
Impact of Mobile Phase Composition on Separation Selectivity of Labeled Dextran Ladder in Hydrophilic Interaction Liquid Chromatography



Abstract
1. Introduction
2. Results and Discussion
2.1. Impact of Organic Phase Selection on Dextran Ladder Separation
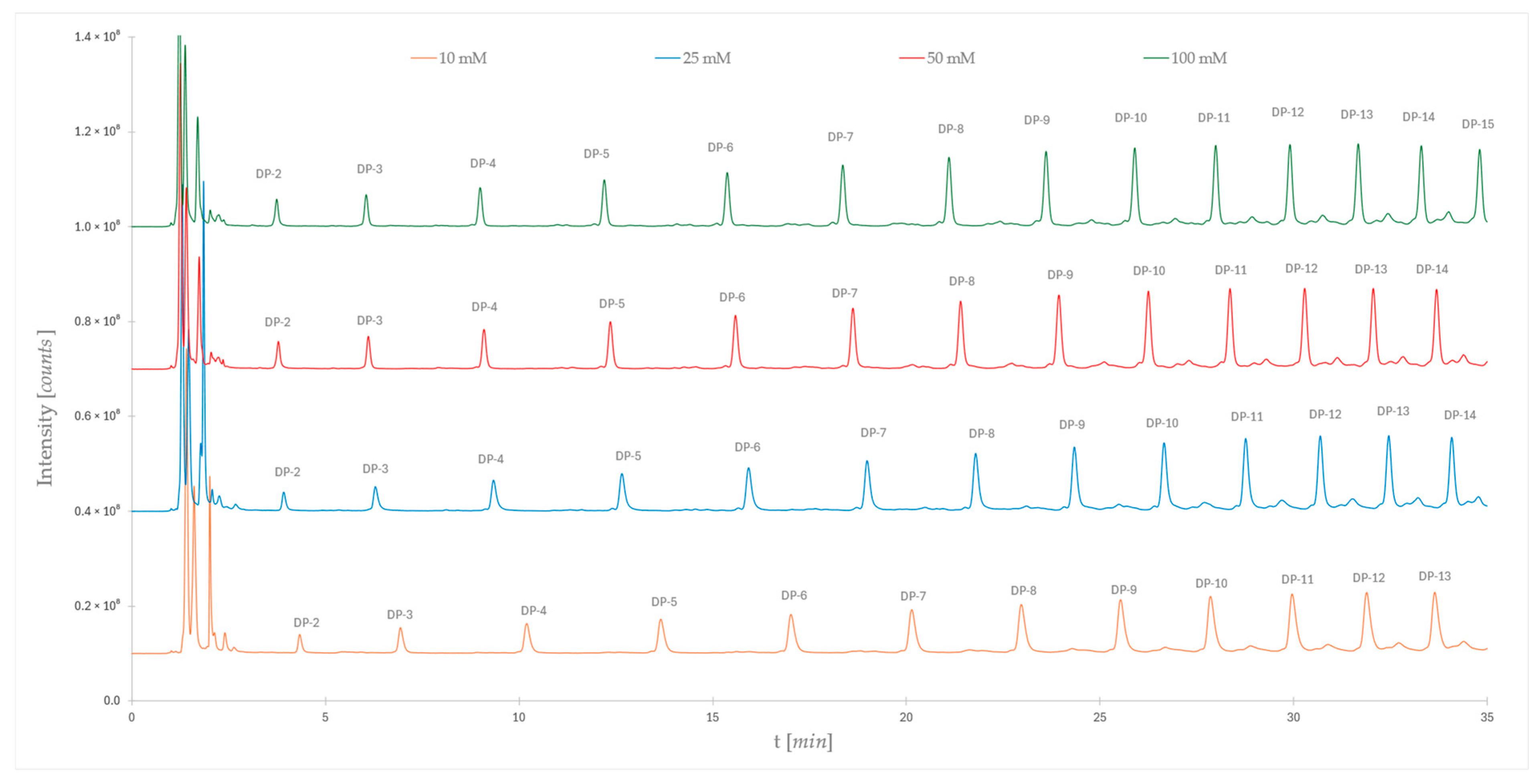
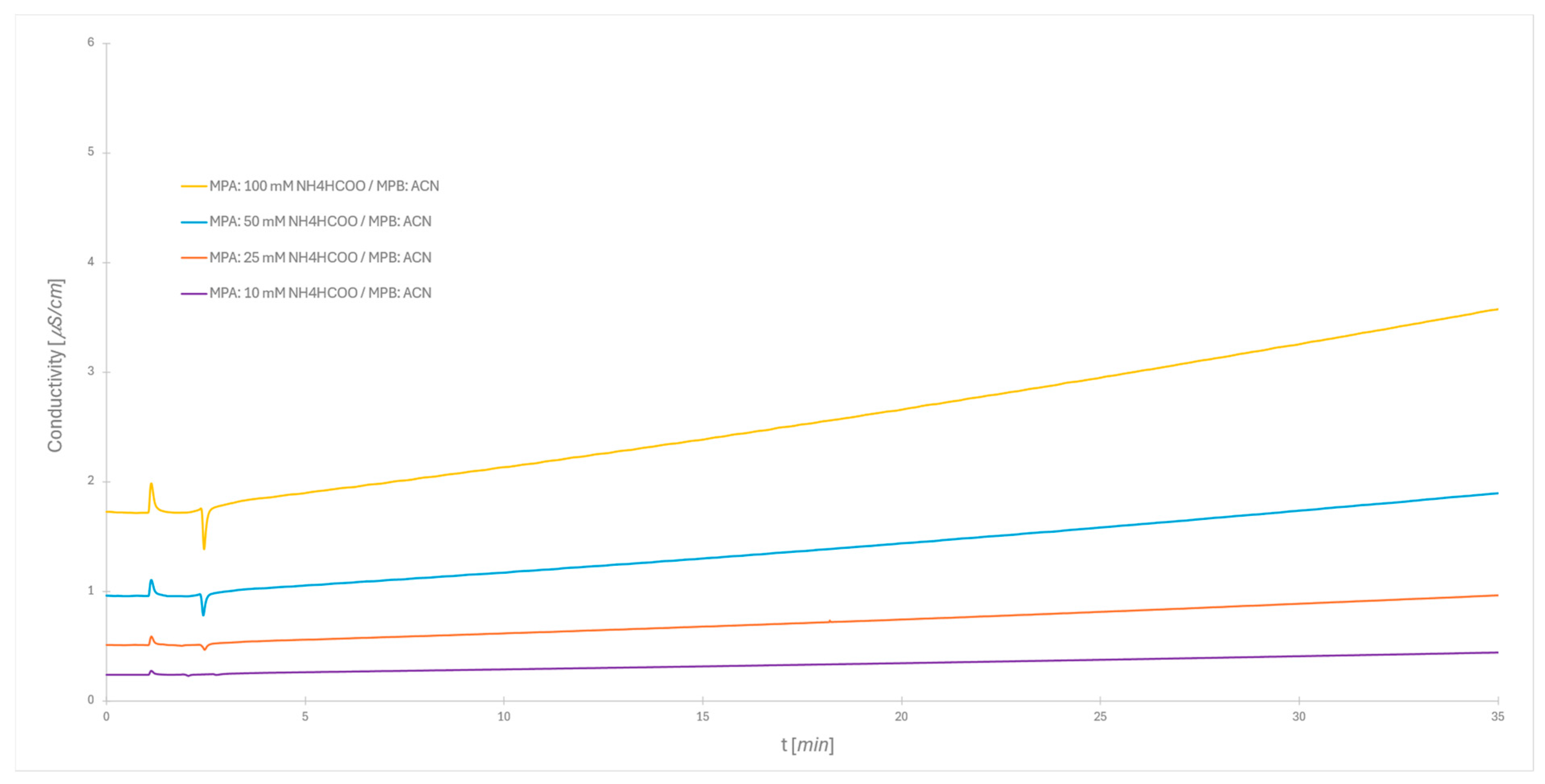
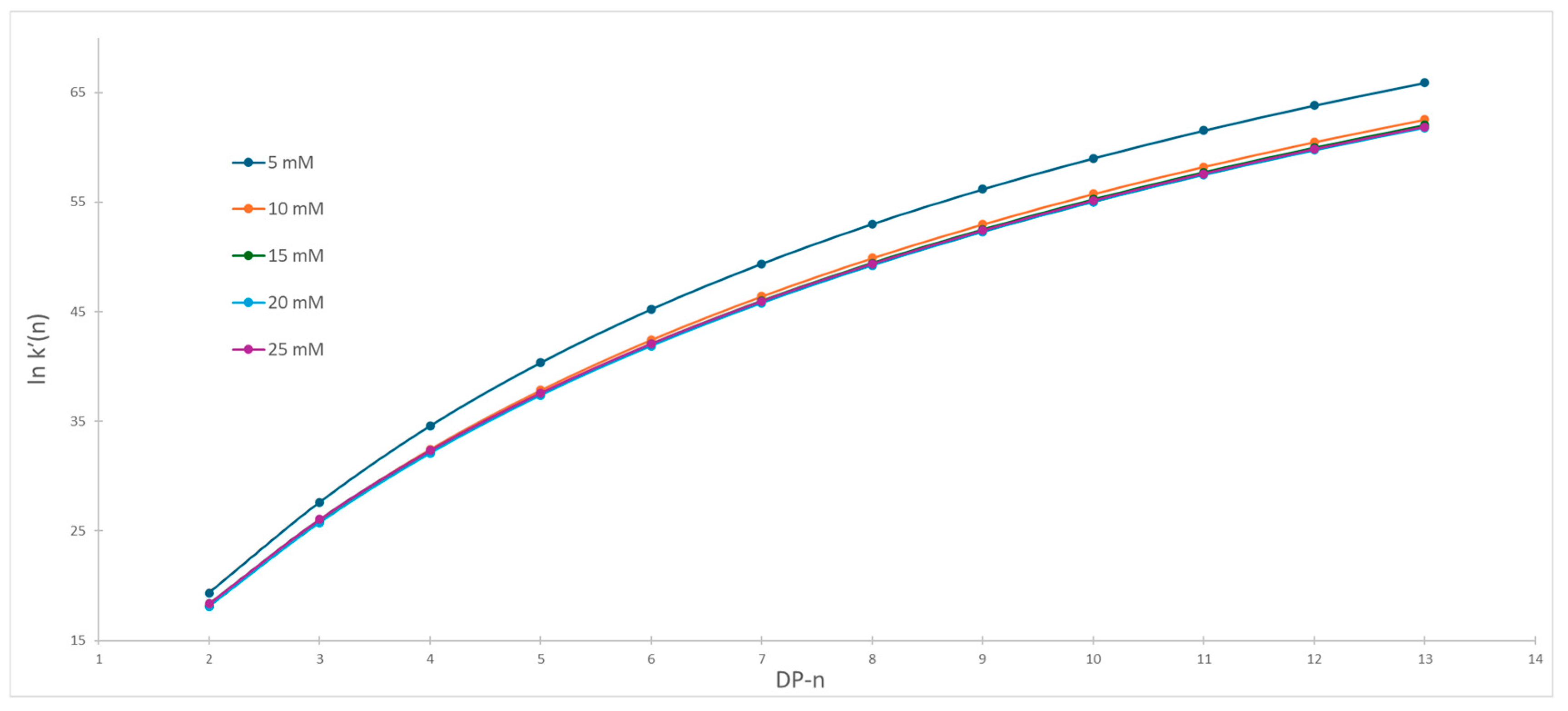
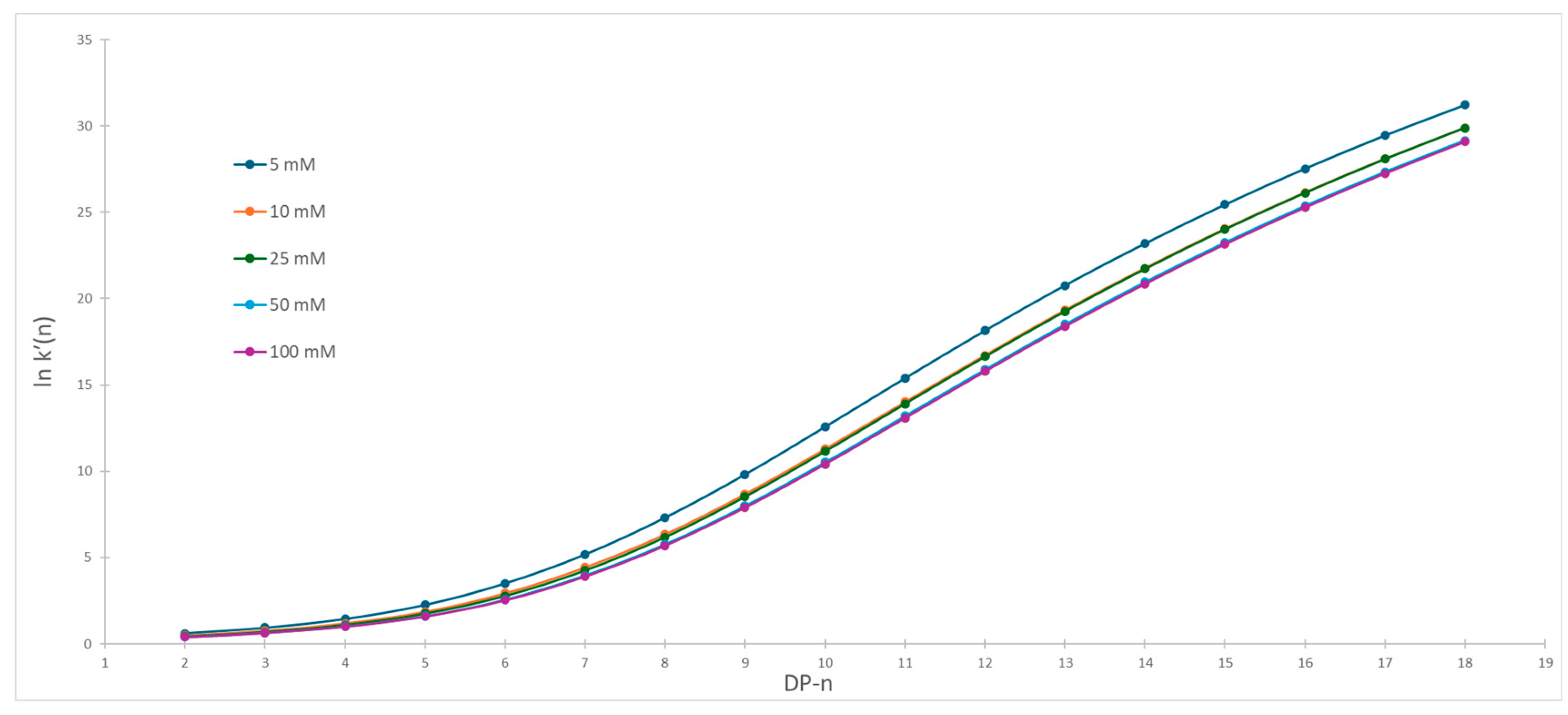
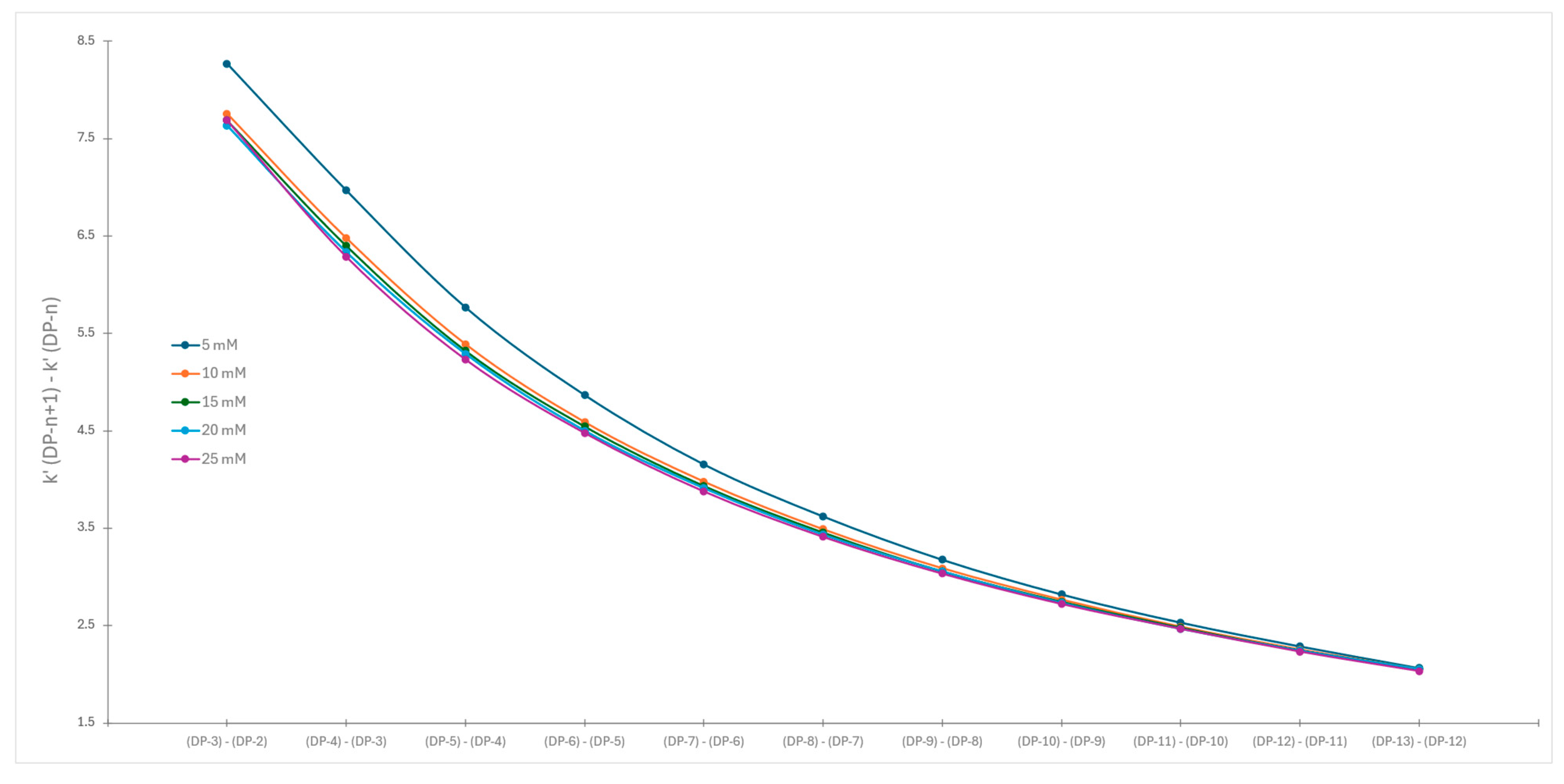
2.2. Impact of Ionic Strength on Dextran Ladder Separation
2.3. The Influence of Organic Modifier on Separation Selectivity
3. Materials and Methods
3.1. Chemicals
3.2. Standard
3.3. Instrument
3.4. Measurement
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gilar, M.; Berthelette, K.D.; Walter, T.H. Contribution of Ionic Interactions to Stationary Phase Selectivity in Hydrophilic Interaction Chromatography. J. Sep. Sci. 2022, 45, 3264–3275. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.-J.; Jeong, H.-C.; Kim, T.-E.; Shin, K.-H. Bioanalytical Method Using Ultra-High-Performance Liquid Chromatography Coupled with High-Resolution Mass Spectrometry (UHPL-CHRMS) for the Detection of Metformin in Human Plasma. Molecules 2020, 25, 4625. [Google Scholar] [CrossRef]
