Abstract
1-Methyl-10-nitro-11-phenyl(2-furyl)-2-aza-13-oxa-tricyclo[7.3.1.03,8]trideca-3,5,7-trienes 3 were synthesized via the reduction of the aromatic 2-nitro group of 2-methyl-5-nitro-6-(2-nitrophenyl)-4-phenyl(2-furyl)tetrahydropyranols 2 and subsequent condensation with their anomeric OH group. As by-products, the corresponding 6-(2-aminophenyl)-2-methyl-5-nitro-4-phenyl(2-furyl)tetrahydropyranols 4 were isolated.
Introduction
Recently we published a new method for the preparation of some tetrahydro-2-pyranols by nitroaldol reaction of γ-nitro ketones with various aromatic aldehydes and subsequent cyclization [1]. The anomeric OH group of these tetrahydro-2-pyranols underwent the Fischer-glycosidation by refluxing in methanol with a catalytic amount of concentrated hydrochloric acid to furnish the corresponding α-methyl glycosides. In this paper, we describe the use of an internal N-glycosidation based on 2-methyl-5-nitro-6-(2-nitrophenyl)-4-phenyl(2-furyl)tetrahydropyranols (2) to prepare new tricyclic compounds.
Results and Discussion
The [2RS-(2α, 4β, 5α, 6β]-(±)-3,4,5,6-tetrahydro-2-methyl-5-nitro-6-(2-nitrophenyl)-4-phenyl(2-furyl)-2H-pyran-2-ols 2a,b were synthesized by treatment of (RS)-(±)-5-nitro-4-phenyl(2-furyl)-2-pentanones (1a,b) with 2-nitrobenzaldehyde in the presence of sodium ethoxide in dry ethanol at room temperature [1]. The reduction of compounds 2 with palladium on charcoal in ethyl acetate yielded mixtures of (1RS, 9RS, 10RS, 11RS)-(±)-1-methyl-10-nitro-11-phenyl(2-furyl)-2-aza-13-oxa-tricyclo-[7.3.1.03,8]trideca-3,5,7-trienes 3a,b and of [2RS-(2α, 4β, 5α, 6β]-(±)-6-(2-aminophenyl)-3,4,5,6-tetrahydro-2-methyl-5-nitro-4-phenyl(2-furyl)-2H-pyran-2-ols 4a,b. By separation with column chromatography we obtained as main products the tricycles 3a and 3b as crystalline solids in 57 and 54% yields (Scheme 1). The 6-(2-aminophenyl)-3,4,5,6-tetrahydro-2-methyl-5-nitro-4-phenyl(2-furyl)-2H-pyran-2-ols 4a,b could be isolated as by-products in yields of 28 and 26%, respectively.
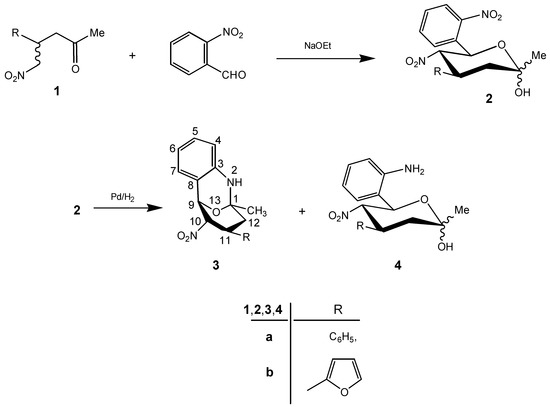
Scheme 1.
Scheme 1.
Although all compounds are racemates only one isomer is shown in Scheme 1. The analytical data of the isolated tricycles 3 are in agreement with these structures. On the other hand, the coupling constants in the 1H NMR spectra and an X-ray crystal structure investigation of compound 3b showed that during the reduction of the aromatic nitro group and the subsequent condensation the pyranoid ring system is forced into a boat conformation (Figure 1, Table 1). To our knowledge tricycles of this kind containing a pyranosidic ring system anellated to an aromatic ring in 1- and 2-position over a NH- function are not yet described in the literature.
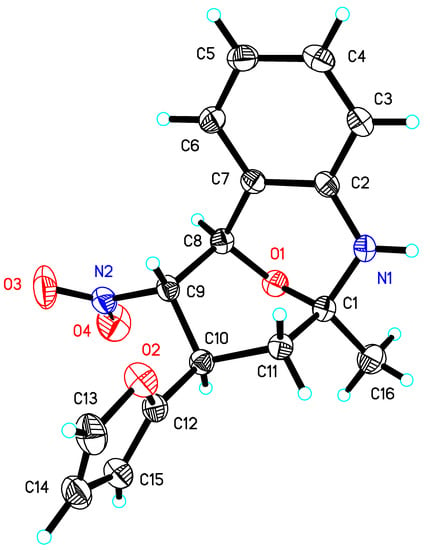
Figure 1.
