
Scoping Review of Extraction Methods for Detecting β-Lactam Antibiotics in Food Products of Animal Origin



Abstract
1. Introduction
2. Methodology
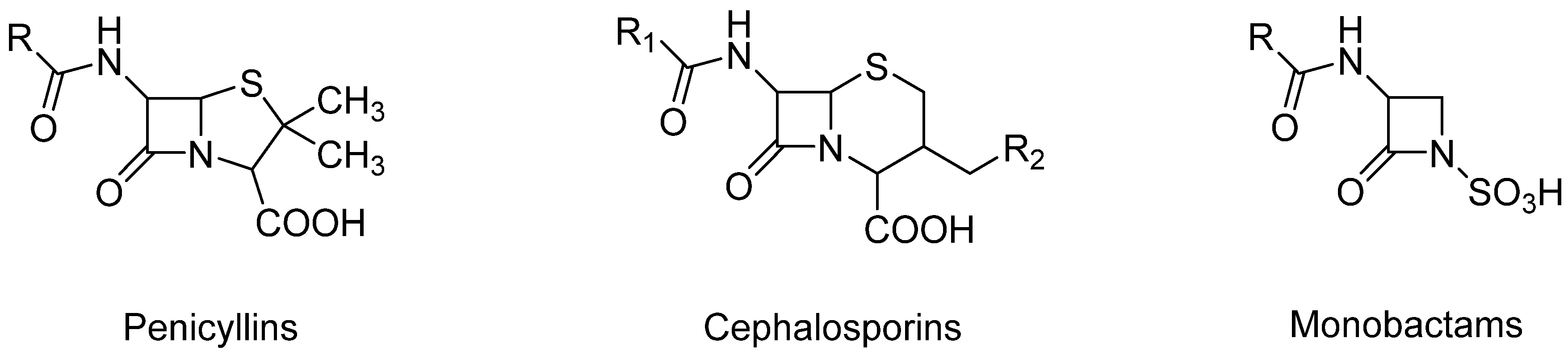
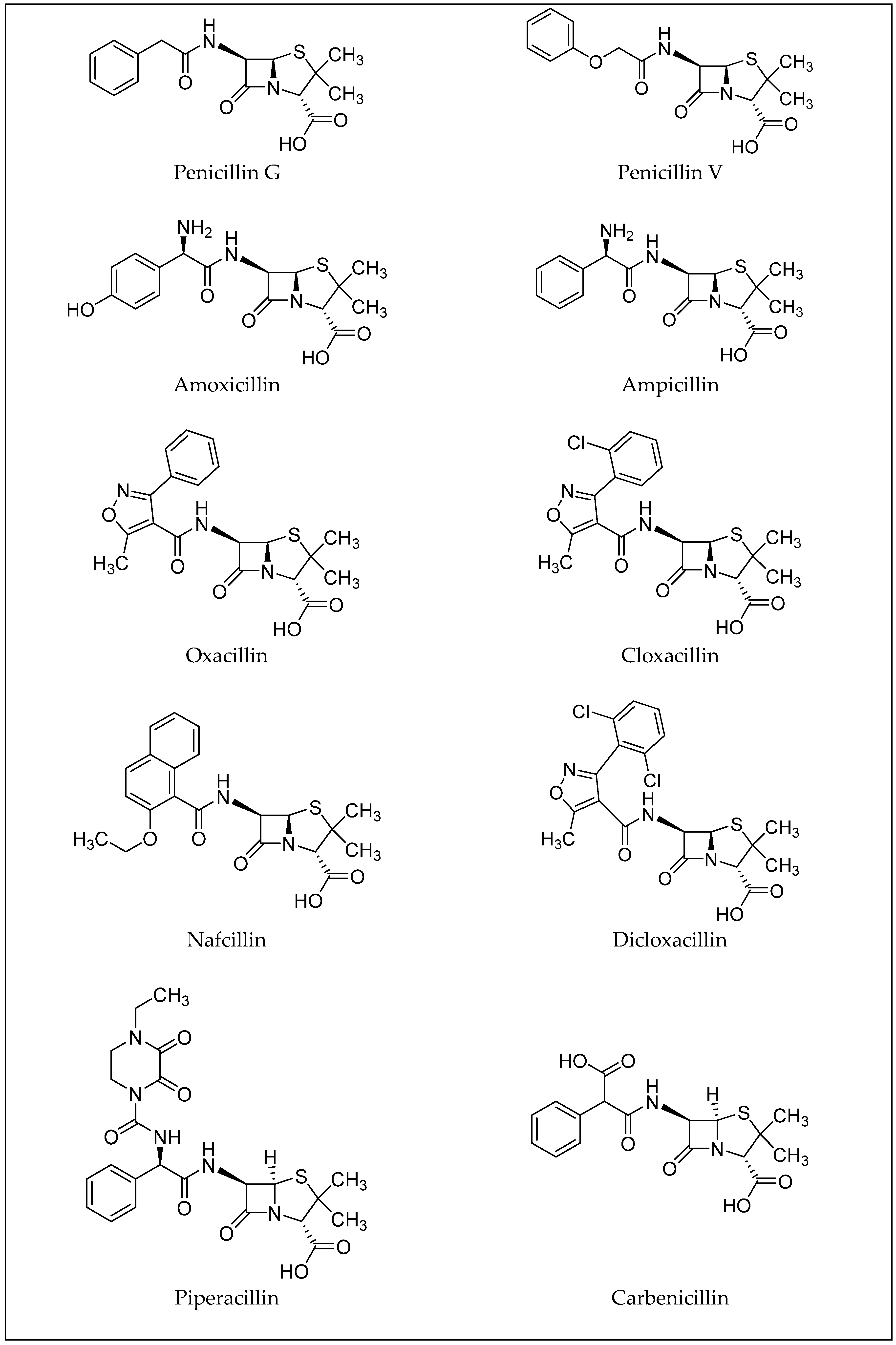
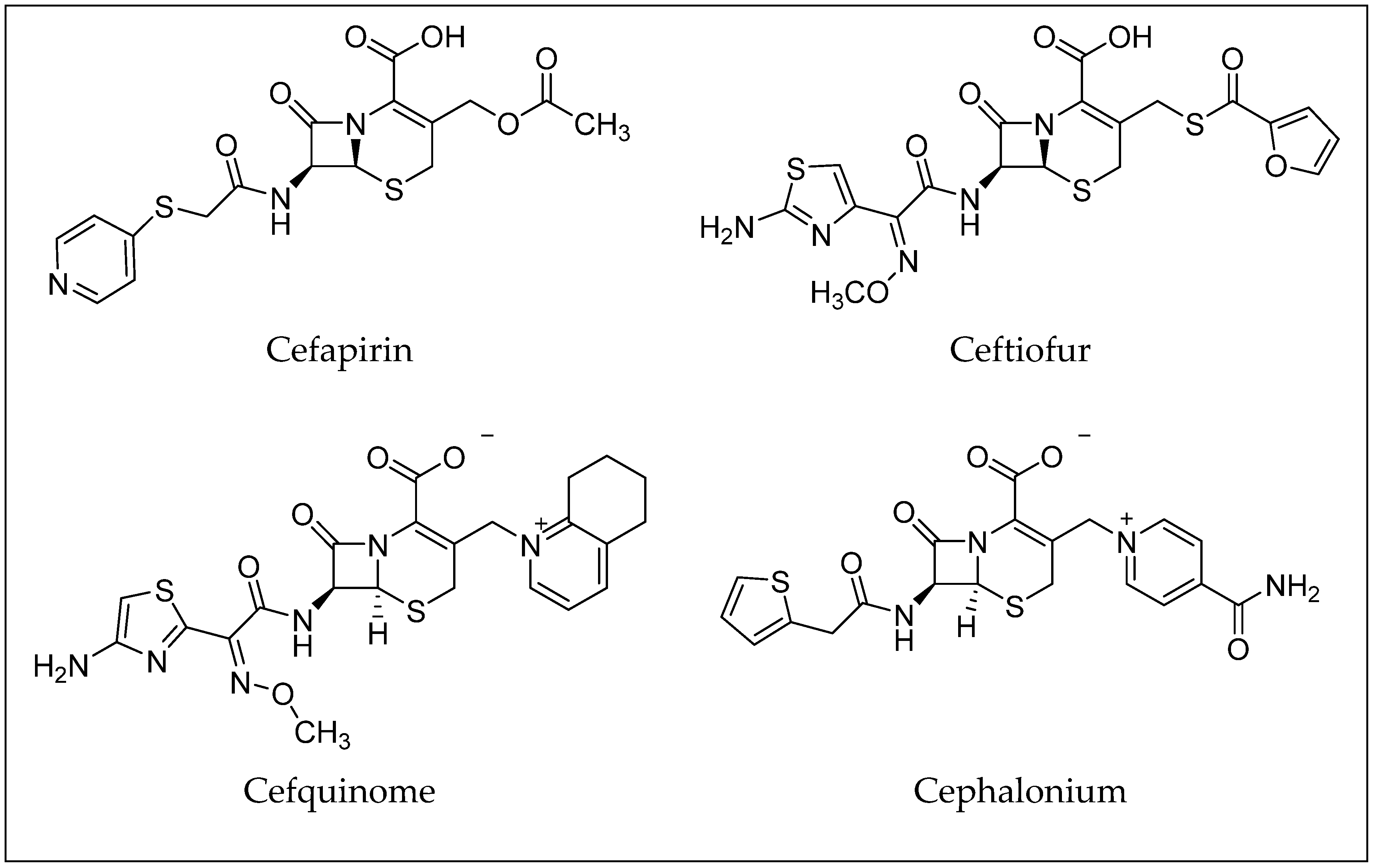
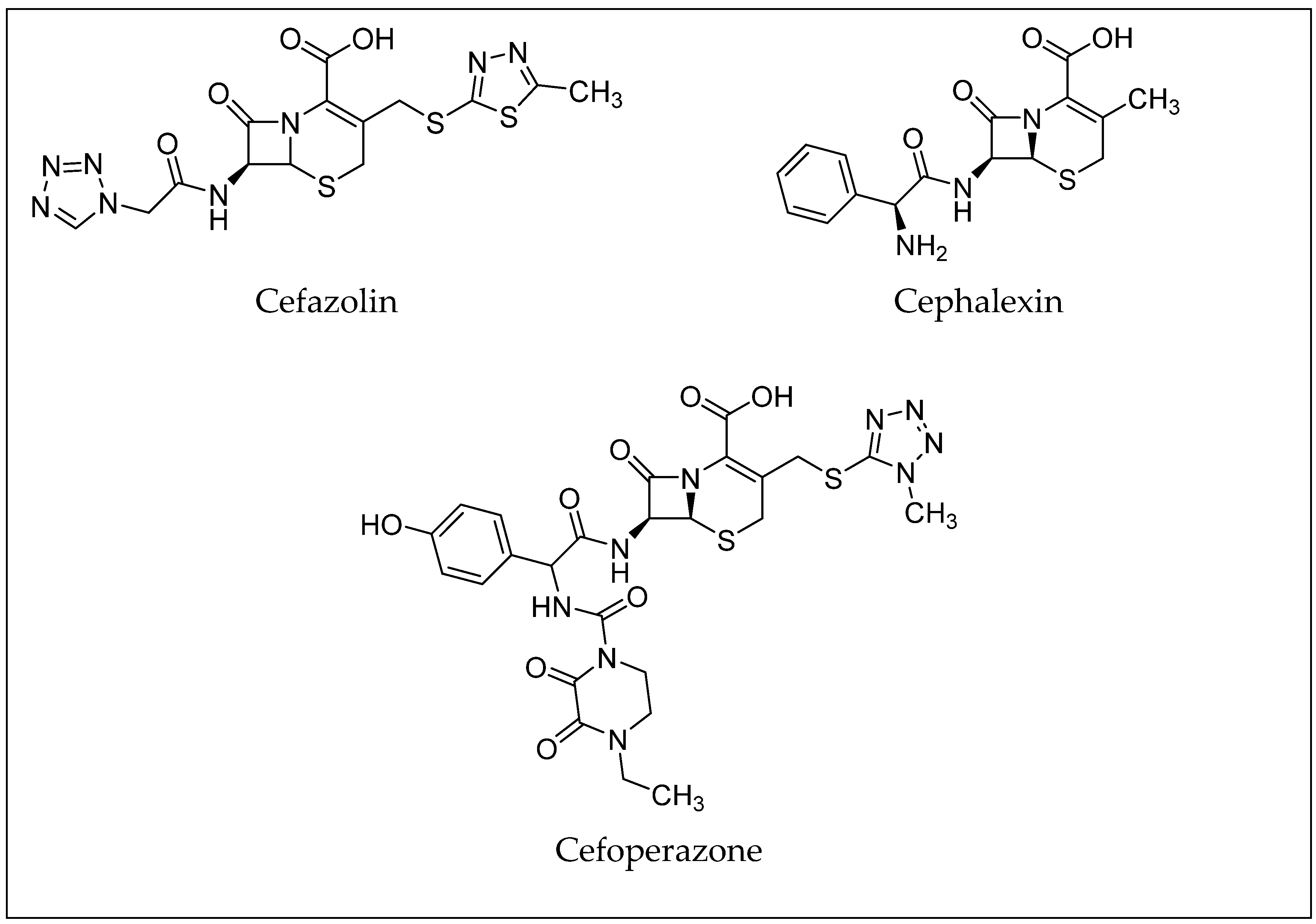
3. β-Lactam Antibiotics
4. Extraction Methods
5. Extraction of β-Lactams
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Moreno-Bondi, M.C.; Marazuela, M.D.; Herranz, S.; Rodriguez, E. An overview of sample preparation procedures for LC-MS multiclass antibiotic determination in environmental and food samples. Anal. Bioanal. Chem. 2009, 395, 921–946. [Google Scholar] [CrossRef]
- Zhang, C.; Deng, Y.; Zheng, J.; Zhang, Y.; Yang, L.; Liao, C.; Su, L.; Zhou, Y.; Gong, D.; Chen, L.; et al. The application of the QuEChERS methodology in the determination of antibiotics in food: A review. TrAC Trends Anal. Chem. 2019, 118, 517–537. [Google Scholar] [CrossRef]
- Rossi, R.; Saluti, G.; Moretti, S.; Diamanti, I.; Giusepponi, D.; Galarini, R. Multiclass methods for the analysis of antibiotic residues in milk by liquid chromatography coupled to mass spectrometry: A review. Food Addit. Contam. Part A 2018, 35, 241–257. [Google Scholar] [CrossRef] [PubMed]
- Ghimpețeanu, O.M.; Pogurschi, E.N.; Popa, D.C.; Dragomir, N.; Drăgotoiu, T.; Mihai, O.D.; Petcu, C.D. Antibiotic Use in Livestock and Residues in Food—A Public Health Threat: A Review. Foods 2022, 11, 1430. [Google Scholar] [CrossRef] [PubMed]
- Cañada-Cañada, F.; Muñoz de la Peña, A.; Espinosa-Mansilla, A. Analysis of antibiotics in fish samples. Anal. Bioanal. Chem. 2009, 395, 987–1008. [Google Scholar] [CrossRef]
- Peris-Vicente, J.; Peris-García, E.; Albiol-Chiva, J.; Durgbanshi, A.; Ochoa-Aranda, E.; Carda-Broch, S.; Bose, D.; Esteve-Romero, J. Liquid chromatography, a valuable tool in the determination of antibiotics in biological, food and environmental samples. Microchem. J. 2022, 177, 107309. [Google Scholar] [CrossRef]
- Pérez-Rodríguez, M.; Pellerano, R.G.; Pezza, L.; Pezza, H.R. An overview of the main foodstuff sample preparation technologies for tetracycline residue determination. Talanta 2018, 182, 1–21. [Google Scholar] [CrossRef]
- Lees, P.; Toutain, P.-L. Pharmacokinetics, Distribution, Bioavailability, and Relationship to Antibiotic Residues. In Chemical Analysis of Antibiotic Residues in Food; Wiley: Hoboken, NJ, USA, 2011; pp. 61–109. [Google Scholar] [CrossRef]
- Kennedy, D.G.; McCracken, R.J.; Cannavan, A.; Hewitt, S.A. Use of liquid chromatography-mass spectrometry in the analysis of residues of antibiotics in meat and milk. J. Chromatogr. A 1998, 812, 77–98. [Google Scholar] [CrossRef]
- Reeves, P.T. Antibiotics: Groups and Properties. In Chemical Analysis of Antibiotic Residues in Food; Wiley: Hoboken, NJ, USA, 2011; pp. 1–60. [Google Scholar]
- Etebu, E.; Arikekpar, I. Antibiotics: Classification and mechanisms of action with emphasis on molecular perspectives. Int. J. Appl. Microbiol. Biotechnol. Res. 2016, 4, 90–101. [Google Scholar]
- Samanidou, V.F.; Evaggelopoulou, E.N. Analytical strategies to determine antibiotic residues in fish. J. Sep. Sci. 2007, 30, 2549–2569. [Google Scholar] [CrossRef]
- Bargańska, Ż.; Namieśnik, J.; Ślebioda, M. Determination of antibiotic residues in honey. TrAC Trends Anal. Chem. 2011, 30, 1035–1041. [Google Scholar] [CrossRef]
- Martín, J.F.; Ullán, R.V.; García-Estrada, C. Regulation and compartmentalization of β-lactam biosynthesis. Microb. Biotechnol. 2010, 3, 285–299. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information. PubChem Compound Summary for CID 5904, Penicillin G. 2025. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Penicillin-G (accessed on 6 April 2025).