- Molnarova, K.; Kozlík, P. Comparison of Different HILIC Stationary Phases in the Separation of Hemopexin and Immunoglobulin G Glycopeptides and Their Isomers. Molecules 2020, 25, 4655. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Liang, T.; Li, Z.; Xu, X.; Ke, Y.; Jin, Y.; Liang, X. Separation of Carbohydrates Using Hydrophilic Interaction Liquid Chromatography. Carbohydr. Res. 2013, 379, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.K.; Owens, J.E.; Lowe, L.E.; Mooney, E.H. Analysis of Sugars and Amino Acids in Aphid Honeydew by Hydrophilic Interaction Liquid Chromatography—Mass Spectrometry. MethodsX 2020, 7, 101050. [Google Scholar] [CrossRef]
- Pismennõi, D.; Kiritsenko, V.; Marhivka, J.; Kütt, M.-L.; Vilu, R. Development and Optimisation of HILIC-LC-MS Method for Determination of Carbohydrates in Fermentation Samples. Molecules 2021, 26, 3669. [Google Scholar] [CrossRef]
- Melnikov, S.M.; Höltzel, A.; Seidel-Morgenstern, A.; Tallarek, U. How Ternary Mobile Phases Allow Tuning of Analyte Retention in Hydrophilic Interaction Liquid Chromatography. Anal. Chem. 2013, 85, 8850–8856. [Google Scholar] [CrossRef]