Molecular structure of 3b.
Figure 1.
Molecular structure of 3b.
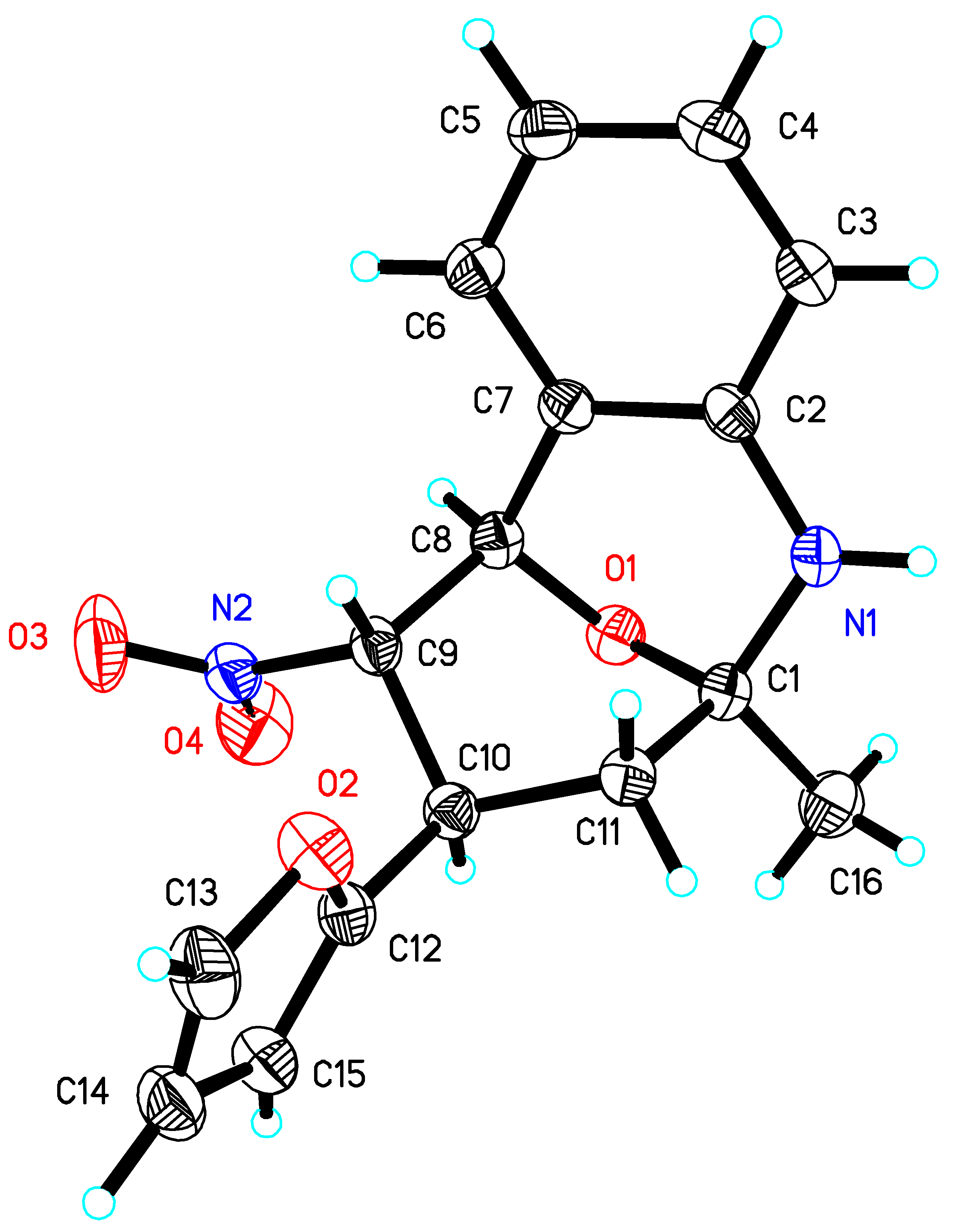

Table 1.
Crystal-structure data.
The corresponding 5-amino-2-methyl-6-(2-nitro-phenyl)-4-phenyl(2-furyl)tetrahydropyranols are interesting owing to the biological activity of a variety of 5-amino-tetrahydro-2-pyranols [2,3,4]. Although there are many literature methods for conversion of a aliphatic nitro group into an amino function the 5-amino-tetrahydro-2-pyranol could not be obtained, neither by means of reducing agents like Zn/hydrochloric acid nor by refluxing with LiAlH4 in tetrahydrofuran [5].
