
Aldehyde-Assisted Alkoxysilane Condensation to Form Siloxane Bond: A New Process for Curing Alkoxy-Functional Silicone Resins




Abstract

1. Introduction
2. Results and Discussion
2.1. Study of the Curing Process of the Alkoxy-Functional Siloxane Resins
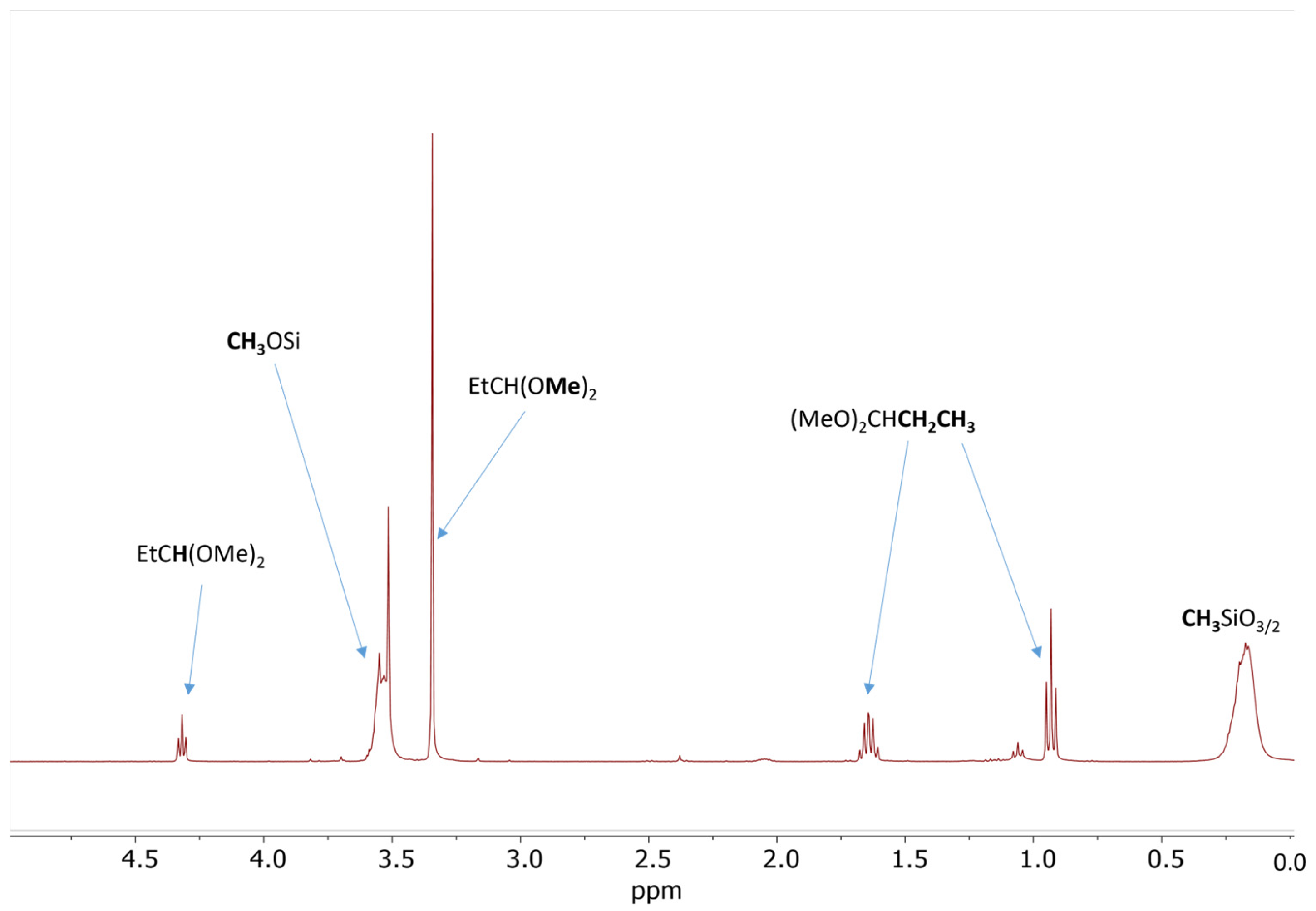
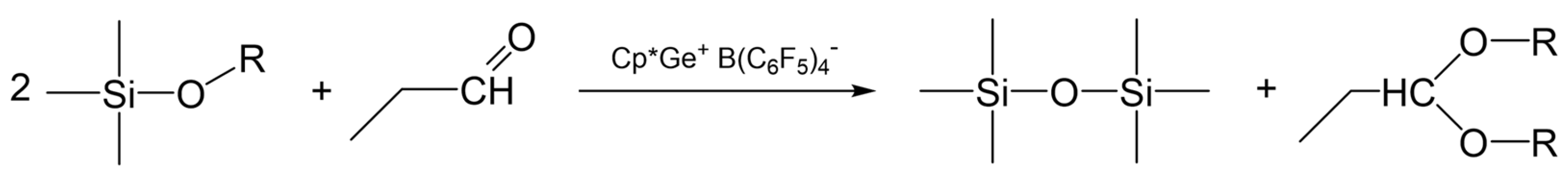
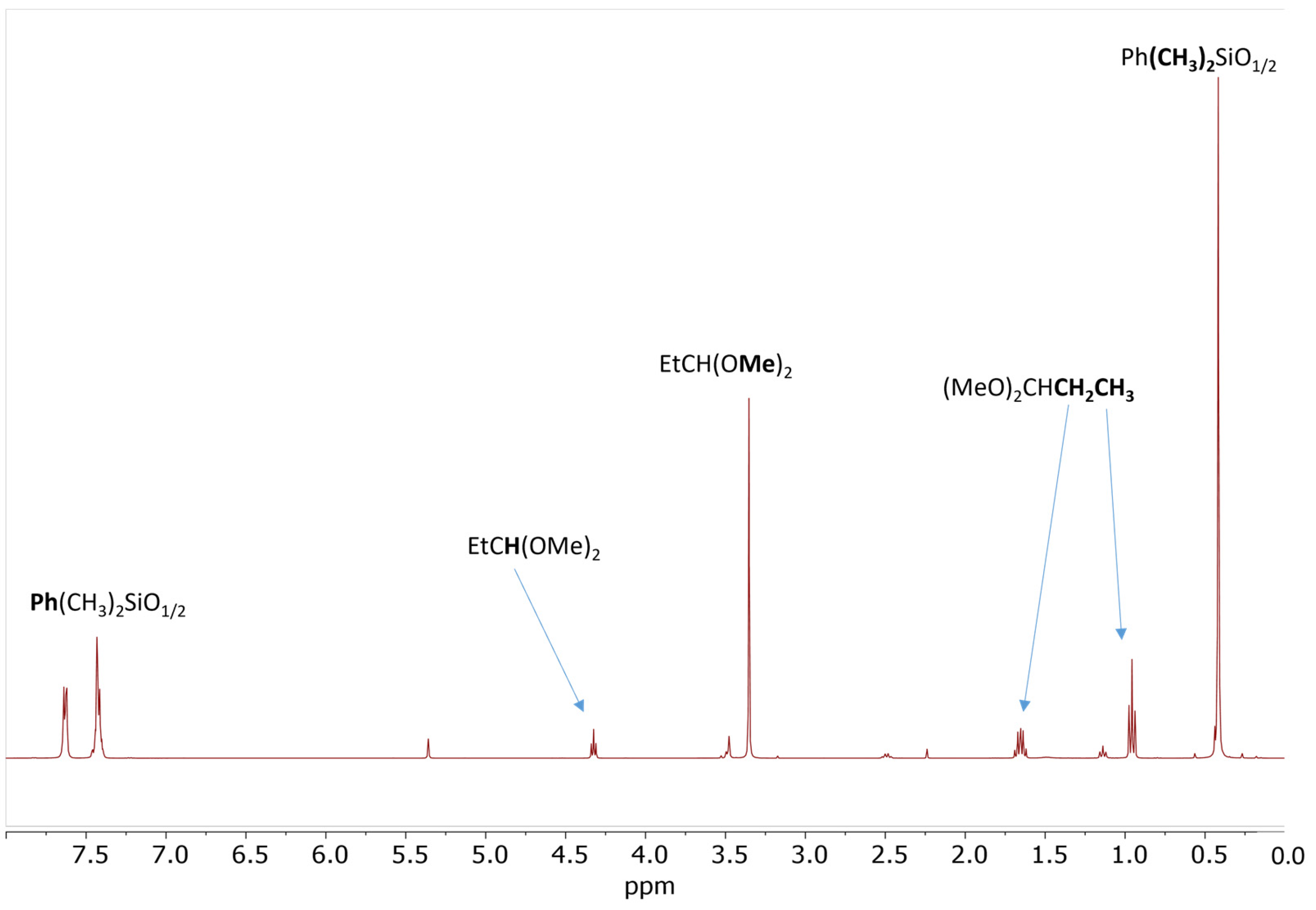
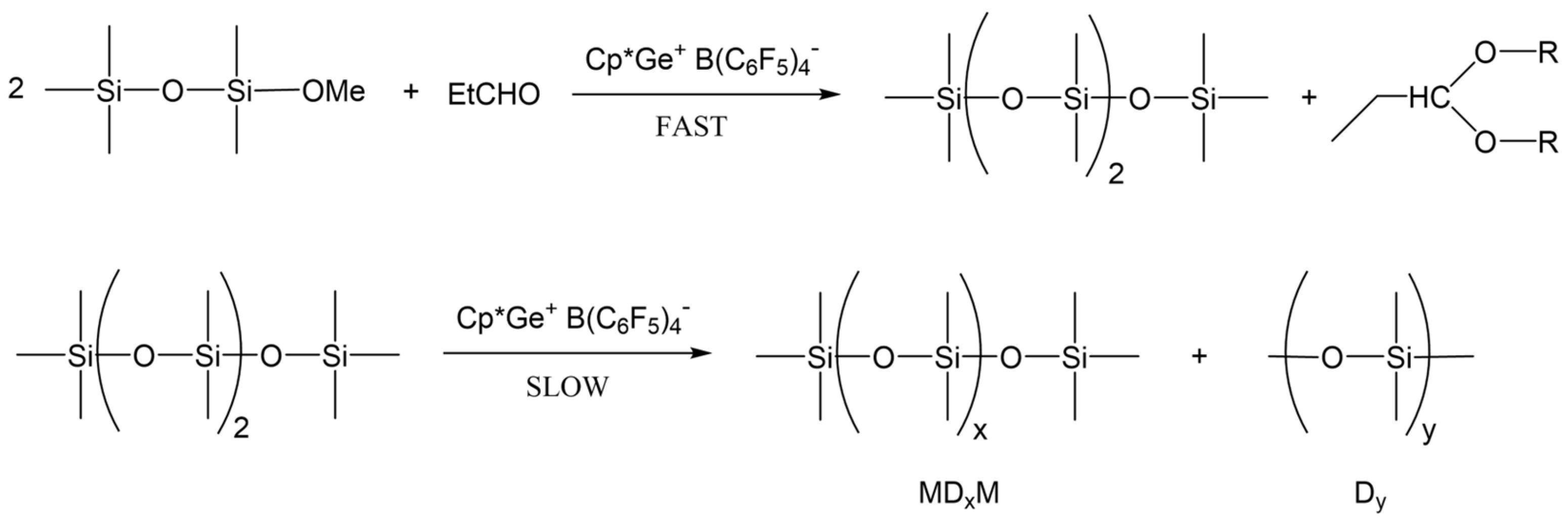
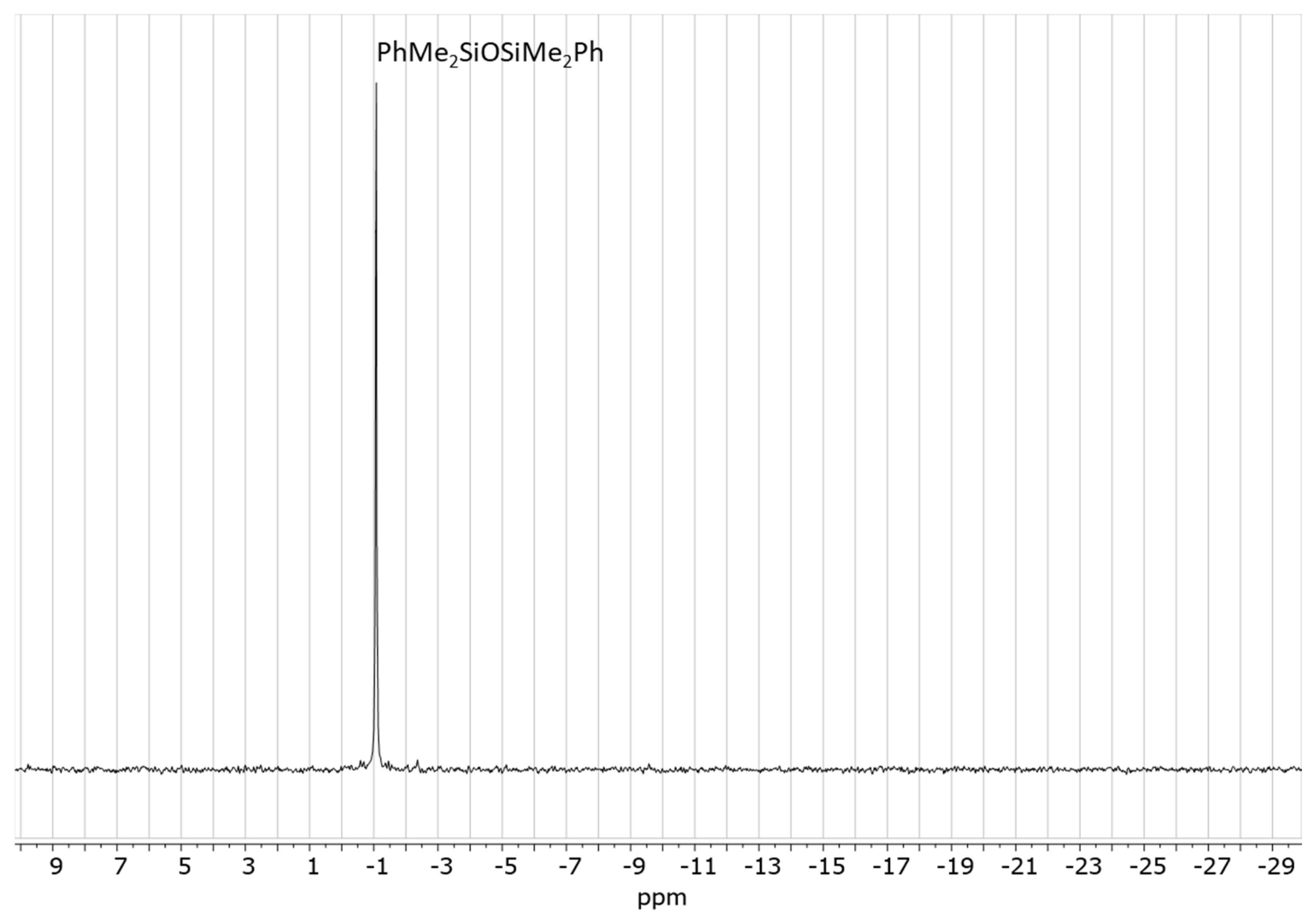
2.2. Model Study of the SiOR + SiOR Condensation Reaction Catalyzed by Cp*Ge+ B(C6F5)4−
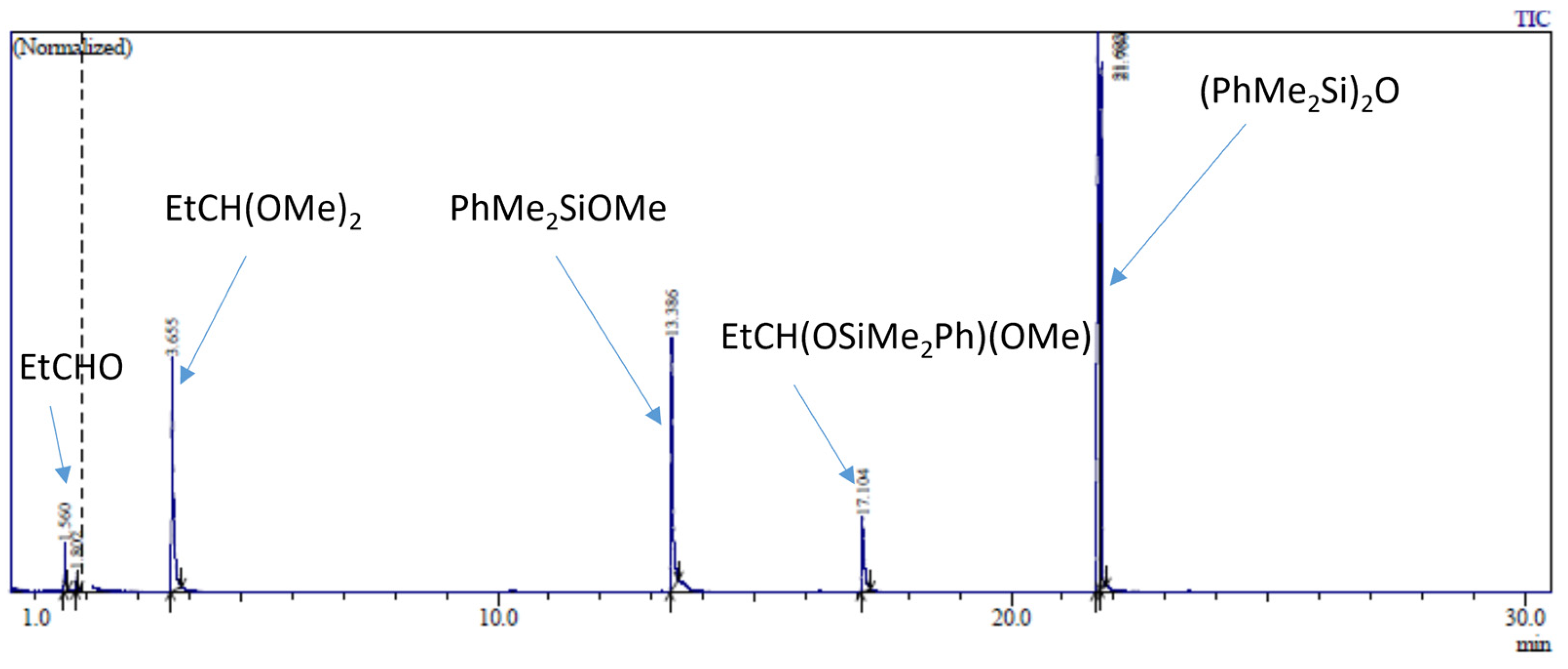
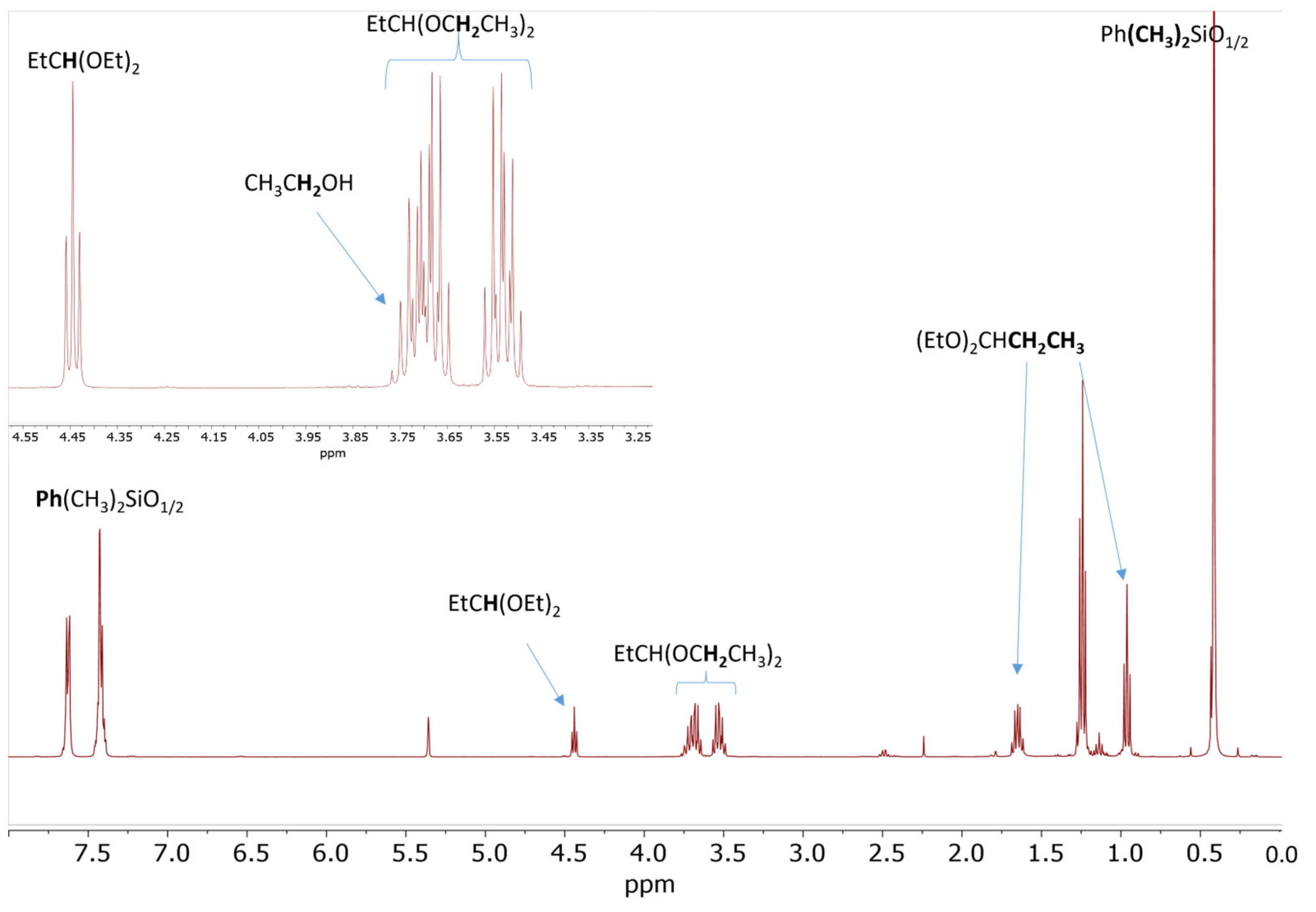
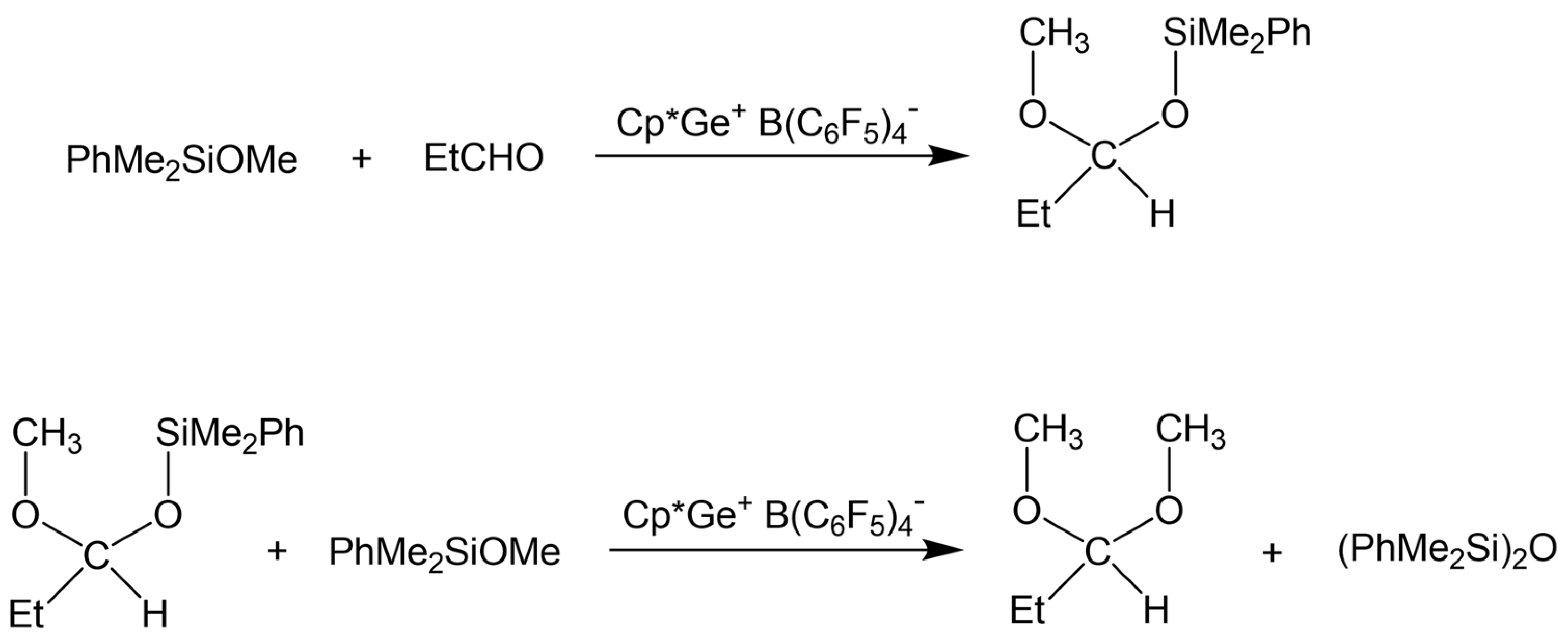
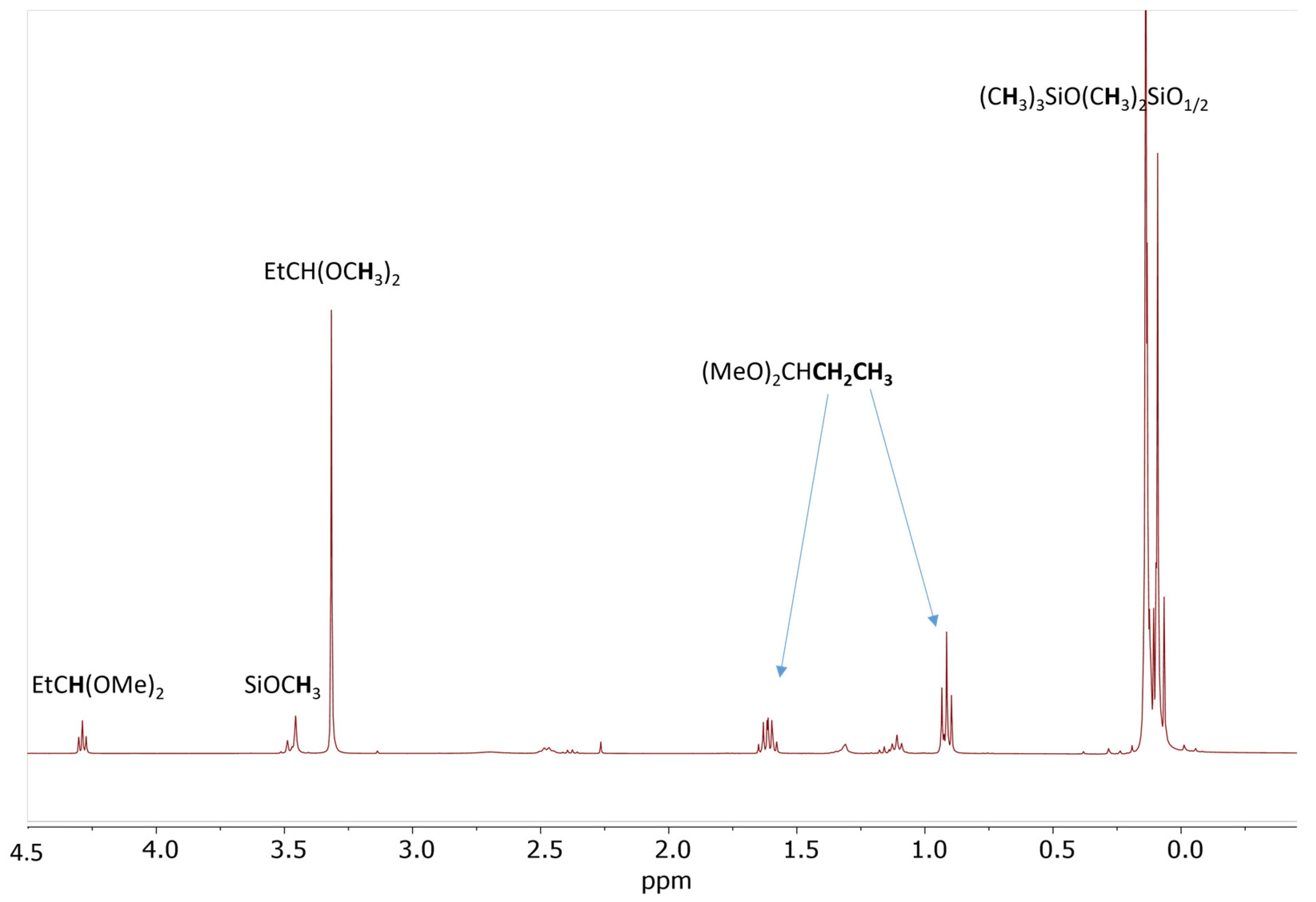
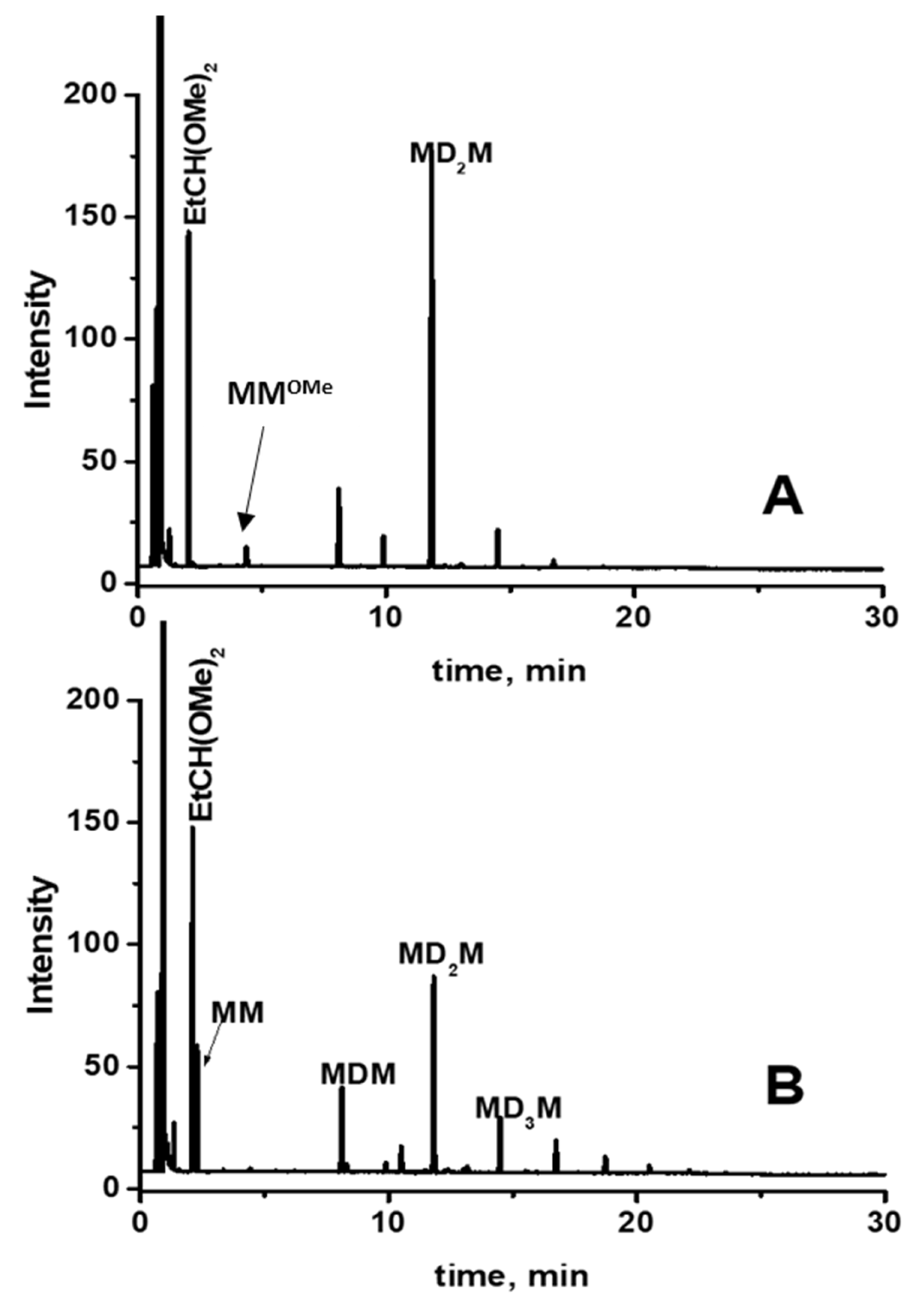
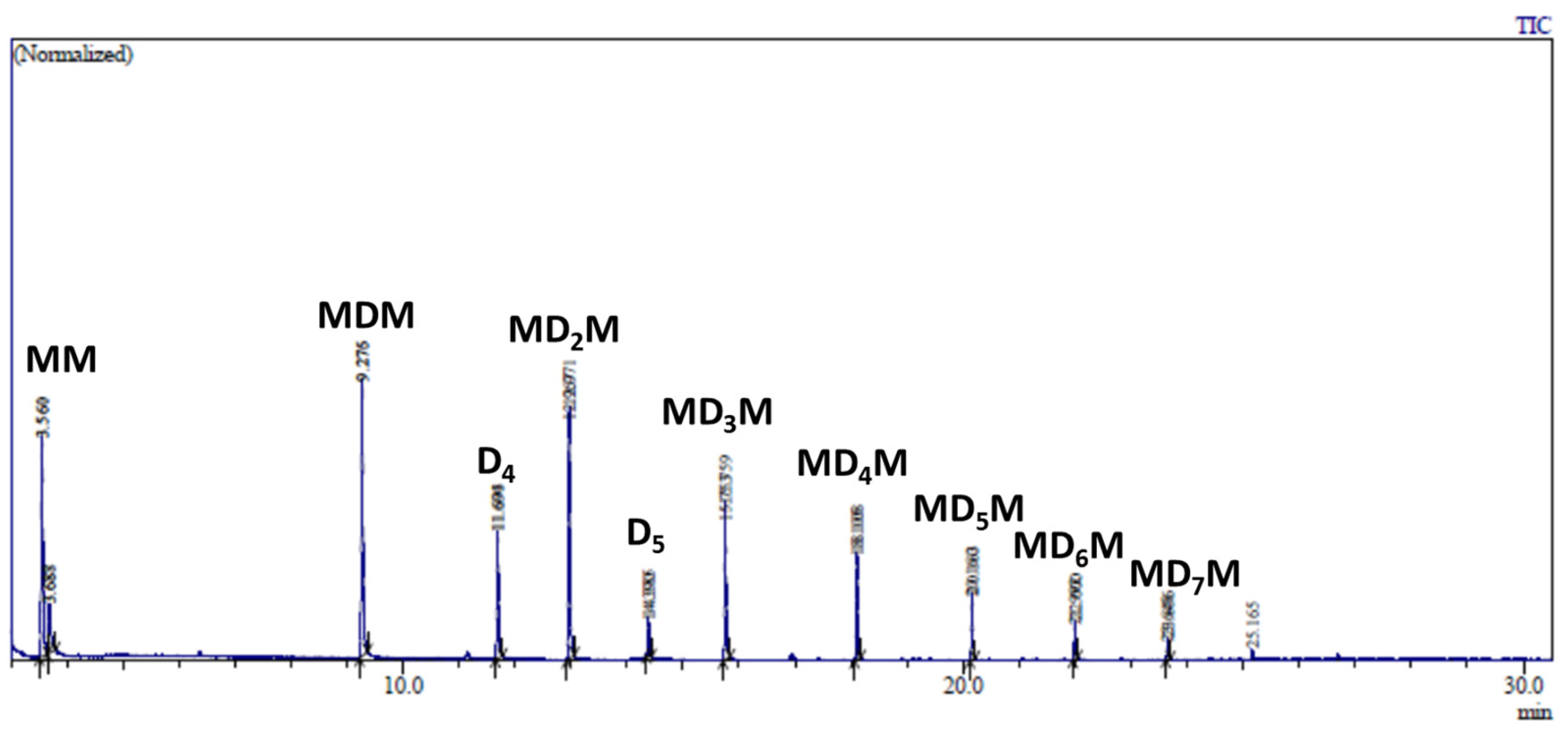
2.3. Study of the SiOR + SiOR Condensation Reaction in the Presence of Paraldehyde Catalyzed by Cp*Ge BArF4
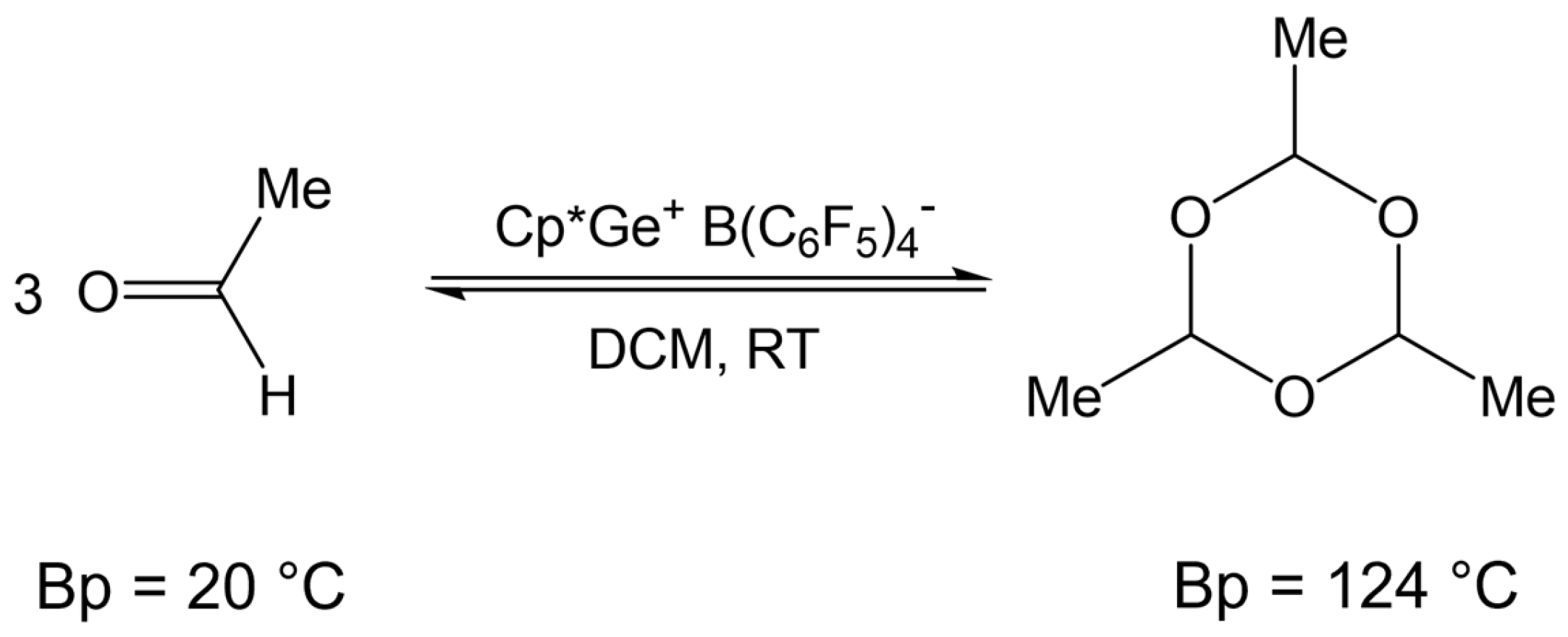
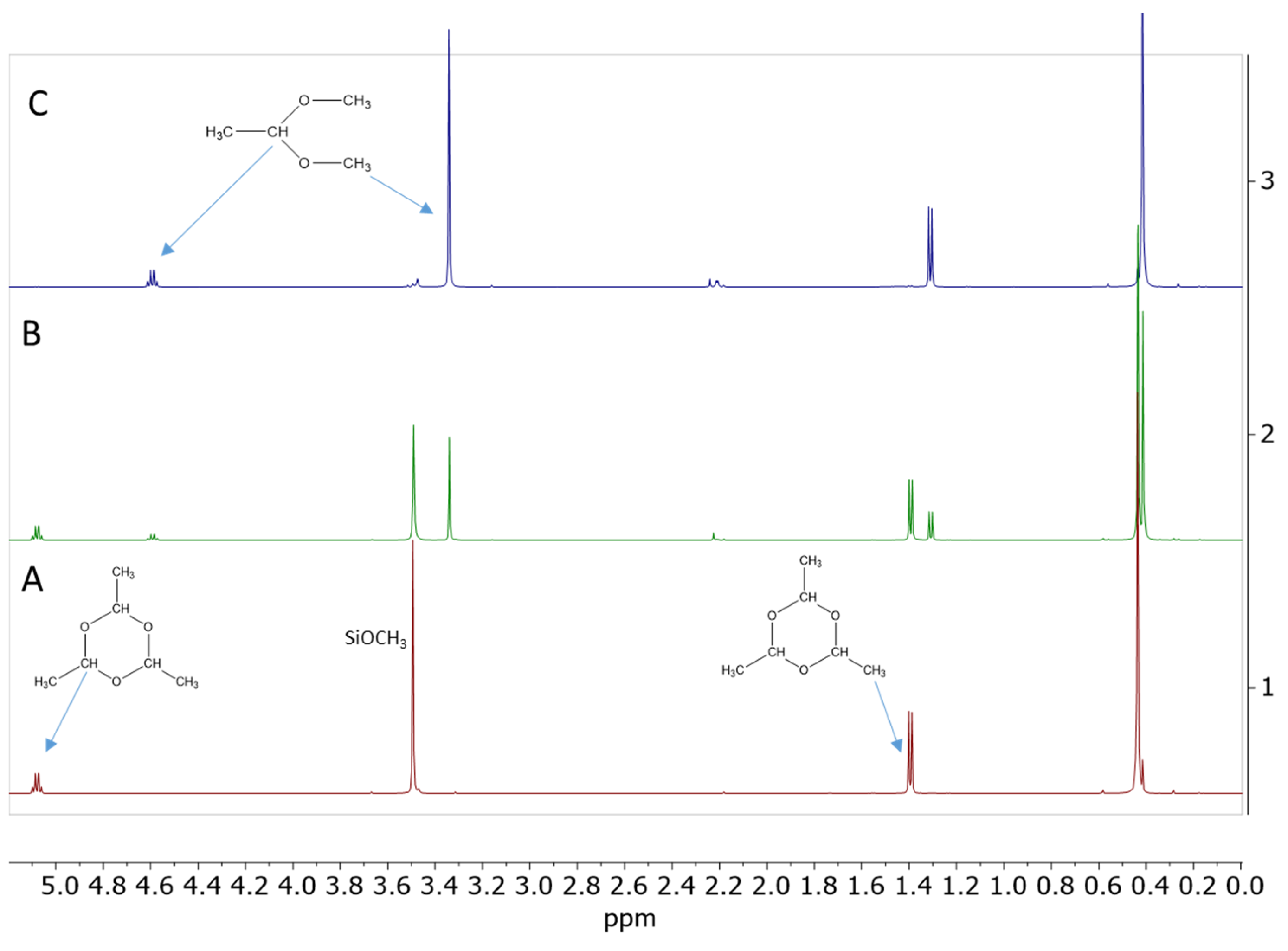
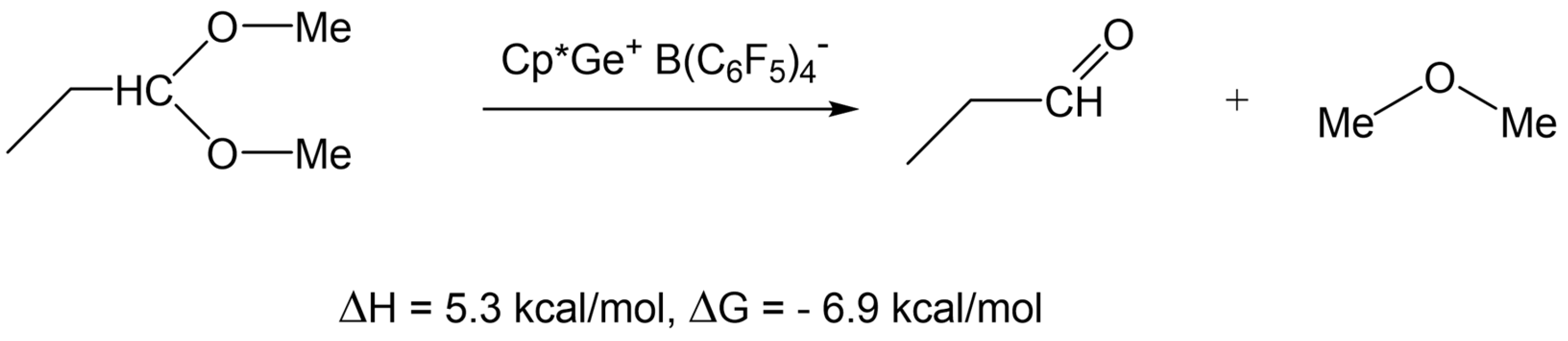
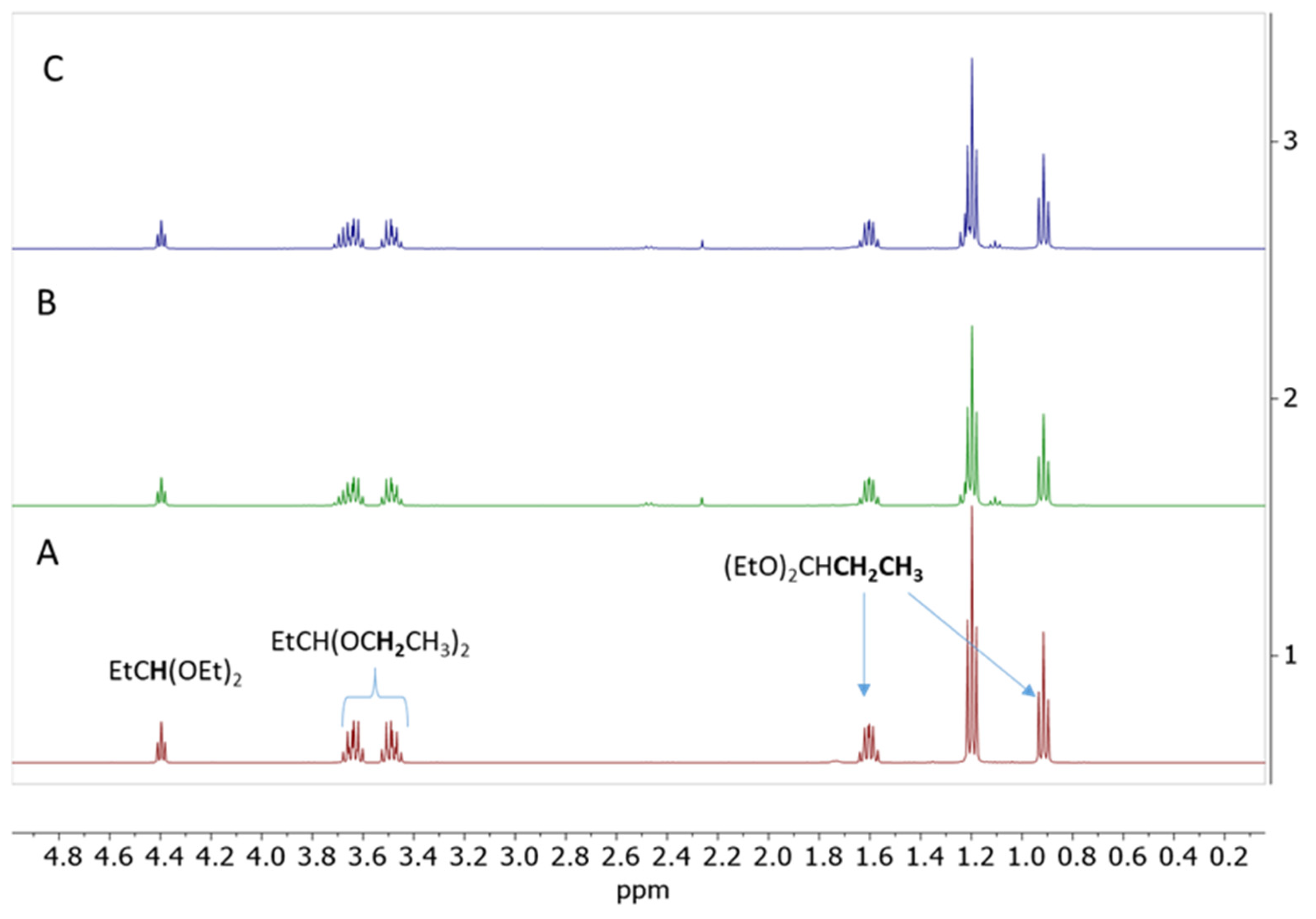