- Kozlik, P.; Goldman, R.; Sanda, M. Hydrophilic Interaction Liquid Chromatography in the Separation of Glycopeptides and Their Isomers. Anal. Bioanal. Chem. 2018, 410, 5001–5008. [Google Scholar] [CrossRef]
- Gritti, F.; Izzo, G.; Schaffer, R. Understanding Retention and Intra-Particle Diffusivity of Alkylsulfobetaine-Bonded Ethylene Bridged Particles with Different Mesopore Sizes for Hydrophilic Interaction Liquid Chromatography Applications. J. Chromatogr. A 2024, 1733, 465232. [Google Scholar] [CrossRef]
- Sheng, Q.; Liu, M.; Lan, M.; Qing, G. Hydrophilic Interaction Liquid Chromatography Promotes the Development of Bio-Separation and Bio-Analytical Chemistry. TrAC Trends Anal. Chem. 2023, 165, 117148. [Google Scholar] [CrossRef]
- Wang, J.; Guo, Z.; Shen, A.; Yu, L.; Xiao, Y.; Xue, X.; Zhang, X.; Liang, X. Hydrophilic-Subtraction Model for the Characterization and Comparison of Hydrophilic Interaction Liquid Chromatography Columns. J. Chromatogr. A 2015, 1398, 29–46. [Google Scholar] [CrossRef] [PubMed]