Due to the 1,3-interaction of the bulky aryl groups the compounds 4, like the corresponding 2-methyl-5-nitro-6-(2-nitro-phenyl)-4-phenyl(2-furyl)tetrahydropyranols 2, possess a 1,4-chair conformation. The J values for the couplings between the protons 3ax-H, 4-H; 4-H, 5-H and 5H, 6-H are 10 - 13 Hz. Therefore, in all known cases the R, NH2 and o-nitrophenyl groups are oriented in the preferred equatorial arrangement. Other diastereomers were not identified. According to the anomeric effect and the long-range coupling of the OH group with an axial proton of the ethylene group observed in the 1H NMR spectra of 4a and further investigated compounds [6] the OH group in 4 should be in axial position.
Conclusion
We have presented a method for the one-pot preparation of 1-methyl-10-nitro-11-phenyl(2-furyl)-2-aza-13-oxa-tricyclo[7.3.1.03,8]trideca-3,5,7-trienes from (2-nitrophenyl)pyran-2-oles by successful reduction of nitro group followed by internal N-glycosidation.
Experimental
General
Melting points were obtained on a Boëtius melting point apparatus. 1H NMR and 13C NMR spectra were recorded on Bruker ARX 300 and AC 250 instruments with DMSO-d6 as solvent. The calibration of spectra was carried out by means of solvent peaks (DMSO-d6: δ 1H= 2.50; δ 13C= 39.7). Signal assignment was confirmed by DEPT and/or 1H,13C COSY experiments. Mass spectra were recorded on an AMD 402/3 spectrometer (AMD Intectra GmbH). TLC was performed on silica gel foils 60 F254 (Merck) with detection by charring with sulphuric acid. For column chromatography silica gel 60 (230-400 mm) (Merck) was used. Elemental analyses were carried out with a Leco CHNS-932 apparatus. Table 1 provides a summary of the crystallographic data of compound 3b. A crystal of 3b was sealed onto a glass fiber and mounted on a Siemens P4 automated four circle diffractometer (Mo-Kα radiation; λ = 0.71073 Å) with graphite monochromator and measured at T = 293 K. The structure was solved by direct methods (Siemens SHELXTL, version 4.2 for MS-DOS, Siemens Analytical Xray Inst. Inc.) and refined by the full-matrix least-squares method of SHELXL-97. Non-H atoms were refined with anisotropic displacement parameters. All hydrogen atoms were placed into theoretical positions and were refined by using the riding model. Crystallographic data (excluding structure factors) reported in this paper for structure 3b have been deposited with the Cambridge Crystallo-graphic Data Centre as Supplementary Publication No. CCDC-139912. Copies of the data can be obtained free of charge on application to The Director, CCDC, 12 Union Road, Cambridge CB2 1EZ, UK (fax: int. code +(1223) 336-033; e-mail:deposit@ccdc.cam.ac.uk).
Method
The [2RS-(2α, 4β, 5α, 6β]-(±)-3,4,5,6-tetrahydro-2-methyl-5-nitro-6-(2-nitrophenyl)-4-phenyl(2-furyl)-2H-pyran-2-ols 2a,b (3 mmol) were dissolved in ethyl acetate (20 mL). A small portion of palladium on charcoal (10% Pd) was added. The vessel was filled three times with hydrogen under stirring at normal pressure. After prolongation of the stirring for 48 h the solution was filtrated over Celite. Elution with ethyl acetate (3x10 mL), solvent evaporation under reduced pressure and column chromatographic separation (toluene/ ethyl acetate = 5 : 1, v/v) afforded compounds 3 and 4.