2.4. Testing Decomposition of Dialkyl Acetal in the Presence of Cp*Ge+ B(C6F5)4−
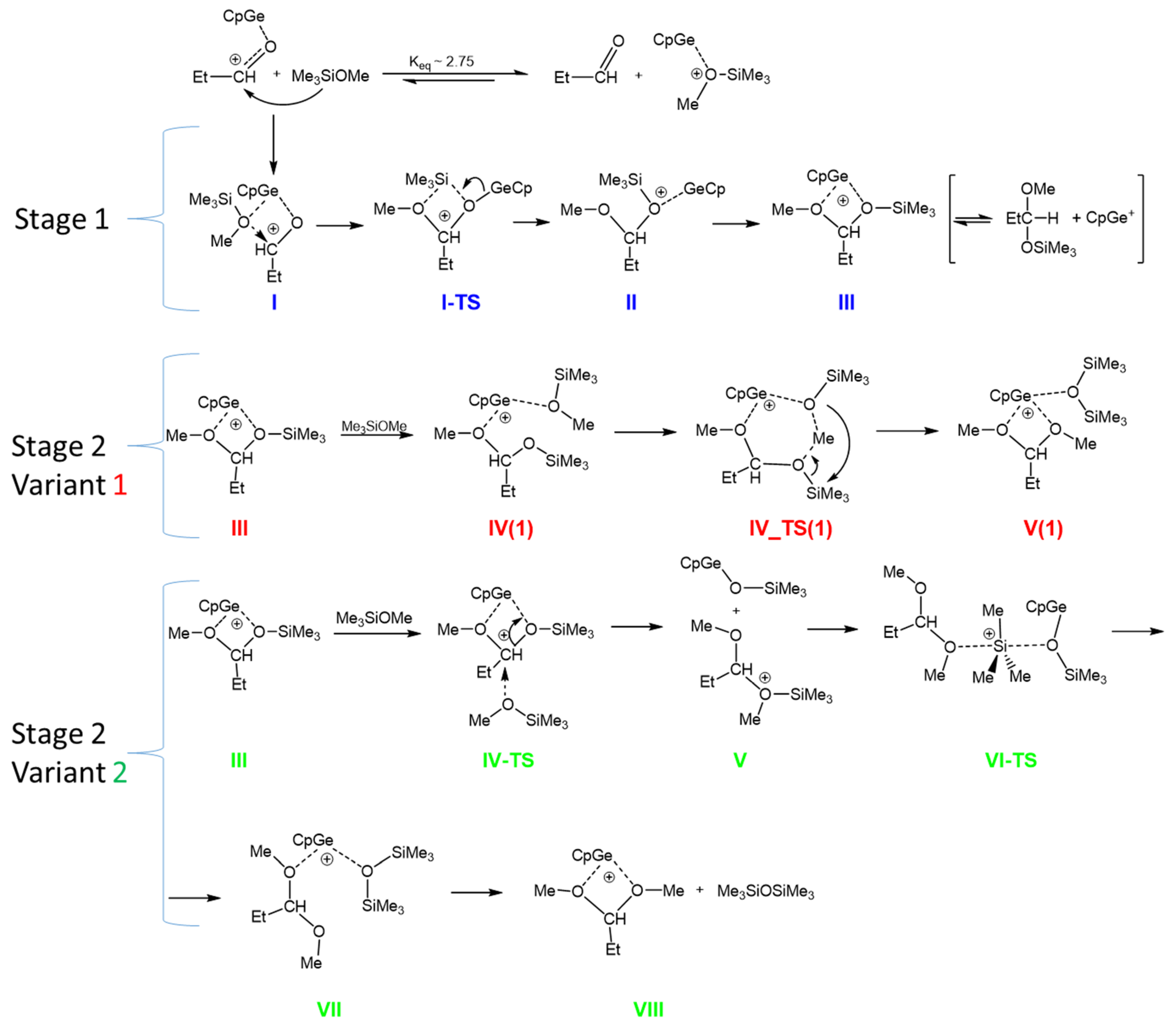
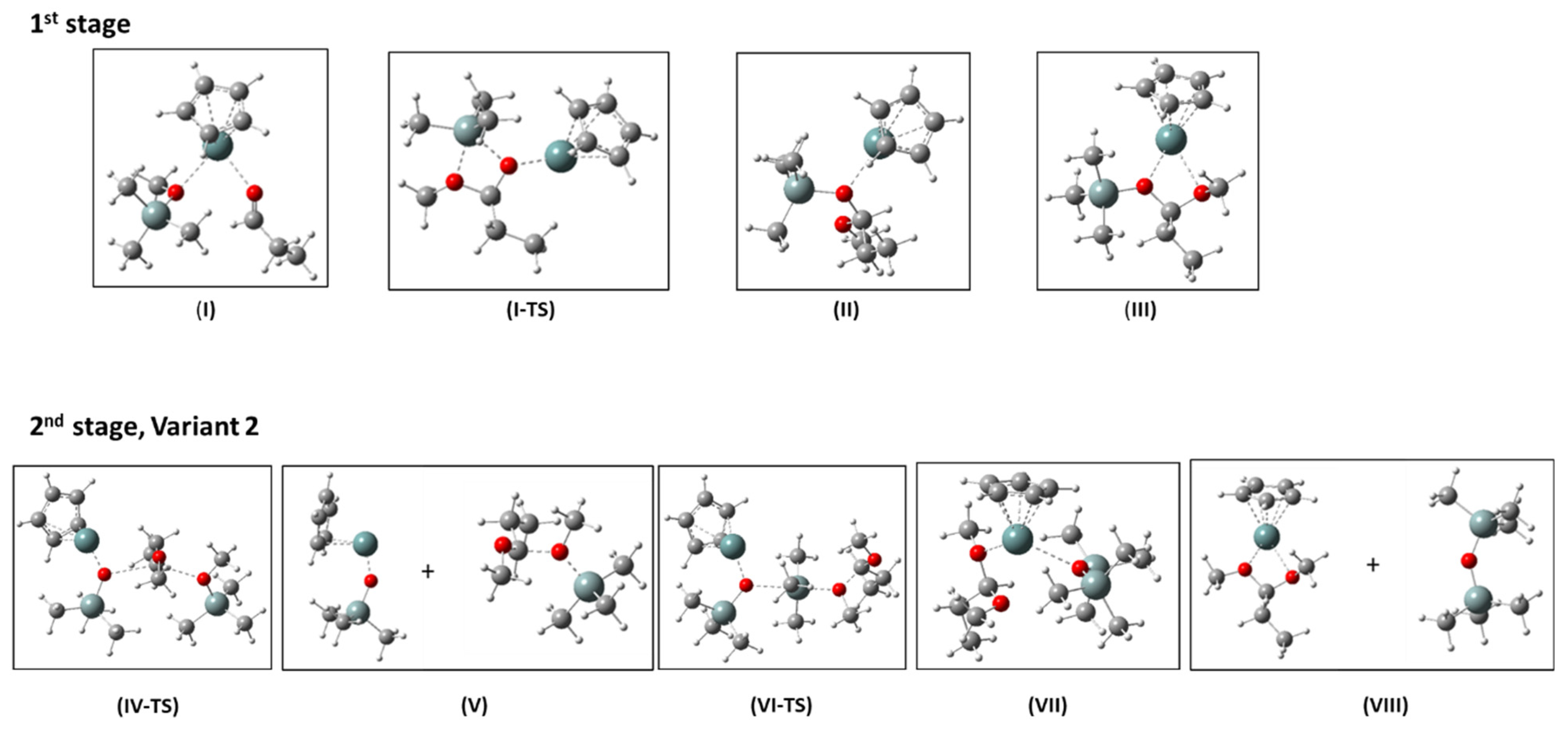
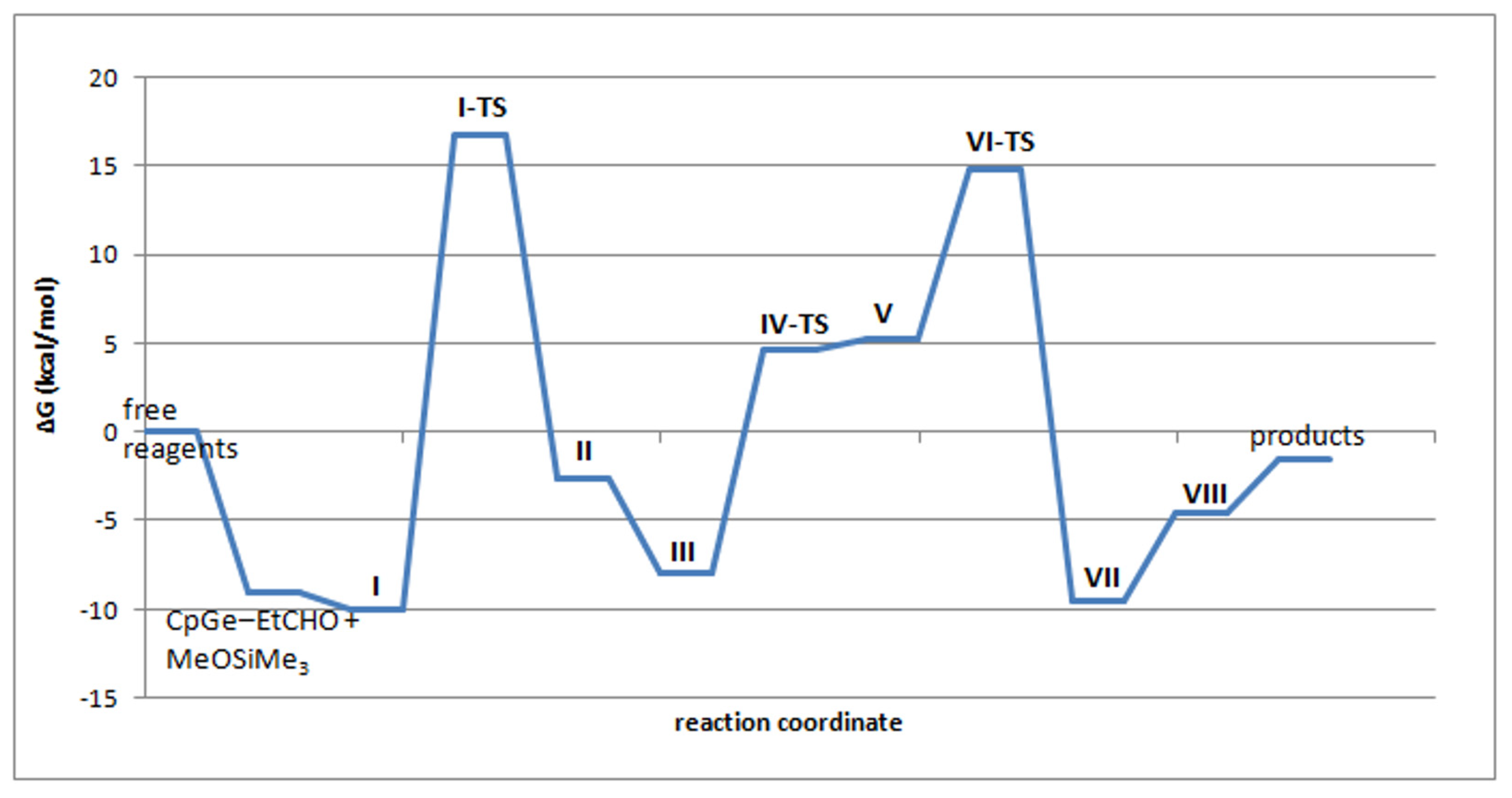
2.5. Mechanism of the De-Alkoxylation Reaction in the Presence of Aldehyde—DFT Calculations
3. Materials and Methods
3.1. Materials
3.2. Synthesis of Alkoxysilanes and Siloxanes
3.3. NMR Spectroscopy
3.4. Gas Chromatography–Mass Spectrometry (GC/MS)
3.5. Gas Chromatography
3.6. Study of the Condensation Cure of the Alkoxy-Functional Silicone Resins
3.7. Study of the Condensation of Model Alkoxysilanes in the Presence of Propionaldehyde
3.8. Theoretical Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kohl, F.X.; Schlüter, E.; Jutzi, P.; Krüger, C.; Wolmershäuser, G.; Hofmann, P.; Stauffert, P. Azinkomplexe des (Pentamethylcyclopentadienyl) germanium-und-zinn-Kations. Chem. Ber. 1984, 117, 1178–1193. [Google Scholar] [CrossRef]
- Jutzi, P. Cyclopentadienyl complexes with main-group elements as central atoms—A decade of research. J. Organomet. Chem. 1990, 400, 1–17. [Google Scholar] [CrossRef]