- National Center for Biotechnology Information. PubChem Compound Summary for CID 6869, Penicillin V. 2025. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Penicillin-V (accessed on 6 April 2025).
- National Center for Biotechnology Information. PubChem Compound Summary for CID 33613, Amoxicillin. 2025. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Amoxicillin (accessed on 6 April 2025).
- National Center for Biotechnology Information. PubChem Compound Summary for CID 6249, Ampicillin. 2025. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Ampicillin (accessed on 6 April 2025).
- National Center for Biotechnology Information. PubChem Compound Summary for CID 6196, Oxacillin. 2025. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Oxacillin (accessed on 6 April 2025).
- National Center for Biotechnology Information. PubChem Compound Summary for CID 6098, Cloxacillin. 2025. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Cloxacillin (accessed on 7 April 2025).
- National Center for Biotechnology Information. PubChem Compound Summary for CID 8982, Nafcillin. 2025. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Nafcillin (accessed on 7 April 2025).
- National Center for Biotechnology Information. PubChem Compound Summary for CID 18381, Dicloxacillin. 2025. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Dicloxacillin (accessed on 7 April 2025).
- National Center for Biotechnology Information. PubChem Compound Summary for CID 43672, Piperacillin. 2025. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Piperacillin (accessed on 7 April 2025).
- National Center for Biotechnology Information. PubChem Compound Summary for CID 20824, Carbenicillin. 2025. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Carbenicillin (accessed on 7 April 2025).
- Samanidou, V.; Nisyriou, S. Multi-residue methods for confirmatory determination of antibiotics in milk. J. Sep. Sci. 2008, 31, 2068–2090. [Google Scholar] [CrossRef] [PubMed]
- Percival, K.M. Antibiotic Classification and Indication Review for the Infusion Nurse. J. Infus. Nurs. Off. Publ. Infus. Nurses Soc. 2017, 40, 55–63. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. PubChem Compound Summary for CID 30699, Cephapirin. 2025. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Cephapirin (accessed on 7 April 2025).
- National Center for Biotechnology Information. PubChem Compound Summary for CID 6328657, Ceftiofur. 2025. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/6328657 (accessed on 7 April 2025).
- National Center for Biotechnology Information. PubChem Compound Summary for CID 5464355, Cefquinome. 2025. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Cefquinome (accessed on 7 April 2025).
- National Center for Biotechnology Information. PubChem Compound Summary for CID 21743, Cefalonium. 2025. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Cefalonium (accessed on 7 April 2025).
- National Center for Biotechnology Information. PubChem Compound Summary for CID 33255, Cefazolin. 2025. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Cefazolin (accessed on 7 April 2025).
- National Center for Biotechnology Information. PubChem Compound Summary for CID 27447, Cephalexin. 2025. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Cephalexin (accessed on 7 April 2025).
- National Center for Biotechnology Information. PubChem Compound Summary for CID 44187, Cefoperazone. 2025. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Cefoperazone (accessed on 7 April 2025).
