Abstract
Herein, we report a cobalt-catalyzed cross-coupling of organoselenides with Grignard reagents using simple CoCl2 as the catalyst. This efficient method accommodates a broad scope of selenides, including vinyl, aryl, heteroaryl, and alkynyl derivatives, under mild and ligand-free conditions. Remarkably, the reaction proceeds efficiently without additives and afforded the desired products in moderate to good yields within just 3 h.
1. Introduction
Cross-coupling reactions represent one of the most powerful tools for constructing complex molecules, particularly for forming Csp2–Csp2 bonds [1]. The wide-ranging application of these reactions in the synthesis of bioactive molecules—particularly in the pharmaceutical and agrochemical industries—was recognized by the 2010 Nobel Prize in Chemistry [2,3]. These transformations typically involve a metal-catalyzed reaction between a nucleophilic organometallic reagent and an electrophilic substrate bearing a halide or pseudohalide leaving group. Prominent examples include the Suzuki–Miyaura [4], Kumada–Tamao–Corriu [5], Sonogashira [6], Stille [7], Negishi [8], and Hiyama [9] reactions.
In the past few years, significant efforts have been devoted to developing sustainable protocols that involve use of various solvents, catalysts, ligands, and additives to accommodate more readily available substrates (C–X) and less reactive organometallic reagents (e.g., Sn, Al, Zn, B, Si) [10]. However, Grignard reagents remain widely used due to their high reactivity and ease of preparation [5]. The application of Grignard reagents has evolved far beyond their classical role in nucleophilic addition, emerging as versatile bases for direct C–H metallation. Notably, their utility in this field is often enhanced by their surprisingly mild and selective basicity compared to stronger alternatives like alkyllithiums, which can lead to over-metallation or substrate decomposition. This strategic use of Grignard reagents as moderate bases has been powerfully demonstrated in recent years for the catalytic functionalization of a range of challenging substrates [11,12,13]. As a result, numerous cross-coupling protocols involving Grignard reagents and various substrates, catalyzed by nickel, palladium, or cobalt, have been reported in recent years [14,15,16].
Organoselenium compounds have attracted considerable attention, primarily due to their diverse biological activities [17,18,19]. These compounds can be selectively introduced into organic substrates through both nucleophilic and electrophilic pathways, often with high chemo-, regio-, and stereo-control [20,21]. Selenium compounds are generally stable in air and in common organic solvents, further enhancing their synthetic utility. Owing to the ability of selenium to exhibit oxidation states ranging from −2 to +6, these compounds display a wide range of structural diversity. Representative classes include diselenides, selenides, selenoxides, selenones, vinyl selenides, and selenoacetylenes [22].
A significant focus in chalcogen chemistry involves heterocyclic systems, where functionalization strategies range from simple reactions with elemental selenium or diselenides to sophisticated transition-metal-catalyzed transformations [23,24,25]. Among the most powerful methods, transition-metal-catalyzed cyclization of simple acyclic precursors stands out as a particularly attractive approach for the direct construction of chalcogen-containing heterocycles [26,27]. Although previous studies have demonstrated promising results, some of them have space of improvement considering the use of precious metals, costly catalysts, ligand, and high reagent loadings. To address these shortcomings, the development of a more sustainable and efficient alternative is urgently needed.
Although organohalides (C–X, where X = I, Br, Cl) and pseudohalides (X = OC(O)R, OTs, OTf, NC(O)R) are the most electrophiles in cross-coupling reactions [28,29]—mainly due to their low bond dissociation energies—organoselenium compounds have emerged as promising alternatives (Scheme 1). This potential is exemplified by the work reported by Zeni and co-authors in 2015, which describes the use of various classes of organoselenium compounds as substrates in palladium-catalyzed cross-coupling reactions, including Negishi, Suzuki, Kumada, and Sonogashira protocols [30]. Further advances include Silveira and co-workers’ iron-catalyzed (Fe(acac)3) cross-coupling of vinylic selenides (both E and Z isomers) and tellurides with Grignard reagents, which proceeds with complete retention of configuration and excellent yields [31]. Most recently, a nickel-catalyzed cross-coupling reaction was developed using an aryl alkyl selenide and an aryl bromide in the presence of 3 equivalents of magnesium [32]. This one-pot transformation proceeds via cleavage of the C–Se bond, affording the desired biaryls in moderate to good yields, while avoiding the pre-preparation of Grignard reagents.

Scheme 1.