- Jutzi, P.; Mix, A.; Rummel, B.; Schoeller, W.W.; Neumann, B.; Stammler, H.-G. The (Me5C5) Si+ cation: A stable derivative of HSi+. Science 2004, 305, 849–851. [Google Scholar] [CrossRef] [PubMed]
- Jutzi, P.; Mix, A.; Neumann, B.; Rummel, B.; Stammler, H.-G. Novel π-complexes of divalent silicon: Mixed substituted neutral sandwich compounds and the half-sandwich cation (iPr5C5) Si+. Chem. Commun. 2006, 3519–3521. [Google Scholar] [CrossRef]
- Leszczyńska, K.; Mix, A.; Berger, R.J.; Rummel, B.; Neumann, B.; Stammler, H.G.; Jutzi, P. The pentamethylcyclopentadienylsilicon (II) cation as a catalyst for the specific degradation of oligo (ethyleneglycol) diethers. Angew. Chem. 2011, 123, 6975–6978. [Google Scholar] [CrossRef]
- Jutzi, P. The Pentamethylcyclopentadienylsilicon (II) Cation: Synthesis, characterization, and reactivity. Chem. Eur. J. 2014, 20, 9192–9207. [Google Scholar] [CrossRef]
- Fritz-Langhals, E. Silicon (II) Cation Cp* Si:+ X−: A New Class of Efficient Catalysts in Organosilicon Chemistry. Org. Process Res. Dev. 2019, 23, 2369–2377. [Google Scholar] [CrossRef]
- Fritz-Langhals, E.; Werge, S.; Kneissl, S.; Piroutek, P. Novel Si (II)+ and Ge (II)+ Compounds as Efficient Catalysts in Organosilicon Chemistry: Siloxane Coupling Reaction. Org. Process Res. Dev. 2020, 24, 1484–1495. [Google Scholar] [CrossRef]
- Fritz-Langhals, E. Main Group Catalysis: Cationic Si (II) and Ge (II) Compounds as Catalysts in Organosilicon Chemistry. Reactions 2021, 2, 442–456. [Google Scholar] [CrossRef]
- Fritz-Langhals, E. [(η5-CpR) Ge:]+[BArF4]−/O2–A Sustainable and Efficient Catalytic System for the Hydrosilylation of Selected Olefins. ChemCatChem 2023, 15, e202300442. [Google Scholar] [CrossRef]
- Butts, M.; Cella, J.; Wood, C.D.; Gillette, G.; Kerboua, R.; Leman, J.; Lewis, L.; Rubinsztajn, S.; Schattenmann, F.; Stein, J.; et al. Silicones. In Encyclopedia of Polymer Science and Technology, 4th ed.; Wiley: New York, NY, USA, 2014; Volume 12, pp. 464–541. [Google Scholar]