- Greco, G.; Grosse, S.; Letzel, T. Study of the Retention Behavior in Zwitterionic Hydrophilic Interaction Chromatography of Isomeric Hydroxy- and Aminobenzoic Acids. J. Chromatogr. A 2012, 1235, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Alpert, A.J. Electrostatic Repulsion Hydrophilic Interaction Chromatography for Isocratic Separation of Charged Solutes and Selective Isolation of Phosphopeptides. Anal. Chem. 2008, 80, 62–76. [Google Scholar] [CrossRef] [PubMed]
- Tyteca, E.; Périat, A.; Rudaz, S.; Desmet, G.; Guillarme, D. Retention Modeling and Method Development in Hydrophilic Interaction Chromatography. J. Chromatogr. A 2014, 1337, 116–127. [Google Scholar] [CrossRef]
- Gritti, F.; Guiochon, G. Mass Transfer Kinetics, Band Broadening and Column Efficiency. J. Chromatogr. A 2012, 1221, 2–40. [Google Scholar] [CrossRef]
- Gritti, F. Quantification of Individual Mass Transfer Phenomena in Liquid Chromatography for Further Improvement of Column Efficiency. LC GC N. Am. 2015, 32, 34–46. [Google Scholar]
- Guo, Y.; Bhalodia, N.; Fattal, B.; Serris, I. Evaluating the Adsorbed Water Layer on Polar Stationary Phases for Hydrophilic Interaction Chromatography (HILIC). Separations 2019, 6, 19. [Google Scholar] [CrossRef]
- Walter, T.H.; Alden, B.A.; Berthelette, K. Evaluation of the Base Stability of Hydrophilic Interaction Chromatography Columns Packed with Silica or Ethylene-Bridged Hybrid Particles. Separations 2022, 9, 146. [Google Scholar] [CrossRef]