- Hu, C.; Zhang, Y.; Zhou, Y.; Liu, Z.-F.; Meng, Q.; Feng, X.-S. A review of pretreatment and analysis of macrolides in food (Update Since 2010). J. Chromatogr. A 2020, 1634, 461662. [Google Scholar] [CrossRef]
- Bitas, D.; Kabir, A.; Locatelli, M.; Samanidou, V. Food Sample Preparation for the Determination of Sulfonamides by High-Performance Liquid Chromatography: State-of-the-Art. Separations 2018, 5, 31. [Google Scholar] [CrossRef]
- Nováková, L.; Vlcková, H. A review of current trends and advances in modern bio-analytical methods: Chromatography and sample preparation. Anal. Chim. Acta 2009, 656, 8–35. [Google Scholar] [CrossRef] [PubMed]
- Stolker, A.A.M.; Danaher, M. Sample Preparation: Extraction and Clean-Up. In Chemical Analysis of Antibiotic Residues in Food; Wiley: Hoboken, NJ, USA, 2011; pp. 125–152. [Google Scholar] [CrossRef]
- Kaufmann, A.; Butcher, P.; Maden, K.; Widmer, M. Quantitative multiresidue method for about 100 veterinary drugs in different meat matrices by sub 2-μm particulate high-performance liquid chromatography coupled to time of flight mass spectrometry. J. Chromatogr. A 2008, 1194, 66–79. [Google Scholar] [CrossRef]
- Gaugain-Juhel, M.; Delepine, B.; Gautier, S.; Fourmond, M.P.; Gaudin, V.; Hurtaud-Pessel, D.; Verdon, E.; Sanders, P. Validation of a liquid chromatography-tandem mass spectrometry screening method to monitor 58 antibiotics in milk: A qualitative approach. Food Addit. Contam. Part A Chem. Anal. Control. Expo. Risk Assess. 2009, 26, 1459–1471. [Google Scholar] [CrossRef]
- Jank, L.; Martins, M.T.; Arsand, J.B.; Hoff, R.B.; Barreto, F.; Pizzolato, T.M. High-throughput method for the determination of residues of β-lactam antibiotics in bovine milk by LC-MS/MS. Food Addit. Contam. Part A 2015, 32, 1992–2001. [Google Scholar] [CrossRef]
- Di Rocco, M.; Moloney, M.; O’Beirne, T.; Earley, S.; Berendsen, B.; Furey, A.; Danaher, M. Development and validation of a quantitative confirmatory method for 30 β-lactam antibiotics in bovine muscle using liquid chromatography coupled to tandem mass spectrometry. J. Chromatogr. A 2017, 1500, 121–135. [Google Scholar] [CrossRef]
- Guidi, L.R.; Santos, F.A.; Ribeiro, A.C.S.R.; Fernandes, C.; Silva, L.H.M.; Gloria, M.B.A. A simple, fast and sensitive screening LC-ESI-MS/MS method for antibiotics in fish. Talanta 2017, 163, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Kinsella, B.; O’Mahony, J.; Malone, E.; Moloney, M.; Cantwell, H.; Furey, A.; Danaher, M. Current trends in sample preparation for growth promoter and veterinary drug residue analysis. J. Chromatogr. A 2009, 1216, 7977–8015. [Google Scholar] [CrossRef]
- Fernandes, J.O.; Ferreira, M.A. Combined ion-pair extraction and gas chromatography–mass spectrometry for the simultaneous determination of diamines, polyamines and aromatic amines in Port wine and grape juice. J. Chromatogr. A 2000, 886, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Carson, M.C. Ion-pair solid-phase extraction. J. Chromatogr. A 2000, 885, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Greenlees, K.J.; Friedlander, L.G.; Boxall, A. Antibiotic Residues in Food and Drinking Water, and Food Safety Regulations. In Chemical Analysis of Antibiotic Residues in Food; Wiley: Hoboken, NJ, USA, 2011; pp. 111–123. [Google Scholar] [CrossRef]
- Nannou, C.; Ofrydopoulou, A.; Heath, D.; Heath, E.; Lambropoulou, D. QuEChERS—A Green Alternative Approach for the Determination of Pharmaceuticals and Personal Care Products in Environmental and Food Samples. In Green Analytical Chemistry: Past, Present and Perspectives; Płotka-Wasylka, J., Namieśnik, J., Eds.; Springer: Singapore, 2019; pp. 395–430. [Google Scholar] [CrossRef]
- Lehotay, S.J. Quick, Easy, Cheap, Effective, Rugged, and Safe Approach for Determining Pesticide Residues. In Pesticide Protocols; Martínez Vidal, J.L., Frenich, A.G., Eds.; Humana Press: Totowa, NJ, USA, 2006; pp. 239–261. [Google Scholar] [CrossRef]
- Bruzzoniti, M.C.; Checchini, L.; De Carlo, R.M.; Orlandini, S.; Rivoira, L.; Del Bubba, M. QuEChERS sample preparation for the determination of pesticides and other organic residues in environmental matrices: A critical review. Anal. Bioanal. Chem. 2014, 406, 4089–4116. [Google Scholar] [CrossRef]
- Wilkowska, A.; Biziuk, M. Determination of pesticide residues in food matrices using the QuEChERS methodology. Food Chem. 2011, 125, 803–812. [Google Scholar] [CrossRef]
- Perestrelo, R.; Silva, P.; Porto-Figueira, P.; Pereira, J.A.M.; Silva, C.; Medina, S.; Câmara, J.S. QuEChERS—Fundamentals, relevant improvements, applications and future trends. Anal. Chim. Acta 2019, 1070, 1–28. [Google Scholar] [CrossRef]
- Chen, Q.; Pan, X.-D.; Huang, B.-F.; Han, J.-L. Quantification of 16 β-lactams in chicken muscle by QuEChERS extraction and UPLC-Q-Orbitrap-MS with parallel reaction monitoring. J. Pharm. Biomed. Anal. 2017, 145, 525–530. [Google Scholar] [CrossRef]
- Pérez-Burgos, R.; Grzelak, E.M.; Gokce, G.; Saurina, J.; Barbosa, J.; Barrón, D. Quechers methodologies as an alternative to solid phase extraction (SPE) for the determination and characterization of residues of cephalosporins in beef muscle using LC–MS/MS. J. Chromatogr. B 2012, 899, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Bessaire, T.; Mujahid, C.; Beck, A.; Tarres, A.; Savoy, M.C.; Woo, P.M.; Mottier, P.; Desmarchelier, A. Screening of 23 β-lactams in foodstuffs by LC-MS/MS using an alkaline QuEChERS-like extraction. Food Addit. Contam. Part A Chem. Anal. Control. Expo. Risk Assess. 2018, 35, 661–673. [Google Scholar] [CrossRef] [PubMed]
- Barker, S.A.; Long, A.R.; Short, C.R. Isolation of drug residues from tissues by solid phase dispersion. J. Chromatogr. A 1989, 475, 353–361. [Google Scholar] [CrossRef]
- Poole, C.F. New trends in solid-phase extraction. TrAC Trends Anal. Chem. 2003, 22, 362–373. [Google Scholar] [CrossRef]
- Poole, C.F.; Poole, S.K. Principles and Practice of Solid-Phase Extraction. Compr. Sampl. Sample Prep. 2012, 2, 273–297. [Google Scholar] [CrossRef]
- Buszewski, B.; Szultka, M. Past, Present, and Future of Solid Phase Extraction: A Review. Crit. Rev. Anal. Chem. 2012, 42, 198–213. [Google Scholar] [CrossRef]
- Becker, M.; Zittlau, E.; Petz, M. Residue analysis of 15 penicillins and cephalosporins in bovine muscle, kidney and milk by liquid chromatography–tandem mass spectrometry. Anal. Chim. Acta 2004, 520, 19–32. [Google Scholar] [CrossRef]
- Heller, D.N.; Nochetto, C.B.; Rummel, N.G.; Thomas, M.H. Development of Multiclass Methods for Drug Residues in Eggs: Hydrophilic Solid-Phase Extraction Cleanup and Liquid Chromatography/Tandem Mass Spectrometry Analysis of Tetracycline, Fluoroquinolone, Sulfonamide, and β-Lactam Residues. J. Agric. Food Chem. 2006, 54, 5267–5278. [Google Scholar] [CrossRef]
- Chiesa, L.M.; Nobile, M.; Panseri, S.; Arioli, F. Antibiotic use in heavy pigs: Comparison between urine and muscle samples from food chain animals analysed by HPLC-MS/MS. Food Chem. 2017, 235, 111–118. [Google Scholar] [CrossRef]
- van Holthoon, F.; Mulder, P.P.; van Bennekom, E.O.; Heskamp, H.; Zuidema, T.; van Rhijn, H.J. Quantitative analysis of penicillins in porcine tissues, milk and animal feed using derivatisation with piperidine and stable isotope dilution liquid chromatography tandem mass spectrometry. Anal. Bioanal. Chem. 2010, 396, 3027–3040. [Google Scholar] [CrossRef]
- Gajda, A.; Nowacka-Kozak, E.; Gbylik-Sikorska, M.; Posyniak, A. Multi-residues UHPLC–MS/MS analysis of 53 antibacterial compounds in poultry feathers as an analytical tool in food safety assurance. J. Chromatogr. B 2019, 1104, 182–189. [Google Scholar] [CrossRef]
- Turnipseed, S.B.; Storey, J.M.; Lohne, J.J.; Andersen, W.C.; Burger, R.; Johnson, A.S.; Madson, M.R. Wide-Scope Screening Method for Multiclass Veterinary Drug Residues in Fish, Shrimp, and Eel Using Liquid Chromatography–Quadrupole High-Resolution Mass Spectrometry. J. Agric. Food Chem. 2017, 65, 7252–7267. [Google Scholar] [CrossRef]
- Hu, M.; Ben, Y.; Wong, M.H.; Zheng, C. Trace Analysis of Multiclass Antibiotics in Food Products by Liquid Chromatography-Tandem Mass Spectrometry: Method Development. J. Agric. Food Chem. 2021, 69, 1656–1666. [Google Scholar] [CrossRef] [PubMed]
- Salis, S.; Rubattu, N.; Rubattu, F.; Cossu, M.; Sanna, A.; Chessa, G. Analytical Approaches in Official Food Safety Control: An LC-Orbitrap-HRMS Screening Method for the Multiresidue Determination of Antibiotics in Cow, Sheep, and Goat Milk. Molecules 2022, 27, 6162. [Google Scholar] [CrossRef]