- Marciniec, B. Comprehensive Handbook on Hydrosilylation; Elsevier: Amsterdam, The Netherlands, 2013. [Google Scholar]
- Fritz-Langhals, E. Process of Preparing Siloxane. WO2022037793A1, 21 August 2020. [Google Scholar]
- Fritz-Langhals, E. Process for Preparing Siloxanes. U.S. Patent 2023/0287018, 14 September 2023. [Google Scholar]
- Noll, W. Chemistry and Technology of Silicones; Elsevier: Amsterdam, The Netherlands, 2012. [Google Scholar]
- Eliel, E.L. Prostereoisomerism (prochirality). In Organic Chemistry; Springer: Berlin/Heidelberg, Germany, 2005; pp. 1–76. [Google Scholar]
- Tsunoda, T.; Suzuki, M.; Noyori, R. A facile procedure for acetalization under aprotic conditions. Tetrahedron Lett. 1980, 21, 1357–1358. [Google Scholar] [CrossRef]
- Sakurai, H.; Sasaki, K.; Hayashi, J.; Hosomi, A. Acetalization of carbonyl compounds with alkoxysilanes catalyzed by iodotrimethylsilane: One-pot allylation reactions of carbonyl compounds to homoallyl ethers using an allylsilane. J. Org. Chem. 1984, 49, 2808–2809. [Google Scholar] [CrossRef]