- Rafique, S.; Yang, S.; Sajid, M.S.; Faheem, M. A Review of Intact Glycopeptide Enrichment and Glycan Separation through Hydrophilic Interaction Liquid Chromatography Stationary Phase Materials. J. Chromatogr. A 2024, 1735, 465318. [Google Scholar] [CrossRef]
- Chapel, S.; Rouvière, F.; Guillarme, D.; Heinisch, S. Reversed HILIC Gradient: A Powerful Strategy for On-Line Comprehensive 2D-LC. Molecules 2023, 28, 3907. [Google Scholar] [CrossRef]
- Bell, D.S. The Impact of Methanol on Hydrophilic Interaction Liquid Chromatography (HILIC) Retention Mechanisms—A Systematic Approach. In Proceedings of the HPLC 2022 Symposium, San Diego, CA, USA, 18–23 June 2022. [Google Scholar]
- Altannak, N.; Bawazeer, S.; Watson, D.G. Exploring the Effect of Buffer Strength on the Retention Time of Weak Acids, Neutral and Weak Bases in Hydrophilic Interaction Liquid Chromatography (HILIC) Mode. Curr. Anal. Chem. 2018, 14, 1–13. [Google Scholar]
- Trbojević-Akmačić, I.; Lageveen-Kammeijer, G.S.M.; Heijs, B.; Petrović, T.; Deriš, H.; Wuhrer, M.; Lauc, G. High-Throughput Glycomic Methods. Chem. Rev. 2022, 122, 15865–15913. [Google Scholar] [CrossRef] [PubMed]