- Stolker, A.A.; Rutgers, P.; Oosterink, E.; Lasaroms, J.J.; Peters, R.J.; van Rhijn, J.A.; Nielen, M.W. Comprehensive screening and quantification of veterinary drugs in milk using UPLC-ToF-MS. Anal. Bioanal. Chem. 2008, 391, 2309–2322. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, B.J.A.; Gerritsen, H.W.; Wegh, R.S.; Lameris, S.; van Sebille, R.; Stolker, A.A.M.; Nielen, M.W.F. Comprehensive analysis of ß-lactam antibiotics including penicillins, cephalosporins, and carbapenems in poultry muscle using liquid chromatography coupled to tandem mass spectrometry. Anal. Bioanal. Chem. 2013, 405, 7859–7874. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, L.; Xu, Y.; Wang, H.; Zeng, Q.; Zhao, Q.; Ren, N.; Ding, L. Determination of β-lactam antibiotics in milk based on magnetic molecularly imprinted polymer extraction coupled with liquid chromatography-tandem mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2010, 878, 3421–3426. [Google Scholar] [CrossRef]
- Baeza, A.N.; Urraca, J.L.; Chamorro, R.; Orellana, G.; Castellari, M.; Moreno-Bondi, M.C. Multiresidue analysis of cephalosporin antibiotics in bovine milk based on molecularly imprinted polymer extraction followed by liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2016, 1474, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Zhao, J.; Wang, X.; Yang, C.; Li, S.; Lu, T.; Li, X.; Wang, X.; Zhu, G. A highly sensitive and selective method for the determination of ceftiofur sodium in milk and animal-origin food based on molecularly imprinted solid-phase extraction coupled with HPLC-UV. Food Chem. 2021, 347, 129013. [Google Scholar] [CrossRef]
- Farooq, S.; Xu, L.; Ullah, S.; Li, J.; Nie, J.; Ping, J.; Ying, Y. Advancements and greenification potential of magnetic molecularly imprinted polymers for chromatographic analysis of veterinary drug residues in milk. Compr. Rev. Food Sci. Food Saf. 2024, 23, e13399. [Google Scholar] [CrossRef]
- Moein, M.M.; Abdel-Rehim, A.; Abdel-Rehim, M. Microextraction by packed sorbent (MEPS). TrAC Trends Anal. Chem. 2015, 67, 34–44. [Google Scholar] [CrossRef]
- Abdel-Rehim, M. Microextraction by packed sorbent (MEPS): A tutorial. Anal. Chim. Acta 2011, 701, 119–128. [Google Scholar] [CrossRef]
- Chiaochan, C.; Koesukwiwat, U.; Yudthavorasit, S.; Leepipatpiboon, N. Efficient hydrophilic interaction liquid chromatography-tandem mass spectrometry for the multiclass analysis of veterinary drugs in chicken muscle. Anal. Chim. Acta 2010, 682, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Junza, A.; Amatya, R.; Barrón, D.; Barbosa, J. Comparative study of the LC–MS/MS and UPLC–MS/MS for the multi-residue analysis of quinolones, penicillins and cephalosporins in cow milk, and validation according to the regulation 2002/657/EC. J. Chromatogr. B 2011, 879, 2601–2610. [Google Scholar] [CrossRef]
- Jank, L.; Hoff, R.B.; Tarouco, P.C.; Barreto, F.; Pizzolato, T.M. β-lactam antibiotics residues analysis in bovine milk by LC-ESI-MS/MS: A simple and fast liquid–liquid extraction method. Food Addit. Contam. Part A 2012, 29, 497–507. [Google Scholar] [CrossRef] [PubMed]
- Macarov, C.A.; Tong, L.; Martínez-Huélamo, M.; Hermo, M.P.; Chirila, E.; Wang, Y.X.; Barrón, D.; Barbosa, J. Multi residue determination of the penicillins regulated by the European Union, in bovine, porcine and chicken muscle, by LC–MS/MS. Food Chem. 2012, 135, 2612–2621. [Google Scholar] [CrossRef]
- Kukusamude, C.; Santalad, A.; Boonchiangma, S.; Burakham, R.; Srijaranai, S.; Chailapakul, O. Mixed micelle-cloud point extraction for the analysis of penicillin residues in bovine milk by high performance liquid chromatography. Talanta 2010, 81, 486–492. [Google Scholar] [CrossRef] [PubMed]
- Kukusamude, C.; Burakham, R.; Chailapakul, O.; Srijaranai, S. High performance liquid chromatography for the simultaneous analysis of penicillin residues in beef and milk using ion-paired extraction and binary water–acetonitrile mixture. Talanta 2012, 92, 38–44. [Google Scholar] [CrossRef]
- Karageorgou, E.G.; Samanidou, V.F.; Papadoyannis, I.N. Ultrasound-assisted matrix solid phase dispersive extraction for the simultaneous analysis of β-lactams (four penicillins and eight cephalosporins) in milk by high performance liquid chromatography with photodiode array detection. J. Sep. Sci. 2012, 35, 2599–2607. [Google Scholar] [CrossRef]
- Maggi, L.; Hurtado de Mendoza, J.; Zalacain, A.; Bonetto, L.; Mocholí, F.A.; Carmona, M. On-line Solid-Phase Extraction Coupled to Liquid Chromatography–Ion Trap Tandem Mass Spectrometry for Determination of Penicillins in Catfish. Food Anal. Methods 2012, 5, 1047–1053. [Google Scholar] [CrossRef]
- Cámara, M.; Gallego-Picó, A.; Garcinuño, R.M.; Fernández-Hernando, P.; Durand-Alegría, J.S.; Sánchez, P.J. An HPLC-DAD method for the simultaneous determination of nine β-lactam antibiotics in ewe milk. Food Chem. 2013, 141, 829–834. [Google Scholar] [CrossRef]
- Dorival-García, N.; Junza, A.; Zafra-Gómez, A.; Barrón, D.; Navalón, A. Simultaneous determination of quinolone and β-lactam residues in raw cow milk samples using ultrasound-assisted extraction and dispersive-SPE prior to UHPLC−MS/MS analysis. Food Control 2016, 60, 382–393. [Google Scholar] [CrossRef]
- Huang, Z.; Pan, X.-D.; Huang, B.-f.; Xu, J.-J.; Wang, M.-L.; Ren, Y.-P. Determination of 15 β-lactam antibiotics in pork muscle by matrix solid-phase dispersion extraction (MSPD) and ultra-high pressure liquid chromatography tandem mass spectrometry. Food Control 2016, 66, 145–150. [Google Scholar] [CrossRef]
- Moretti, S.; Dusi, G.; Giusepponi, D.; Pellicciotti, S.; Rossi, R.; Saluti, G.; Cruciani, G.; Galarini, R. Screening and confirmatory method for multiclass determination of 62 antibiotics in meat. J. Chromatogr. A 2016, 1429, 175–188. [Google Scholar] [CrossRef]
- Karageorgou, E.; Christoforidou, S.; Ioannidou, M.; Psomas, E.; Samouris, G. Detection of β-Lactams and Chloramphenicol Residues in Raw Milk—Development and Application of an HPLC-DAD Method in Comparison with Microbial Inhibition Assays. Foods 2018, 7, 82. [Google Scholar] [CrossRef]
- Lehotay, S.J.; Lightfield, A.R. Simultaneous analysis of aminoglycosides with many other classes of drug residues in bovine tissues by ultrahigh-performance liquid chromatography-tandem mass spectrometry using an ion-pairing reagent added to final extracts. Anal. Bioanal. Chem. 2018, 410, 1095–1109. [Google Scholar] [CrossRef] [PubMed]
- Giusepponi, D.; Paoletti, F.; Barola, C.; Moretti, S.; Saluti, G.; Ianni, F.; Sardella, R.; Galarini, R. Transfer of a Multiclass Method for over 60 Antibiotics in Food from High Resolution to Low Resolution Mass Spectrometry. Molecules 2019, 24, 2935. [Google Scholar] [CrossRef]
- Moretti, S.; Cruciani, G.; Romanelli, S.; Rossi, R.; Saluti, G.; Galarini, R. Multiclass method for the determination of 62 antibiotics in milk. J. Mass Spectrom. JMS 2016, 51, 792–804. [Google Scholar] [CrossRef]
- Di Rocco, M.; Moloney, M.; Haren, D.; Gutierrez, M.; Earley, S.; Berendsen, B.; Furey, A.; Danaher, M. Improving the chromatographic selectivity of β-lactam residue analysis in milk using phenyl-column chemistry prior to detection by tandem mass spectrometry. Anal. Bioanal. Chem. 2020, 412, 4461–4475. [Google Scholar] [CrossRef] [PubMed]
- Amelin, V.G.; Bol’shakov, D.S.; Podkolzin, I.V. Rapid Screening and Determination of Residual Amounts of β-Lactam Antibiotics in Foods by Ultrahigh-Performance Liquid Chromatography–High-Resolution Quadrupole Time-of-Flight Mass Spectrometry. J. Anal. Chem. 2020, 75, 1177–1188. [Google Scholar] [CrossRef]
- Paoletti, F.; Sdogati, S.; Barola, C.; Giusepponi, D.; Moretti, S.; Galarini, R. Development and validation of a multiclass confirmatory method for the determination of over 60 antibiotics in eggs using liquid-chromatography high-resolution mass spectrometry. Food Control 2021, 127, 108109. [Google Scholar] [CrossRef]
- Lakew, A.; Assefa, T.; Woldeyohannes, M.; Megersa, N.; Chandravanshi, B.S. Development and validation of liquid chromatography method for simultaneous determination of multiclass seven antibiotic residues in chicken tissues. BMC Chem. 2022, 16, 5. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Guo, E.; Wang, M.; Wang, K.; Ma, L.; Lian, K. Determination of β-lactam antibiotics in animal derived foods by modified QuEChERS coupled with ultra performance liquid chromatography-tandem mass spectrometry. J. Food Compos. Anal. 2023, 122, 105437. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibiotic | Molecular Formula | CAS Number | LD50 (mg/kg) (Oral, Rat) | PubChem CID |
|---|---|---|---|---|
| Penicillin G | C16H18N2O4S | 61-33-6 | 8000 | 5904 [15] |
| Penicillin V | C16H18N2O5S | 87-08-1 | >2220 | 6869 [16] |
| Amoxicillin | C16H19N3O5S | 26787-78-0 | >15,000 | 33613 [17] |
| Ampicillin | C16H19N3O4S | 69-53-4 | >5000 | 6249 |
| Oxacillin | C19H19N3O5S | 66-79-5 | 10,000 | 6196 [19] |