- Bezlepkina, K.A.; Milenin, S.A.; Vasilenko, N.G.; Muzafarov, A.M. Ring-opening polymerization (ROP) and catalytic rearrangement as a way to obtain siloxane mono-and telechelics, as well as well-organized branching centers: History and prospects. Polymers 2022, 14, 2408. [Google Scholar] [CrossRef]
- Belowich, M.E.; Roberts, J.M.; Peterson, T.H.; Bellinger, E.; Syverud, K.; Sidle, T. Lewis Acids As Highly Active Silanol Polycondensation Catalysts Affording Low Levels of Cyclosiloxanes. Macromolecules 2020, 53, 7487–7495. [Google Scholar] [CrossRef]
- Carey, F.A.; Sundberg, R.J. Advanced Organic Chemistry: Part A: Structure and Mechanisms; Springer Science & Business Media: New York, NY, USA, 2007. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 16 Rev. C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Barone, V.; Cossi, M. Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experiment # | Atmosphere | Propionaldehyde mol% vs. SiOMe | Time to Gel |
|---|---|---|---|
| 1 | Dry nitrogen | none | Liquid after 1 week |
| 2 | Air, RH = 74% | none | 8 h |
| 3 | Nitrogen, RH = 90% | none | 4 h |
| 4 | Dry oxygen | none | Liquid after 1 week |
| 5 | Dry nitrogen | 5 | Liquid after 1 week |
| 6 | Dry nitrogen | 14 | 1.5 h |
| 7 | Dry nitrogen | 18 | 20 min |
| 8 | Dry nitrogen | 24 | 10 min |
| ΔH | ΔG | |
|---|---|---|
| CpGe+---Nu | ||
| EtCHO | −16.6 | −9.1 |
| MeOSiMe3 | −18.9 | −9.7 |
| Me3SiOSiMe3 | −17.5 | −5.3 |
| Cp*Ge+---Nu | ||
| EtCHO | −12.7 | −4.9 |
| MeOSiMe3 | −14.0 | −4.0 |
| Me3SiOSiMe3 | −12.6 | −0.7 |
| ΔHrel | ΔGrel | |
|---|---|---|
| CpGe+ + EtCHO + MeOSiMe3 | 0 | 0 |
| CpGe+–EtCHO + MeOSiMe3 | −16.6 | −9.1 |
| CpGe+–MeOSiMe3 + EtCHO | −18.9 | −9.7 |
| I | −30.7 | −10.0 |
| I-TS | −8.5 | 16.7 |
| II | −26.1 | −2.6 |
| III | −31.6 | −8.0 |
| CpGe+ + EtCH(OMe)(OSiMe3) | −8.3 | 5.3 |
| IV-TS | −30.9 | 4.6 |
| V | −29.8 | 5.9 |
| VI | −19.7 | 5.2 |
| VI-TS | −24.6 | 14.8 |
| VII | −46.5 | −9.5 |
| VIII, CpGe+–EtCHO + Me3SiOSiMe3 | −19.4 | −4.6 |
| CpGe+ + EtCH(OMe)2 + Me3SiOSiMe3 | −14.9 | −1.6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rubinsztajn, S.; Mizerska, U.; Kurjata, J.; Kwiatkowska, M.; Cypryk, M. Aldehyde-Assisted