Metal-catalyzed cross-coupling of organoselenides and Grignard Reagents [26,27,28].
As part of our ongoing interest in organoselenium chemistry and in the development of new synthetic methodologies [33,34,35,36,37,38,39,40], we herein report a cobalt-catalyzed cross-coupling of diverse organoselenides (vinyl, heteroaryl, aryl, and alkynyl) with Grignard reagents using CoCl2 as a simple, efficient catalyst.
2. Results & Discussion
For the optimization, (Z)-butyl(styryl)selenide 1a and freshly prepared (4-methoxyphenyl) magnesium bromide 2a (1 M solution) were selected as model substrates (Table 1). Initially, 2a (2 equiv.) were added dropwise to a solution of 1a (0.25 mmol) in THF (1 mL) at 0 °C. After 2 h, product 3a was isolated in 50% yield (entry 1). We then evaluated common co-solvents for Grignard cross-coupling reactions, but THF/NMP (2:1) and THF/Et3N (2:1) mixtures did not improve the yield (entries 2–3). Similarly, conducted reaction at room temperature proved ineffective (entries 4–5), with incomplete consumption of 1a observed in all cases.

Table 1.
Optimization of the cross-coupling conditions [a].
Notably, using 5 mol% of catalyst at 0 °C led to a significant increase in the yield of product 3a, whereas 2 mol% proved insufficient (entries 6–7). As expected, in the absence of the cobalt catalyst, only the starting material was recovered (Table 1, entry 8). Further optimization revealed that extending the reaction time to 3 h provided 3a in 73 and 83% yield for 2 mol% and 5 mol % catalytic loading, respectively (entry 9–10). Prolonging the reaction beyond 3 h did not further improve the yield. Temperature control was critical, as performing the reaction at room temperature substantially reduced the yield (entry 11). The use of an excess of Grignard reagent (2 equiv.) was necessary due to the competitive formation of a biaryl byproduct alongside product 3a. Although 1.5 equivalents of 2a appeared to be sufficient, we opted to maintain the use of 2 equivalents for consistency.
With the optimized conditions in hand (Table 1, entry 10), we next explored the scope and limitations of the method by applying it to a range of substrates (Table 2). First, we examined various aryl Grignard reagents (freshly prepared) with vinyl selenide 1a. Aryl Grignard reagents bearing electron-donating groups (2a and 2b) reacted efficiently, providing higher yields compared to phenylmagnesium bromide (2c) (Table 2, entries 1–3). In contrast, the presence of an electron-withdrawing substituent, such as fluorine, diminished reactivity, affording product 3b in only 63% yield (Table 2, entry 4). Notably, alkyl Grignard reagents generated multiple byproducts with similar polarity, preventing isolation of the desired products. Similarly, when substrate 1b was employed, product 3e and the corresponding biaryl byproduct co-formed an inseparable mixture, making purification and yield estimation challenging (Table 2, entry 5). However, when Grignard reagent 2b was used, product 3f was isolated in 87% yield (Table 2, entry 6).
Table 2.
Scope of the Reaction Between Vinyl Selenides and Grignard Reagents [a].
Other vinyl selenides, such as 1c, 1d, and 1e, also reacted satisfactorily with reagent 2b (Table 2, entries 7–9). Interestingly, the reaction also proved efficient for the coupling between a divinylic selenide and Grignard reagents. In this case, the use of 4 equivalents of Grignard reagent 2a led to the formation of product 3a in 73% yield (Table 2, entry 10). The protocol tolerated functional groups such as alcohols, substrate 1g reacted successfully with 2a, albeit requiring 3 equivalents of the Grignard reagent (Table 2, entry 11).
Considering that heterocycle-based organoselenium compounds represent one of the most important classes of selenium-containing molecules, we investigated the reactivity of pyridine derivative 4a with various Grignard reagents under our optimized conditions. As shown in Scheme 2, both electron-rich and electron-deficient Grignard reagents successfully coupled with 4a, yielding products 5a–5c in good yields. Notably, Grignard reagents bearing electron-donating groups consistently provided superior yields compared to their electron-withdrawing counterparts.
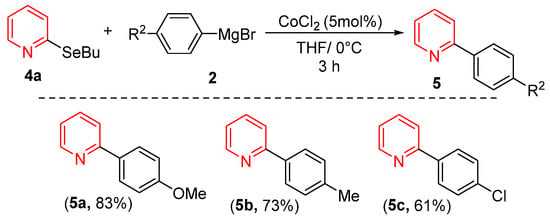
Scheme 2.