- Waters Corporation. RapiFluor-MS Dextran Calibration Ladder; Waters Corporation: Milford, CT, USA, 2015. [Google Scholar]
- Waters Corporation. ACQUITY UPLC Glycan BEH Amide, 130 Å, 1.7 Μm Columns, ACQUITY Premier Glycan BEH Amide, 130 Å, 1.7 Μm Columns, and Glycan Performance Test Standards; Waters Corporation: Milford, CT, USA, 2021. [Google Scholar]
- Morrone, S.R.; Francesconi, A.Z. A Model for Excess Volumes of Salty Water–Acetonitrile Mixtures at 298.15 K. Fluid Phase Equilib. 2012, 313, 52–59. [Google Scholar] [CrossRef]
- Thawarkar, S.; Khupse, N.D.; Shinde, D.R.; Kumar, A. Understanding the Behavior of Mixtures of Protic-Aprotic and Protic-Protic Ionic Liquids: Conductivity, Viscosity, Diffusion Coefficient and Ionicity. J. Mol. Liq. 2019, 276, 986–994. [Google Scholar] [CrossRef]
- Thompson, J.W.; Kaiser, T.J.; Jorgenson, J.W. Viscosity Measurements of Methanol–Water and Acetonitrile–Water Mixtures at Pressures up to 3500 Bar Using a Novel Capillary Time-of-Flight Viscometer. J. Chromatogr. A 2006, 1134, 201–209. [Google Scholar] [CrossRef]