Alkoxysilane Condensation to Form Siloxane Bond: A New Process for Curing Alkoxy-Functional Silicone Resins. Molecules 2025, 30, 714. https://doi.org/10.3390/molecules30030714
Rubinsztajn S, Mizerska U, Kurjata J, Kwiatkowska M, Cypryk M. Aldehyde-Assisted Alkoxysilane Condensation to Form Siloxane Bond: A New Process for Curing Alkoxy-Functional Silicone Resins. Molecules. 2025; 30(3):714. https://doi.org/10.3390/molecules30030714
Chicago/Turabian StyleRubinsztajn, Sławomir, Urszula Mizerska, Jan Kurjata, Małgorzata Kwiatkowska, and Marek Cypryk. 2025. "Aldehyde-Assisted Alkoxysilane Condensation to Form Siloxane Bond: A New Process for Curing Alkoxy-Functional Silicone Resins" Molecules 30, no. 3: 714. https://doi.org/10.3390/molecules30030714
APA StyleRubinsztajn, S., Mizerska, U., Kurjata, J., Kwiatkowska, M., & Cypryk, M. (2025). Aldehyde-Assisted Alkoxysilane Condensation to Form Siloxane Bond: A New Process for Curing Alkoxy-Functional Silicone Resins. Molecules, 30(3), 714. https://doi.org/10.3390/molecules30030714