(1RS, 9RS, 10RS, 11RS)-(±)-1-Methyl-10-nitro-11-phenyl-2-aza-13-oxa-tricyclo[7.3.1.03,8]trideca-3,4,5- triene (3a)
Yield 0.53 g (57%).- m.p. 193-194oC (ether).- 1H NMR (250.1 MHz, DMSO-d6): δ/ppm = 7.32-7.10 (m, 7H, C6H5, H-5, H-7), 6.80 (m, 1H, H-6), 6.79 (s, 1H, NH), 6.70 (m, 1H, H-4), 5.45 (br s, 1H, H-9), 4.93 (dd, 1H, J10,11 = 10.4 Hz, J9,10 = 0.9 Hz, H-10), 3.35 (ddd, 1H, H-11), 2.05 (dd, 1H, J12,12• =14.3 Hz, J11,12 = 4.3 Hz, H-12), 1.66 (dd, 1H, J11,12• = 14.0 Hz, H-12´), 1.58 (s, 3H, CH3).- 13C NMR (62.9 MHz, DMSO-d6): δ/ppm = 140.1(C-3), 139.7 (i-C6H5), 128.8 (m-C6H5), 128.4, 127.4, 126.1 (C-5, C-7, p-C6H5), 127.6 (o-C6H5), 121.9 (C-8), 118.4 (C-6), 117.0 (C-4), 95.5 (C-10), 81.3 (C-1), 72.5 (C-9), 39.4 (C-12), 38.6 (C-11), 26.6 (CH3).- IR(nujol), /cm-1 = 3402 (NH), 1548, 1378 (NO2). - MS (70 eV, EI): m/z (%) = 310 (100, M+·). For C18H18N2O3 (310.1) calcd.: C 69.65 H 5.85 N 9.03; found C 69.43 H 5.81 N 8.87.
(1RS, 9RS, 10RS, 11RS)-(±)-11-(2-Furyl)-1-methyl-10-nitro-2-aza-13-oxa-tricyclo[7.3.1.03,8]trideca- 3,4,5-triene (3b)
Yield 0.48 g (54%).- m.p. 137-138oC (ether).- 1H NMR (250.1 MHz, DMSO-d6): δ/ppm = 7.45 (dd, 1H, J4-Fur,5-Fur = 1.8 Hz, J3-Fur,5-Fur = 0.6 Hz, H-5-Fur), 7.16 (m, 1H, H-7), 7.08 (m, 1H, H-5), 6.74 (m, 1H, H-6), 6.73 (s, 1H, NH), 6.62 (m, 1H, H-4), 6.26 (dd, 1H, J4-Fur,3-Fur = 3.4 Hz, H-4-Fur), 6.16 (dd, 1H, H-3-Fur), 5.45 (br s, 1H, H-9), 4.94 (dd, 1H, J10,11 = 8.9 Hz, J9,10 = 0.9 Hz, H-10), 3.60 (ddd, 1H, H-11), 2.23 (dd, 1H, J12,12• = 14.5 Hz, J11,12 = 4.6 Hz, H-12), 1.71 (dd, 1H, J11,12• = 12.3 Hz, H-12´), 1.55 (s, 3H, 13 CH3) ).- 13C NMR (62.9 MHz, DMSO-d6): δ/ppm = 152.8 (C-2-Fur), 142.4 (C-5-Fur), 140.4 (C-3), 128.4, 125.9 (C-5, C-7), 120.7 (C-8), 118.1 (C-6), 116.5 (C-4), 110.5 (C-4-Fur), 105.5 (C-3-Fur), 91.8 (C-10), 80.7 (C-1), 72.3(C-9), 36.3 (C-12), 26.9 (C-11), 25.9 (CH3).- IR(nujol), /cm-1 = 3409 (NH), 1548, 1367 (NO2). - MS (70 eV, EI): m/z (%) = 310 (100, M+·). For C16H16N2O4 (300.1) calcd. C 63.98 H 5.37 N 9.33; found C 63.98 H 5.21 N 9.25.
[2RS-(2α, 4β, 5α, 6β]-(±)-6-(2-Aminophenyl)-3,4,5,6-tetrahydro-2-methyl-5-nitro-4-phenyl-2H-pyran- 2-ol (4a)
Yield 0.27 g (28%).- m.p. 147-148oC (ethanol).- 1H NMR (250.1 MHz, DMSO-d6): δ/ppm = 7.30- 7.15 (m, 5H, C6H5), 7.07-6.94 (m, 2H, H-4-C6H4, H-6-C6H4), 6.71 (dd, 1H, J3-C6H4,4-C6H4 = 8.0 Hz, J3-C6H4,5-C6H4 = 0.9 Hz, H-3-C6H4), 6.50 (ddd, 1H, J5-C6H4,6-C6H4 = J5-C6H4,3-C6H4 = 7.5 Hz, H-5-C6H4), 6.42 (d, 1H, J3ax,OH = 1.5 Hz, OH), 5.50-5.30 (m, 2H, H-5, H-6), 5.05 (s, 2H, NH2), 3.85 (ddd, 1H, J3ax,4 = 13.0 Hz, J4,5 = 10.7 Hz, J3eq,4 = 4.0 Hz, H-4), 2.18 (ddd, 1H, H-3ax), 1.93 (dd, 1H, J3ax,3eq = 13.5 Hz, H-3eq), 1.44 (s, 3H, CH3).- 13C NMR (62.9 MHz, DMSO-d6): δ/ppm = 147.2 (C-2-C6H4), 139.6 (i-C6H5), 129.5, 129.0 (C-4-C6H5, C-6-C6H5), 128.8 (m-C6H5), 128.3 (p-C6H5), 127.5 (o-C6H5), 118.9 (C-1-C6H4), 116.3, 116.1 (C-3-C6H4, C-5-C6H4), 95.6 (C-2), 88.5 (C-5), 72.7 (C-6), 42.6 (C-4), 41.4 (C-3), 28.4 (CH3).- IR(KBr), /cm-1 = 3395, 3321 (NH2), 1545, 1336 (NO2). - MS (70 eV, EI): m/z (%) = 328 (88, M+·). For C18H20N2O4 (328.1) calcd. C 65.83 H 6.14 N 8.53; found C 65.60 H 5.94 N 8.41.