- Antoniou, E.; Tsianou, M.; Alexandridis, P. Solvent Modulation of Polysaccharide Conformation. In Proceedings of the AIChE Annual Meeting, Conference Proceedings, Philadelphia, PA, USA, 16–21 March 2008. [Google Scholar]
- Brini, E.; Fennell, C.J.; Fernandez-Serra, M.; Hribar-Lee, B.; Lukšič, M.; Dill, K.A. How Water’s Properties Are Encoded in Its Molecular Structure and Energies. Chem. Rev. 2017, 117, 12385–12414. [Google Scholar] [CrossRef]
- Steffè, A.; Milano, F.; Reyes, S.G.; Buco, F.; Leonetti, R.; Roque-Diaz, Y.; Zuffi, S.; Di Gianvincenzo, P.; Cortese, A.R.; Ritacco, H.; et al. Supramolecular Dextran/Polyamine Phosphate Nanocapsules with Smart Responsiveness for Encapsulation of Therapeutics. J. Colloid Interface Sci. 2025, 683, 620–630. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time [min] | Flow [mL/min] | Mobile Phase A [%] | Mobile Phase B [%] |
|---|---|---|---|
| 0 | 0.4 | 25 | 75 |
| 35 | 0.4 | 46 | 54 |
| 36.5 | 0.2 | 100 | 0 |
| 39.5 | 0.2 | 100 | 0 |
| 43.1 | 0.2 | 75 | 25 |
| 47.6 | 0.4 | 75 | 25 |
| 55 | 0.4 | 75 | 25 |
| Time [min] | Flow [mL/min] | Mobile Phase A [%] | Mobile Phase B [%] |
|---|---|---|---|
| 0 | 0.4 | 0 | 100 |
| 60 | 0.4 | 40 | 60 |
| 61.5 | 0.2 | 100 | 0 |
| 64.5 | 0.2 | 100 | 0 |