| Cloxacillin | C19H18ClN3O5S | 61-72-3 | n.a. | 6098 [20] |
| Nafcillin | C21H22N2O5S | 147-52-4 | n.a. | 8982 [21] |
| Dicloxacillin | C19H17Cl2N3O5S | 3116-76-5 | n.a. | 18381 [22] |
| Piperacillin | C23H27N5O7S | 61477-96-1 | n.a. | 43672 [23] |
| Carbenicillin | C17H18N2O6S | 4697-36-3 | n.a. | 20824 [24] |
| Antibiotic | Molecular Formula | CAS Number | LD50 (mg/kg) (Rat) | PubChem CID |
|---|---|---|---|---|
| Cefapirin | C17H17N3O6S2 | 21593-23-7 | 16,356 (oral) | 30699 [27] |
| Ceftiofur | C19H17N5O7S3 | 80370-57-6 | 1250 (intramuscular) | 6328657 [28] |
| Cefquinome | C23H24N6O5S2 | 84957-30-2 | n.a. | 5464355 [29] |
| Cephalonium | C20H18N4O5S2 | 5575-21-3 | 2750 (intraperitoneal) | 21743 [30] |
| Cefazolin | C14H14N8O4S3 | 25953-19-9 | n.a. | 33255 [31] |
| Cephalexin | C16H17N3O4S | 15686-71-2 | >20,000 (oral) | 27447 [32] |
| Cefoperazone | C25H27N9O8S2 | 62893-19-0 | n.a. | 44187 [33] |
| Amount of Compounds | Matrix | Sample Preparation | Analytical Technique | Reference | |
|---|---|---|---|---|---|
| Equipment | Separation | ||||
| 8 β-lactam antibiotics | porcine muscle and kidney tissue milk | Samples (2 g) were homogenized with phosphate buffer (2 mL, 200 mM, pH = 6.0). Internal standard mixture was added, incubated (30 min, room temperature), phosphate buffer (100 mM, pH = 8.0, 40 mL) was added, vortexed (30 s), piperidine (300 µL) was added, mixed (5 min), phosphoric acid (100 µL) was added, vortexed, heated in a water bath (85 °C, 5 min), and cooled. The samples were centrifuged (2700× g, 10 min). Supernatants were filtered through a cotton wool into tubes, purified (Oasis® HLB 60 mg, 3 cm3), and conditioned (2 mL methanol, 2 mL water and 2 mL phosphate buffer 100 mM, pH = 8.0). The samples were loaded (10 mL) and subsequently washed (2 × 2 mL of water), dried, and eluted (3 mL methanol/water; 80/20, v/v). Then, evaporated (dryness, 60 °C in a stream of nitrogen) and redissolved (500 μL 2% acetonitrile). The porcine tissue method was applied with a few modifications. Internal standards were added to milk samples (4 g). Phosphate buffer (100 mM, pH = 8.0, 40 mL) was added, vortexed (30 s), piperidine (300 µL) was added, mixed (5 min), and phosphoric acid (100 µL) was added and vortexed. The samples were centrifuged (2700× g, 10 min). Supernatants were filtered through a cotton wool into tubes. After conditioning the SPE column, extract was applied (20 mL). Final residue was redissolved in 2% methanol (200 µL). | LC-MS/MS (ESI−) | Column Waters Symmetry C18 column (3.0 × 150 mm; 5 µm) Mobile phase Gradient: A: 0.2% formic acid in water B: 0.2% formic acid in acetonitrile/water (9/1, v/v) | [62] |
| 3 β-lactam antibiotics | bovine milk | MMIPs (100 mg) were placed into a conical flask and conditioned (3.0 mL methanol, 3.0 mL water). Supernatant was isolated and separated and removed. Milk sample (2.0 mL) and hydrochloric acid aqueous solution (18.0 mL) at pH 5 were added. Ultrasound for 5 min. β-Lactam antibiotics were isolated. Supernatant solution was discarded. The MMIPs were washed with water (3.0 mL). The antibiotics were eluted from the MMIPs (3 × 1.0 mL methanol solution with 5.0% acetic acid). Ultrasound for 30 s during each elution step. The eluate merged and evaporated (nitrogen gas at 40 °C). The residue was reconstituted (1.0 mL of 0.1% formic acid methanol solution). The eluate was filtered (0.45 μm) and injected into the LC-MS/MS system for analysis. | LC-MS/MS (ESI+) | Column Waters Symmetry C18 column (150 mm × 4.6 mm, 5 µm) Mobile Phase Isocratic: 0.1% formic acid in water: methanol (40:60, v/v) | [69] |
| 3 β-lactam antibiotics | Chicken muscles | Sample (5 g) was weighed into a centrifuge tube, and extraction solution (10 mL; 2% trichloroacetic acid aqueous solution mixed with acetonitrile in a 1:1 volume ratio) was added and vortexed for 30 s. The sample was mechanically shaken (10 min) and centrifuged (3400 rpm, 5 min). Supernatant was transferred to a centrifuge tube. Hexane (5 mL) was added. The mixture was vortexed (1 min) and centrifuged (2400 rpm, 5 min). The hexane layer was discarded. The sample (200 μL) was transferred to a centrifuge tube and diluted with 10% formic acid (800 μL) in water–acetonitrile (1:9, v/v). The extract was filtered (0.2 μm nylon syringe filter) for LC separation. | LC-MS/MS ESI+ | Column SeQuant ZIC-HILIC column (2.1 mm × 100 mm; 3.5 µm) Mobile phase Gradient: A: 50 mM ammonium formate in water (pH 2.5) B: acetonitrile | [75] |
| 14 β-lactam antibiotics | cow milk | Sample (2 g) was weighed. Phosphate solution 0.2 M at pH 10 (0.5 mL) was added. Sample was centrifuged, and the SPE process was performed (HLB cartridges). Samples were activated with methanol (1 mL), water (1 mL), and 0.1 M phosphate solution at pH 10 (1 mL). Cartridge was washed with water (3 mL). The analytes were eluted with methanol (2 mL). Extract was injected into the LC-MS/MS system. | LC-MS/MS (ESI+) UPLC-MS/MS (ESI+) | Column (LC-MS/MS) Agilent Zorbax Eclipse XDB-C8 column (5 µm, 4.6 × 150 mm) Akady Kromasil C8 precolumn (4.6 × 15 mm; 5 µm) Column (UPLC-MS/MS) Waters Acquity UPLC BEH Shield RP 18 column (1.7 µm, 2.1 mm × 50 mm) Mobile phase Gradient: A: 0.1% formic acid in water B: 0.1% formic acid in acetonitrile | [76] |
| 6 β-lactam antibiotics | milk | Milk sample (2 mL) was extracted with 4 mL of acetonitrile. Acetonitrile (1 mL) was added to the sample and vortexed (10 s). This step was repeated three more times. Then, it was mixed (20 min), sodium chloride (1.0 g) was added, mixed (20 min), and centrifuged (5 min, 5000× g, 5 °C). 1 mL of the supernatant was collected for LC-MS/MS analysis. | LC-MS/MS (ESI+) | Column Phenomenex Synergy C18 column (3.0 × 150 mm; 4 µm) Phenomenex security guard system C18 (3.0 × 4.0 mm; 5 µm) Mobile phase Gradient: A: 0.1% formic acid in water B: 0.1% formic acid in acetonitrile | [77] |
| 7 β-lactam antibiotics | beef muscle | Muscle sample (4 g) was homogenized. Internal standard mixture was added, kept in the dark (30 min). A mixture of acetonitrile and water was added (15 mL; 80:20, v/v), vortexed (2 min), and centrifuged (5 min, 3500 rpm). The clean-up using QuEChERS method: Extract solutions (10 mL) were added to dispersive SPE kits (150 mg PSA sorbent, 150 mg of C18 sorbent, and 900 mg of MgSO4), shaken (5 min), and centrifuged (5 min, 3500 rpm). 5 mL was collected and evaporated, then redissolved in water (200 µL). The clean-up using the SPE method: Saturated NaCl solution (4 mL) was added to the acetonitrile extract then dried under a stream of nitrogen. Phosphate solution (pH 5) was added. Final volume 30 mL. ENV+ cartridges conditioned: Methanol (2 mL), water (2 mL), and 0.05 M sodium dihydrogen phosphate solution (2 mL, pH 5). Sample was loaded and washed with phosphate solution (3 mL, pH 5) and water (1 mL). Eluted with 4 mL of acetonitrile:methanol:water mixture (45:45:10, v:v:v). Evaporated and redissolved in water (200 μL). | LC-MS/MS (ESI+) | Column Agilent Technologies Zorbax Eclipse XDB-C8 column (4.6 × 150 mm; 5 µm) Akady precolumn Kromasil C8 (4.6 × 15 mm; 5 µm) Mobile phase Gradient: A: 0.1% formic acid in water B: 0.1% formic acid in acetonitrile | [53] |
| 9 β-lactam antibiotics | bovine, porcine, and chicken muscle | Samples (4 g) were homogenized, placed into a 50 mL centrifuge tube, mixed with penicillin solutions and an internal standard, and allowed to sit (15 min). Water was added (2 mL), vortexed (1 min), and acetonitrile was added (20 mL), vortexed (1 min), and centrifuged (5 min, 3500 rpm, 25°C). Saturated NaCl solution (2 mL) was added to the acetonitrile extract then dried under a stream of nitrogen. 50 mM phosphate buffer was added (25 mL, pH 5–8.5). Purification was performed (isolute ENV+, 200 mg, 3 mL), then it was conditioned (2 mL methanol, 2 mL water and 2 mL phosphate buffer pH = 5.0). The samples were loaded and subsequently washed (3 mL phosphate buffer pH = 5.0, 1 mL water), dried, and eluted (2 mL methanol and 2 mL acetonitrile). They were evaporated and redissolved (200 μL of water), then centrifuged (13,000 rpm, 5 min). | LC-MS/MS (ESI+) | Column Agilent Technologies Zorbax Eclipse XDB-C8 column (4.6 × 150 mm) Akady precolumn Kromasil C8 (4.5 × 20 mm) Mobile phase Gradient: A: 0.1% formic acid in water B: 0.1% formic acid in acetonitrile | [78] |