[2RS-(2α, 4β, 5α, 6β]-(±)-6-(2-Aminophenyl)-4-(2-furyl)-3,4,5,6-tetrahydro-2-methyl-5-nitro-2H-pyran- 2-ol (4b)
Yield 0.24 g (26%).- m.p. 153-154oC (ether).- 1H NMR (250.1 MHz, DMSO-d6): δ/ppm = 7.58 (dd, 1H, J5-Fur,4-Fur = 2.2 Hz, J5-Fur,3-Fur = 0.8 Hz, H-5-Fur), 7.05 (ddd, 1H, H-4-C6H4), 6.92 (dd, 1H, J6-C6H4,5-C6H4 = 7.6 Hz, J6-C6H4,4-C6H4 = 1.5 Hz, H-6-C6H4), 6.71 (dd, 1H, J3-C6H4,4-C6H4 = 8.0 Hz, J3-C6H4,5-C6H4 = 0.9 Hz, H-3-C6H4), 6.51 (ddd, 1H, J5-C6H4,4-C6H4 = 7.6 Hz, H-5-C6H4), 6.48 (br, 1H, OH), 6.39 (dd, 1H, J3-Fur,4-Fur = 3.4 Hz, H-4-Fur), 6.27 (dd, 1H, H-3-Fur), 5.33-5.22 (m, 2H, H-5, H-6), 5.02 (br, 2H, NH2), 4.00 (m, 1H, H-4), 2.20-2.00 (m, 2H, H-3ax, H-3eq), 1.46 (s, 3H, CH3).- 13C NMR (62.9 MHz, DMSO-d6): δ/ppm = 152.9 (C-2-Fur), 147.2 (C-2-C6H4), 142.8 (C-5-Fur), 129.6, 129.2 (C-4-C6H4, C-6-6H4), 118.6 (C-1-6H4), 116.4, 116.3 (C-3-6H4, C-5-6H4), 110.6 (C-4-Fur), 106.6 (C-3-Fur), 95.4 (C-2), 86.8 (C-5), 72.8 (C-6), 38.7 (C-3), 35.9 (C-4), 28.2 (CH3).- IR(nujol), /cm-1 = 3391, 3321 (NH2), 1546, 1367 (NO2). - MS (70 eV, EI): m/z (%) = 316 (18, M+·). For C16H16N2O5 (316.1) calcd. C 60.74 H 5.10 N 8.86; found C 60.86 H 5.03 N 8.79.
Acknowledgment:
We would like to thank the Deutsche Forschungsgemeinschaft and the Fonds der Chemischen Industrie for financial support. José Quincoces is grateful to FAPESP, Sao Paulo, Brasil, for financial support.
References and Notes
- Peseke, K.; Götze, L.; Reinke, H.; Cedeno, Q. A.; Suarez, J. Q.; Andreu, M. G.; Castro, H. V. Synthesis of Substituted Tetrahydro-2-pyranols. J. prakt. Chem. 1997, 339, 656–659. [Google Scholar] [CrossRef]
- Kuhn, R. Aminozucker. Angew. Chem. 1957, 69, 23–33. [Google Scholar] [CrossRef]
- Balazs, E. A.; Jeanloz, R. W. The Amino Sugars; Academic Press: New York, 1966. [Google Scholar]
- Brimacombe, J. S. Synthesen von Antibiotica-Zuckern. Angew. Chem. 1971, 83, 261–300. [Google Scholar] [CrossRef]
- Gibson, M. S. The chemistry of functional groups; The chemistry of the amino group; Patai, S., Feuer, H., Eds.; Wiley & Sons: New York, 1986; Vol. 1, Chapter 2; p. 37. [Google Scholar]
- Götze, L.; Peseke, K.; Michalik, M. unpublished results.
- Samples Availability: Available from the authors.
© 2000 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.