| 68.1 | 0.2 | 0 | 100 |
| 72.6 | 0.4 | 0 | 100 |
| 80 | 0.4 | 0 | 100 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grčman, M.; Pompe, N.R.; Kočar, D.; Pompe, M. Impact of Mobile Phase Composition on Separation Selectivity of Labeled Dextran Ladder in Hydrophilic Interaction Liquid Chromatography. Molecules 2025, 30, 1327. https://doi.org/10.3390/molecules30061327
Grčman M, Pompe NR, Kočar D, Pompe M. Impact of Mobile Phase Composition on Separation Selectivity of Labeled Dextran Ladder in Hydrophilic Interaction Liquid Chromatography. Molecules. 2025; 30(6):1327. https://doi.org/10.3390/molecules30061327
Chicago/Turabian StyleGrčman, Matjaž, Niko R. Pompe, Drago Kočar, and Matevž Pompe. 2025. "Impact of Mobile Phase Composition on Separation Selectivity of Labeled Dextran Ladder in Hydrophilic Interaction Liquid Chromatography" Molecules 30, no. 6: 1327. https://doi.org/10.3390/molecules30061327
APA StyleGrčman, M., Pompe, N. R., Kočar, D., & Pompe, M. (2025). Impact of Mobile Phase Composition on Separation Selectivity of Labeled Dextran Ladder in Hydrophilic Interaction Liquid Chromatography. Molecules, 30(6), 1327. https://doi.org/10.3390/molecules30061327