| 3 β-lactam antibiotics | beef, milk | In the first step, proteins and fats were removed. Beef samples (10 g) were homogenized and mixed with acetone–acetonitrile solution (20 mL; 4:1, v/v). Milk samples (3 mL) were mixed with an acetone–acetonitrile solution (6 mL; 5:1, v/v) [28]. The samples were centrifuged (4500 rpm, 20 min) and evaporated (37 °C in a nitrogen atmosphere). Then, they were mixed with phosphate buffer (1 mL, pH 8) and 100 mmoll−1 tetrabutylammonium bromide (600 µL). Saturated ammonium sulfate and acetonitrile (2 mL) were added, and the volume was topped up to 10 mL with water. They were mixed and left until the phases were completely separated. The upper layer was directly injected into the HPLC. | HPLC-PDA | Column Waters Xbridge™ C18 column (250 mm × 4.6 mm, 5 µm) Mobile phase Isocratic (75:25, v/v) A: 5 mmoll−1 phosphate buffer (pH 6.6) B: acetonitrile | [79,80] |
| 12 β-lactam antibiotics | milk | Conditioned Oasis HLB columns (2 mL methanol, 2 mL water). Sorbent was transferred to a glass beaker, and milk sample (500 µL), standard solution (500 µL), and half the quantity of a QuEChERS tube (125 mg) were added, mixed, and sonicated (10 min) in an ultrasonic bath. Samples were transferred to an empty cartridge reservoir, compressed, dried under vacuum, washed (5 mL of water (7% acetone)), and eluted (1 mL methanol, 2 mL of acetonitrile). They were evaporated to dryness under a nitrogen stream in a water bath (35 °C), then dissolved (500 µL water). A 20 µL aliquot was taken and injected into the HPLC. | HPLC-PDA | Column MZ-Analysentechnik Inertsil ODS-3 colum (5 µm, 250 × 4 mm) Mobile phase Gradient: A: 0.05 M ammonium acetate B: acetonitrile | [81] |
| 8 β-lactam antibiotics | catfish | Sample (4 g) was minced and weighed in an 80 mL centrifuge tube. 20 mL acetonitrile/water (9:1) was added. Sample was homogenized. Extraction was performed in an orbital shaker (5 min). Sample was centrifuged (4000× g, 15 min, at 4 °C). The supernatant was filtered (nylon 0.45 µm). Filtrate (0.25 mL) was combined with McIlvaine buffer (4.75 mL, pH 7.6) and filtered with a 0.45 µm pore diameter. The online SPE system was used. C18 cartridges were conditioned with methanol (2 mL; 5 mL min−1 flow rate) and water (2 mL; 2 mL min−1). The filtrate was diluted (1:100) and inserted into the C18 cartridge (1 mL). The analytes were eluted (2.5 mL of water). | On-line SPE LC-MS/MS (ESI−) | Column Phenomenex Synergy Max column (150 × 4.6 mm; 4 μm) Mobile phase Gradient A: 2 mM ammonium formate B: 0.1% formic acid in water and acetonitrile | [82] |
| 22 β-lactam antibiotics | poultry muscle | Samples (2.5 g) were homogenized and mixed with an internal standard (200 μL). Borate solution (10 mL) and piperidine (500 μL) were added, shaken (5 s), incubated in a water bath (1 h, 60 °C), and cooled (10 min, room temperature), and n-hexane (10 mL) was added. After shaking (5 min), the sample was centrifuged (3500× g, 15 min). The aqueous layer was transferred into a clean tube and neutralized (pH 7.2) using acetic acid (25%) and/or ammonia (2.5%). Phenomenex Strata-X (200 mg, 6 mL) was conditioned (5 mL methanol, 5 mL water). The samples were loaded and subsequently washed (5 mL methanol/water; 1:9, v/v), dried (5 min), eluted (5 mL methanol/acetonitrile; 50:50, v/v), evaporated (45 °C), and redissolved (500 μL 1% piperidine in water). | LC-MS/MS (ESI+) | Column Waters Acquity UPLC CSH C18 (2.1 × 100 mm, 1.7 μm) Mobile phase Gradient: A: 0.0032% ammonia in water B: 0.0032% ammonia in water/acetonitrile (1:9, v/v) | [68] |
| 9 β-lactam antibiotics | ewe milk | Samples (5 mL) were homogenized and transferred into a test tube (15 mL). Acetonitrile was added (5 mL), agitated (1 min), and allowed to settle (10 min, room temperature). Then, they were centrifuged (3500 rpm, 20 min, 4 °C). The supernatant was filtered through a syringe membrane filter (0.45 μm, Millex-HN). Spe-ed SPE C18 was conditioned (5 mL acetonitrile, 5 mL of water, each for two cycles under vacuum for 15 s). The analytes retained on the cartridge were eluted (2 × 3 mL phosphate buffer (pH 3.4) and acetonitrile mixture (30:70, v/v)), evaporated, and reconstituted (500 μL of deionized water). 100 µL was taken for analysis. | HPLC-DAD | Column Scharlau Supelcosil LC 18 DB 5 µm (15 cm × 4.6 mm) Mobile phase Gradient: A: 25 mM phosphate buffer solution (pH 3.4) B: acetonitrile | [83] |
| 14 β-lactam antibiotics | bovine milk | Sample was weighed (2 mL) and placed into a centrifuge tube (50 mL). Analytes were extracted with 4 mL of acetonitrile, added in three steps (2 mL, 2 mL, 1 mL), and manually mixed. Analytes were kept in a horizontal mixer (15 min). Tubes were centrifuged (5 min at 4000× g; 5 °C). Supernatant was transferred to a tube (50 mL) containing C18 bulk (at least 100 mg). Tubes were mixed with a vortex (15 s) and centrifuged (5 min at 4000× g). Supernatant was transferred to a clean tube (15 mL), kept in a freezer (−17 °C, 20 min). Tubes were centrifuged (10 min at 4000× g, 0 °C). Supernatant was transferred to a 50 mL tube and evaporated (water bath, ≤45 °C) with a N2 stream. Volume of the solvent was reduced to ~500 µL. The final amount was −1 mL. Extract was transferred into a HPLC vial. | LC-MS/MS (ESI+) | Column Angela Technologies DuraShell RP column (C18, 100 × 2.1 mm, 3 µm, 150 Å) Phenomenex SecurityGuard Cartridge C18 (4 × 3.0 mm) Mobile phase Gradient: A: 0.1% formic acid B: 0.1% formic acid in methanol | [40] |
| 14 β-lactam antibiotics | cow milk | Samples of milk underwent freeze-drying. Samples (1 g) were mixed with standard solution. McIlvaine buffer (3 mL, pH 6.0) was added and vortexed (1 min). An acetonitrile:methanol mixture (12 mL, 70:30; v/v) was added, vortexed (2 min), sonicated (20 min), and centrifuged (3 min at 4109× g). Supernatant was transferred to a tube containing 300 mg of dispersive sorbent (PSA) and 900 mg of anhydrous MgSO4, shaken (1 min), and centrifuged (1 min, 4109× g). It was evaporated (40 °C) and redissolved in 1 mL of a solution of ammonium formate (50 mM, pH 4.0) and methanol (80:20; v/v). It was then centrifuged (15 min; 16,300× g) and injected into the LC system. | UHPLC-MS/MS (ESI+) | Column Waters Acquity UPLC BEHTM C18 column (1.7 µm; 2.1 × 100 mm) Mobile phase Gradient: A: ammonium formate 50 mM, pH 4.0 B: methanol | [84] |
| 6 β-lactam antibiotics | milk | Samples of milk (30 g) were mixed with standard solution and centrifuged (30 min, 10,800× g, 35 °C). 1 g of the defatted milk (middle fraction) was taken and diluted to 10 mL (0.05 M phosphate buffer, pH 7.5). MIPs (20 mg) were loaded to solid-phase extraction cartridges. Cartridges were equilibrated (10 mL of methanol and 10 mL of 0.05 M phosphate buffer at pH 7.5). Samples (10 mL) were applied and passed through the cartridges (steady flow rate of 0.50 mL/min using a peristaltic pump). Cartridges were rinsed (5 mL of a mixture of methanol and 0.1 M HEPES buffer (pH 7.5, 2:98 v/v)). The analytes were eluted (1 mL of the mixture of methanol containing 0.1% trifluoroacetic acid). The eluate (200 µL) was taken and diluted (800 µL of water) and injected into the UHPLC-MS/MS system for analysis. | UHPLC-MS/MS (ESI+) | Column Waters Acquity UPLC BEHTM C18 column (1.7 µm; 1.0 × 150 mm) Mobile phase Isocratic: water:acetonitrile:formic acid (74.9:25.0:0.1, v/v/v) | [70] |
| 16 β-lactam antibiotics | pork muscle | Sample (2 g) was put into a glass grinder. The dispersion absorbent (Oasis HLB) was preconditioned (10 mL acetonitrile, distilled water, 5 mL 2% NaCl solution). The sample was blended with the dispersion absorbent (3 g) and put into a glass column. The column was compressed and washed (6 mL of n-hexane). Analytes were eluted (8 mL acetonitrile and water, both containing 0.1% formic acid; 50:50). Ethyl acetate (6 mL) was added and mixed (1 min), then centrifuged (3000× g; 10 min). Dispersion was repeated twice. The supernatant was dried (nitrogen dryer, 50 °C). Residue was dissolved (2 mL, acetonitrile/water; 10:90, v/v). | UPLC-MS/MS (ESI+) | Column ACQUITY UPLC HSS T3 C18 column (1.7 mm, 2.1 × 100 mm) Mobile phase Gradient: A: 0.1% formic acid in water B: 0.1% formic acid in acetonitrile | [85] |
| 17 β-lactam antibiotics | muscles | Sample (0.5 g) was weighed into a tube. 0.1 M of EDTA (100 μL) was added. The muscle was extracted with acetonitrile and water (3 mL, 80/20, v/v). The sample was shaken and centrifuged. A second extraction was performed with acetonitrile (3 mL). Collected extracts were evaporated and redissolved (1.5 mL ammonium acetate 0.2 M). Ultracentrifugation was performed. Sample was injected. | LC-HRMS/MS ESI+ | Column Agilent Technologies Poroshell 120 EC-C18 column (100 × 3.0 mm; 2.7 μm) Poroshell (2.1 × 5 mm) guard column Mobile phase Gradient: A: aquenous solution with 0.1% (v/v) formic acid B: methanol | [86] |
