google Scholar]
- Vinciguerra, M.; Foti, M. PTEN at the crossroad of metabolic diseases and cancer in the liver. Ann. Hepatol 2008, 7, 192–199. [Google Scholar]
- Hay, N. Akt isoforms and glucose homeostasis - the leptin connection. Trends Endocrinol. Metab 2011, 22, 66–73. [Google Scholar]
- Leoni, S.; Spagnuolo, S.; Massimi, M.; Terenzi, F.; Conti Devirgiliis, L. Amino acid uptake regulation by cell growth in cultured hepatocytes isolated from fetal and adult rats. Biosci. Rep 1992, 12, 135–141. [Google Scholar]
- Pastore, A.; Federici, G.; Bertini, E.; Piemonte, F. Analysis of glutathione: implication in redox and detoxification. Clin. Chim. Acta 2003, 333, 19–39. [Google Scholar]
- Babich, H.; Borenfreund, E. Cytotoxicity of T-2 toxin and its metabolites determined with the neutral red cell viability assay. Appl. Environ. Microbiol 1991, 57, 2101–2103. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | SD | HFD/HF |
|---|---|---|
| Body weight (g) | 140.8 ± 25.8 | 143.6 ± 23.2 |
| Triglycerides (mg/dL) | 92 ± 13.5 | 104 ± 19.4 |
| Total cholesterol (mg/dL) | 39.5 ± 5.8 | 44.0 ± 6.2 |
| ALT (U/L) | 22.3 ± 3.6 | 25.4 ± 4.5 |
| Glucose (mg/dL) | 65.0 ± 7.5 | 73.4 ± 10.1 |
| Insulin (ng/mL) | 0.23 ± 0.04 | 0.25 ± 0.03 |
| HOMA-IR | 0.92 ± 0.08 | 1.13 ± 0.15 |
| Parameters | SD | HFD/HF | SD+emodin | HFD/HF+emodin |
|---|---|---|---|---|
| Body weight (g) | 295.6 ± 34.2 | 355.6 ± 28.7 * | 308.1 ± 30.5 | 393.6 ± 31.4 † |
| Liver weight (g) | 11.2 ± 1.3 | 15.2 ± 1.5 * | 12.0 ± 0.5 | 12.4 ± 0.9 † |
| Liver weight/Body weight | 3.7 ± 0.11 | 4.27 ± 0.09 * | 3.8 ± 0.19 | 3.2 ± 0.2 ‡ |
| Triglycerides (mg/dL) | 105.1 ± 18.6 | 138.0 ± 16.3 ** | 110.4 ± 13.1 | 109.4 ± 15.7 ‡ |
| Total cholesterol (mg/dL) | 45.7 ± 7.2 | 48.5 ± 8.1 | 39.9 ± 10.2 | 48.0 ± 12.3 |
| ALT (U/L) | 25.2 ± 5.0 | 37.4 ± 3.9 ** | 26.0 ± 4.4 | 25.9 ± 5.8 ‡ |
| Glucose (mg/dL) | 69.4 ± 4.4 | 81.9 ± 5.7 ** | 68.3 ± 7.2 | 70.8 ± 9.6 ‡ |
| Insulin (ng/mL) | 0.24 ± 0.05 | 0.41 ± 0.09 ** | 0.25 ± 0.07 | 0.26 ± 0.03 ‡ |
| HOMA-IR | 1.02 ± 0.05 | 2.07 ± 0.18 ** | 1.05 ± 0.06 | 1.13 ± 0.19 ‡ |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Alisi, A.; Pastore, A.; Ceccarelli, S.; Panera, N.; Gnani, D.; Bruscalupi, G.; Massimi, M.; Tozzi, G.; Piemonte, F.; Nobili, V. Emodin Prevents Intrahepatic Fat Accumulation, Inflammation and Redox Status Imbalance During Diet-Induced Hepatosteatosis in Rats. Int. J. Mol. Sci. 2012, 13, 2276-2289. https://doi.org/10.3390/ijms13022276
Alisi A, Pastore A, Ceccarelli S, Panera N, Gnani D, Bruscalupi G, Massimi M, Tozzi G, Piemonte F, Nobili V. Emodin Prevents Intrahepatic Fat Accumulation, Inflammation and Redox Status Imbalance During Diet-Induced Hepatosteatosis in Rats. International Journal of Molecular Sciences. 2012; 13(2):2276-2289. https://doi.org/10.3390/ijms13022276
Chicago/Turabian StyleAlisi, Anna, Anna Pastore, Sara Ceccarelli, Nadia Panera, Daniela Gnani, Giovannella Bruscalupi, Mara Massimi, Giulia Tozzi, Fiorella Piemonte, and Valerio Nobili. 2012. "Emodin Prevents Intrahepatic Fat Accumulation, Inflammation and Redox Status Imbalance During Diet-Induced Hepatosteatosis in Rats" International Journal of Molecular Sciences 13, no. 2: 2276-2289. https://doi.org/10.3390/ijms13022276
APA StyleAlisi, A., Pastore, A., Ceccarelli, S., Panera, N., Gnani, D., Bruscalupi, G., Massimi, M., Tozzi, G., Piemonte, F., & Nobili, V. (2012). Emodin Prevents Intrahepatic Fat Accumulation, Inflammation and Redox Status Imbalance During Diet-Induced Hepatosteatosis in Rats. International Journal of Molecular Sciences, 13(2), 2276-2289. https://doi.org/10.3390/ijms13022276