| 30 β-lactam antibiotics | bovine muscles | Samples were weighed (2 g ± 0.01 g) into a polypropylene centrifuge tube (50 mL). Internal standard solution (100 μL) and working standard solution were added. The samples were left for 15 min. Water (1.9 mL) and acetonitrile (8 mL) were added. The samples were homogenized (20 s) and centrifuged (15 min; 2842× g, 4°C). The supernatant was transferred to a polypropylene tube containing 500 mg of C18 sorbent, vortexed (40 s), centrifuged (15 min, 2842× g, 4°C), and evaporated under nitrogen (TurboVap, 40 °C). The final volume was filled up with water to 1 mL. The extracts were vortexed (10 s) and filtered (0.2 μm PTFE syringe filters). The extracts were transferred into autosampler vials. | UHPLC-MS/MS ESI+ | Column CHS C18 column (2.1 × 100 mm, 1.7 µm) Mobile phase Gradient: A: 0.01% formic acid and 0.2 mM ammonium acetate in water B: 0.01% formic acid in acetonitrile | [41] |
| 16 β-lactam antibiotics | chicken muscle | Sample (2 g) was weighed, added to a glass tuber, and mixed. Then, tissues were placed in QuEChERS extraction tubes, and acetonitrile containing 0.1% formic acid (15 mL) was added, shaken, and centrifuged (3000× g, 10 min). An acetonitrile layer was placed into a QuEChERS clean-up tube, shaken, and centrifuged (3000× g, 5 min). Obtained supernatant was put into a glass tube and dried using nitrogen, then it was reconstituted (2 mL of acetonitrile/water; 10:90, v/v). Obtained solution was filtered through a 0.22 μm nylon membrane and analyzed. | UPLC-Q-Orbitrap-MS (ESI+) | Column Waters Corporation HSS T3 C18 column (1.7 μm, 2.1 × 100 mm) Mobile phase Gradient: A: 0.1% formic acid in water B: 0.1% formic acid in acetonitrile | [52] |
| 8 β-lactam antibiotics | fish, shrimp, and eel | Tissue (2.0 g) was placed into a tube. Spiking standard mixes were added and left to stand (5 min). Tissue was extracted (8 mL of 0.2% p-toluenesulfonic acid monohydrate and 2% glacial acetic acid in acetonitrile), vortexed (30 min, 2500 rpm), and centrifuged (7 min, 4 °C (min. 17,000 RCF (g)). The extract (3 mL) was transferred to an Oasis PRiME HLB 6 cc (200 mg) extraction cartridge. The samples were drained through the cartridges. The remaining extract was evaporated to near dryness (N2, 55 °C). The extract was reconstituted (400 μL of 10% acetonitrile in water (v/v)), mixed, and centrifuged (min. 28,900 RCF (g), 7 min). 300 μL of the extract was erased and placed into a LC vial. | LC-Q-Orbitrap HRMS (HESI+) | Column Supelco Ascentis Express C18 (7.5 cm × 2.1 mm, 2.7 μm) column Mobile phase Gradient: A: 0.1% formic acid in water B: acetonitrile | [64] |
| 7 β-lactam antibiotics | heavy pigs’ urine and muscle | Urine: Samples (5 mL) were centrifuged (2500× g, 4°C, 5 min) and spiked. Compounds were extracted under vacuum (Oasis HLB cartridges; 3 mL, 60 mg). The cartridges were preconditioned (3 mL methanol, 3 mL 0.5 M HCI, 3 mL water). The samples were loaded. The cartridges were washed (3 mL water; 3 mL methanol:water, 20:80, v/v). The analytes were eluted (5 mL methanol). The eluate was evaporated. The extract was reconstituted (200 μL methanol:water, 10:90, v/v). Muscle: Samples (1 g) were spiked. The analytes were extracted (5 mL Mcllvaine buffer, pH 4.0). Trichloroacetic acid (100 μL, 20% w/v) was added. The samples were vortexed, sonicated (10 min), then centrifuged (2500× g, 4 °C, 10 min). The supernatant was transferred to a centrifuge tube and defatted (2 × 3 mL n-hexane). The n-hexane layer was removed after each centrifugation (2500× g, 5 min). The samples were purified and extracted by vacuum with Oasis HLB cartridges. The cartridges were preconditioned (3 mL methanol, 3 mL water). The samples were loaded. The cartridges were washed (2 × 3 mL methanol:water, 5:95, v/v). The analytes were eluted (5 mL methanol), transferred to a polypropylene tube, and evaporated. The extract was reconstituted (200 μL methanol:water, 10:90, v/v). | HPLC-MS/MS (ESI+/−) | Column Synergi Hydro-RP column (150 × 2.0 mm, 4 μm) Phonomenex C18 guard column (4 × 3.0 mm) Mobile phase Gradient: A: 0.1% aqueous formic acid B: methanol | [61] |
| 23 β-lactam antibiotics | foods of animal origins: eggs, raw milk, processed dairy ingredients, infant formula, and meat- and fish-based products, including baby food | Samples (1 g) were weighed into polypropylene tubes (50 mL). Phosphate buffer (15 mL, 500 mm, pH 9.2) was added. Samples were homogenized with a ceramic blender. Tubes were mechanically shaken (3 min). Acetonitrile (30 mL) was added. Tubes were shaken again (1.5 min). QuEChERS salt mixtures were added. Tubes were shaken (3 min). The samples were centrifuged (10 min, 4000× g, RT). The supernatants (8 mL) were transferred to 15 mL polypropylene tubes containing dispersive SPE sorbents. The tubes were shaken (1.5 min) and centrifuged (4000× g, 5 min, RT). The supernatants (5 mL) were evaporated under nitrogen (35 ± 2 °C). Obtained volume: 0.5 mL. Water (0.6 mL) was added. The mixture was evaporated. Final volume: 0.5 mL. Extracts were transferred to polypropylene tubes and centrifuged (17,000× g, 10 min, 4 °C), then filtered through a 0.45 µm PTFE filter. Further analysis was performed using the LC-MS/MS method. | LC-MS/MS (ESI+/−) | Column Waters Acquity BEH VanGuard precolumn (2.1 × 5 mm, 1.7 μm) Waters Acquity BEH C18 column (2.1 × 100 mm, 1.7 μm) Mobile phase Gradient: A: 0.5 mM ammonium formate and 0.1% formic acid in water B: 0.5 mM ammonium formate and 0.1% formic acid in methanol | [54] |
| 5 β-lactam antibiotics | fish | Homogenized samples (2 g) were weighed into a centrifuge tube. Internal standard (200 μL) and deionized water (800 μL) were added. The samples were vortexed (30 s) and left at room temperature (10 min). 5% trichloroacetic acid (8 mL) was added. The samples were homogenized (20 s) in ultra-turrax, placed in a shaker (10 min), and centrifuged (2700× g, 4 °C, 12 min). The extract was filtered (Millipore PVDF membrane, 4 μm pore size) before LC-MS/MS analysis. | LC-MS/MS (ESI+) | Column Agilent Technologies Zorbax Eclipse XDB C18 (150 × 4.6 mm, 1.8 μm) column Mobile phase Gradient: A: 0.1% heptafluorobutyric acid in water B: acetonitrile | [39,42] |
| 8 β-lactam antibiotics | raw milk | SPE sorbent material was preconditioned with water (2 mL) and methanol (2 mL), and transferred into a glass beaker. Milk (500 μg) and a standard solution of the antibiotics (500 μL) with 125 mg of a QuEChERS tube were added. Homogenization with sonication in an ultrasound bath was performed (10 min). Samples were transferred to an empty cartridge and compressed. The sorbent bed was cleaned (5 mL water, 1% acetone) twice. The analytes were eluted (2 mL methanol). Samples were filtered (PVDF Durapore syringe filters; 13 mm × 0.45 μm) and evaporated to dryness (nitrogen stream). The residues were dissolved (500 μL water). The obtained samples (100 μL) were transferred to the HPLC system. | HPLC-DAD | Column MZ-Analysentechnik Perfectsil ODS-2 (5 μm, 250 × 4 mm) Mobile phase Gradient: A: 0.05 M CH3COONH4 B: acetonitrile | [87] |
| 11 β-lactam antibiotics | bovine tissues: kidney, liver, and muscle | Samples (2 g) were weighed. Acetonitrile/water (4/1; v/v; 10 mL) was added. The solution was placed in a shaker (5 min). The samples were centrifuged (4150 rpm, 3 min, room temperature). 407 μL of the extract was placed into autosampler vial. Aqueous 146.5 mM 1-heptanesulfonate reagent solution (273 μL) was added. 7/3 (v/v) acetonitrile/water (50 μL) was added. | LC-MS/MS (ESI+/−) | Column Waters Acquity HSS T3 analytical column (10 × 2.1, 1.8 μm) Waters Vanguard column (0.5 cm) Mobile phase Gradient: A: 0.1% formic acid in water B: 0.1% formic acid in acetonitrile: methanol 1:1 (v:v) | [88] |
| 17 β-lactam antibiotics | muscle and milk | Samples (1.50 g) were weighed into a Falcon tube. 0.1 M of EDTA (100 μL) was added to muscle samples. The samples were extracted with acetonitrile/water (3 mL, 80/20, v/v). The milk samples were extracted with 0.1 M of EDTA (1 mL) and acetonitrile (3 mL). Both matrices were extracted for the second time with acetonitrile (3 mL). The samples were centrifuged. The extracts were evaporated and solubilized (1.5 mL, ammonium acetate 0.2 M) and injected into the LC system (10 μL). | LC-MC/MC (ESI+) | Column Agilent Technologies Poroshell 120 EC-C18 column (3.0 × 100 mm; 2.7 μL) Agilent Technologies Poroshell guard column (2.1 × 5 mm) Mobile phase Gradient: A: aqueous solution 0.1% (v/v) formic acid B: methanol | [86,89,90] |
| 15 β-lactam antibiotics | chicken feathers | Samples (0.2 g) were placed into a polypropylene tube. Internal standards were added. 0.02 M oxalic acid (1 mL, pH 4) was added. The samples were vortexed (3 min). 0.1 M Na2EDTA (0.5 mL) was added. The samples were mixed (3 min). Acetonitrile (8 mL) was added, shaken on a rotary tumbler (30 min), and centrifuged (10 min, 4200× g). The supernatants were put into Oasis HLB cartridges. Extracts were evaporated to dryness (N2, 40 °C). The residue was redissolved (0.025% HFBA; 0.5 mL). The samples were filtered (PVDF filter; 0.22 µm). UHPLC-MS/MS analysis was performed. | UHPLC-MS/MS (ESI+) | Column Agilent Technologies ZORBAX SB-C18 (50 mm × 2.1 mm × 1.8 μm) Phenomenex octadecyl guard column (2 × 4 mm) Mobile phase Gradient: A: acetonitrile B: 0.025% HFBA | [63] |
| 32 β-lactam antibiotics | milk | Samples (2 g) were weighed into polypropylene tubes. Standard solution was added, mixed, and left to stand for 15 min. Water (1 mL) and acetonitrile (7 mL) were added, then vortexed for 1 min. Samples were centrifuged (15 min, 2842× g, 4°C). Supernatant was transferred to a 50 mL d-SPE polypropylene tube containing 500 mg endcapped C18 sorbent, vortexed (40 s), and centrifuged (15 min, 2842× g, 4 °C). Supernatant was transferred (15 mL tube), evaporated in a TurboVap (under nitrogen, 40°C) to <1 mL, and adjusted to 1 mL with water. Extracts were vortexed (10 s) and centrifuged (15 min, 2842× g, 4 °C). The extract (400 µL) was filtered through syringeless Mini-UniPrep PTFE devices and injected into the UHPLC-MS/MS system for analysis. | UHPLC-MS/MS (ESI+) | Column Agilent Zorbax Eclipse Plus Phenyl-Hexyl Rapid Resolution HD analytical column (3.0 × 100 mm, 1.8 μm) fitted with an in-line filter (0.2 μm) Mobile phase Gradient: A: 0.01% formic acid with 0.2 mM ammonium acetate in water B: 0.01% formic acid in acetonitrile | [91] |
| 19 β-lactam antibiotics | meat, kidneys, liver, bacon, milk, and eggs honey | Option I: Sample (1 g), acetonitrile (2 g), NaCl (0.5 g), and EDTA (40 mg) were put into a centrifuge tube (15 mL). The samples were stirred (5 min) and centrifuged (5 min at 2700 rpm). The acetonitrile layer was collected and evaporated to dryness (40 °C) under nitrogen. Methanol (50 μL) and water (950 μL) were added to the residue, stirred (5 min), and filtered through a membrane filter for chromatography. Option II: Honey sample (1 g) was dissolved in water (1 mL). The sample, succinic acid (12 mg), EDTA (40 mg), and water (2 mL) were put into a centrifuge tube (15 mL) and stirred manually. Acetonitrile (2 mL) and ammonium sulfate (2 mg) were added, stirred (5 min), and centrifuged (5 min at 2700 rpm). The acetonitrile layer was collected and evaporated to dryness (40 °C) under nitrogen. Methanol (50 μL) and water (950 μL) were added to the residue, stirred (5 min), and filtered through a membrane filter for chromatography. | UHPLC-Q-TOF | Column Waters Acquity UPLC®BEN C18 (30 × 2.1 mm) Mobile phase Gradient: A: 0.1% formic acid in water B: 0.1% formic acid in acetonitrile | [92] |
| 1 β-lactam antibiotic | milk bovine, porcine, and chicken muscle | Samples of milk (10 g) were mixed with a standard solution, and acid-acetonitrile (4 mL, pH = 4.0) was added and centrifuged (10 min, 3000 rpm). Supernatant was evaporated and dissolved (10 mL of water). Muscle samples of chicken, pork, and beef (5 g) were mixed with a standard solution, and acetonitrile (3 mL) was added. Samples were mixed and centrifuged (10 min, 3000 rpm). Supernatants were evaporated and dissolved (10 mL of water). MIPs (120 mg) were packed into solid-phase extraction cartridges. MISPE cartridges were conditioned (10 mL methanol, 10 mL water). Prepared aqueous sample solutions (10 mL) were loaded onto the cartridges and eluted (1.5 mL, methanol:water 70:30, v/v). Eluents were analyzed using HPLC-UV. | HPLC-UV | Column C18 column (4.6 × 150 mm) Mobile phase Isocratic: acetonitrile:water contained 0.2% acetic acid (30/70, v/v) | [71] |
| 16 β-lactam antibiotics | eggs | Samples (1.5 g) were weighed into a polypropylene tube. The samples were spiked. 0.1 M Na2EDTA (0.5 mL) and acetonitrile:H2O 4:1 (v/v; 3 mL) with 0.05% formic acid were added, shaken, and centrifuged. The supernatant was transferred to a polypropylene tube. Extraction was repeated again with acetonitrile (3 mL), shaken, centrifuged, and sonicated (10 min). Supernatants were reunited and evaporated (40 °C; N2). Resuspension (1.5 mL ammonium acetate 0.2 M) and centrifugation (14,000 g, 30 min, 4 °C) were performed. 5 μL was injected into the LC-HRMS system. | LC-HRMS | Column Agilent Technologies Poroshell 120 EC-C18 32k (100 × 3 mm; 2.7 μm) Agilent Technologies Poroshell guard column (2.1 × 5 mm) Mobile phase [86] Gradient: A: aqueous solution with 0.1% (v/v) formic acid B: methanol | [93] |
| 4 β-lactam antibiotics | cereals, meat, eggs, milk, vegetables, and fruits | Samples were weighed in a polypropylene centrifuge tube (1.0 ± 0.1 g dry weigh) and spiked. The samples were ultrasonically extracted (10 mL acetonitrile:water (4:1, v/v) with 0.1% formic acid, 10 min). The sampes were extracted by a vertical oscillator (30 min) and centrifuged (9000 rpm, 10 min, 3 ± 1°C). Second extraction: DisQue salt pack and 10 mL of acetonitrile:water (4:1, v/v) with 0.2% formic acid. Third extraction: 10 mL of acetonitrile:water (4:1, v/v) with 0.2% formic acid. Oasis PRiME HLB cartridge was preconditioned (10 mL of methanol). Extracts were concentrated to 6 mL and transferred to the cartridge (5–10 mL/min). The eluate was collected, evaporated to dryness (nitrogen, Turbovap, 35 °C), and reconstituted with methanol (0.5 mL) in a glass vial (2 mL), then stored at −20 °C until analyzed. | UHPLC-MS/MS (ESI+) | Column Waters BEH C8 column (1.7 μm × 2.1 × 100 mm) Mobile phase Gradient: A: 0.1% formic acid in water B: methanol | [65] |
| 14 β-lactam antibiotics | milk: cow, sheep, and goat | Samples (1 g) were placed into a polypropylene tube (25 mL). EDTA 0.1 M (100 μL) and ACN with 2% formic acid (4 mL) were added. The samples were vortexed (30 s) and centrifuged (6000 rpm, 5 °C, 10 min). Supernatant was loaded on an Oasis HLB PRiME cartridge, which had been preconditioned (3 mL, acetonitrile). Purified extract (100 μL) was placed into a vial, diluted (900 μL solution of ammonium acetate 0.2 M:methanol 9:1 (v/v)), and analyzed. | LC-Orbitrap-HRMS | Column Agilent Technologies Poroshell 120 EC-C18 (100 × 3 mm; 2.7 μm) Agilent Technologies Poroshell guard column (2.1 × 5 mm) Mobile phase Gradient: A: 0.1% formic acid in water B: methanol | [66] |
| 3 β-lactam antibiotics | chicken tissue | Samples (2 g) were placed into a polypropylene centrifuge tube (50 mL), spiked, shaken manually (1 min), and left at a room temperature (20 min). Extraction solvent (10 mL, acetonitrile/methanol (10:20, v/v)) was added. The mixture was vortexed (1000 rpm, 1 min), sonicated (15 min), and centrifuged (3500 rpm, 5 min). Top supernatant was transferred to a flask (1 mL) through a syringe filter (0.2 μm, nylon) and evaporated to dryness (35°C, rotary evaporator). The residues were redissolved with methanol (1 mL). Residue (20 μL) was injected into the LC-UV system. | LC-UV | Column Phenomenex Hypersil BDS-C18 (3 μm, 100 mm × 4 mm) Mobile phase Isocratic 0.05 M Na2HPO4:acetonitrile:methanol (70:10:20), pH 8 | [94] |
| 52 β-lactam antibiotics | meat and poultry, aquatic products, milk, and eggs | Samples (1 g) were placed into a 50 mL polypropylene centrifuge tube, and acetonitrile:water mixture (12 mL, 75:25, v/v) was added and vortexed (3 min). Solution was centrifuged (5 min, 10,000 rpm, 4 °C), and supernatant (9 mL) was transferred into a 15 mL polypropylene tube containing 300 mg C18 sorbent. Samples were vortexed (30 s) and centrifuged (5 min, 10,000 rpm, 4 °C). Supernatant (6 mL) was transferred to a 15 mL polypropylene tube and evaporated (under a nitrogen stream, 40 °C). Residue was dissolved (1 mL, acetonitrile:0.1% formic acid (1:9, v/v)). Extract was vortexed (10 s) and filtered (0.2 µm PTFE syringe filter). Extract was injected into the UPLC-MS/MS system. | UPLC-MS/MS (ESI+) | Column Agilent ZORBAX SB-Aq column (2.1 × 150 mm, 3.5 µm) Mobile phase Gradient: A: 0.4% formic acid in water B: acetonitrile | [95] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pacyńska, J.; Niedzielski, P. Scoping Review of Extraction Methods for Detecting β-Lactam Antibiotics in Food Products of Animal Origin. Molecules 2025, 30, 1937. https://doi.org/10.3390/molecules30091937
Pacyńska J, Niedzielski P. Scoping Review of Extraction Methods for Detecting β-Lactam Antibiotics in Food Products of Animal Origin. Molecules. 2025; 30(9):1937. https://doi.org/10.3390/molecules30091937
Chicago/Turabian StylePacyńska, Joanna, and Przemysław Niedzielski. 2025. "Scoping Review of Extraction Methods for Detecting β-Lactam Antibiotics in Food Products of Animal Origin" Molecules 30, no. 9: 1937. https://doi.org/10.3390/molecules30091937
APA StylePacyńska, J., & Niedzielski, P. (2025). Scoping Review of Extraction Methods for Detecting β-Lactam Antibiotics in Food Products of Animal Origin. Molecules, 30(9), 1937. https://doi.org/10.3390/molecules30091937