Cross-Coupling of 2-(buthylselanyl)pyridine with Grignard Reagents.
To further explore the synthetic utility of and demonstrate the potential of alkynyl and aryl selenide derivatives as starting materials, we evaluated their reactivity in cobalt-catalyzed cross-coupling reactions. Under standard conditions, compound 6a coupled with Grignard reagent 2a to afford product 7a in 35% yield (Scheme 3A). More encouragingly, the reaction of aryl selenide 8a furnished the corresponding biaryl 9a in 61% yield (Scheme 3B), demonstrating the particular effectiveness of this substrate class.
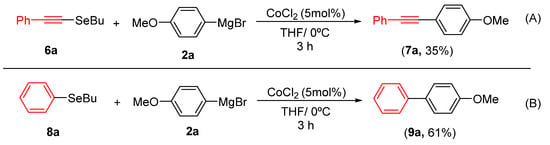
Scheme 3.
Reactivity of Grignard Reagent 2a. (A) with 6a; (B) with 8a.
Building upon the foundational work by Oshima and co-workers [41,42] on cobalt-catalyzed cross-coupling reactions involving Grignard reagents, and later advancements by Hayashi [43], we were encouraged to propose a detailed mechanism (Scheme 4). In these studies, a complex formed in situ from cobalt salts and at least 4 equivalents of PhMgBr drastically altered the reaction outcome, with the coupling product being preferentially obtained over biphenyl. In our proposal, CoCl2 is first reduced to a cobalt(0) complex by 4 equivalents of aryl Grignard reagent 2. This complex then undergoes oxidative addition to form a bis-aryl cobalt(III) intermediate B, which subsequently undergoes reductive elimination to give the product 3 and monoaryl-ate complex C. Finally, the reaction of C with 2 regenerates di-ate complex A.
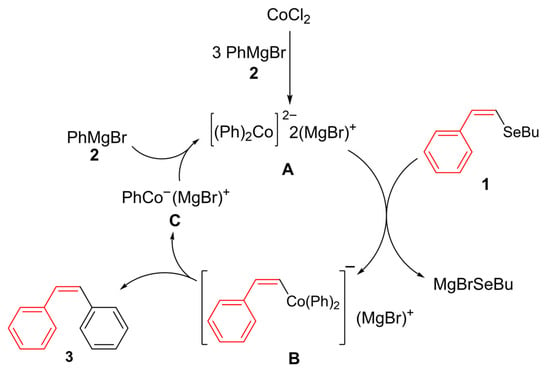
Scheme 4.
A possible mechanism.
3. Materials and Methods
The reagents used were purchased from Aldrich Chemical Co. (Cajamar, Brazil) and used without purification. All the solvents used were of PA grade and used without purification. Analytical thin-layer chromatography analysis was carried out using Merck DC Kieselgel 0 F254 (Merck, Darmstadt, Germany) (230–400 mesh) aluminum plates coated with treated silica. For visualization, TLC plates were either placed under ultraviolet light, or stained with iodine vapor, or acidic vanillin. Most reactions were monitored by TLC for disappearance of starting material. Chromatographic columns (LabGlass, Sao Paulo, Brazil) were filled with silica gel 60 (230–400 mesh ASTM). Nuclear magnetic resonance (NMR) spectra were recorded using a Bruker Ascend 500TM spectrometer or Bruker DPX 300 NMR spectrometer (Bruker, Rheinstetten, Germany), with the chemical shifts (δ) set in parts per million (ppm), referenced by the residual solvent peak. The multiplicity of each peak is designated by the following abbreviations: s (singlet), d (doublet), dd (doublet of doublet), t (triplet), quint (quintet), sext (sextet) and m (multiplet). The coupling constants (J) are reported in Hertz (Hz).
General Procedure for the Cobalt-Catalyzed Cross-Coupling Reaction of Organoselenides with Organomagnesium Reagents
A mixture of the organoselenide (0.20 mmol) and CoCl2 (0.05 equiv.) was dissolved in THF (1 mL). Subsequently, freshly prepared organomagnesium reagent (2 equiv., 1 M solution) was then added. The mixture was stirred for 3 h at 0 °C. Afterward, the reaction mixture was allowed to warm to room temperature, diluted with ethyl acetate (3 mL), and washed with a saturated solution of NH4Cl (20 mL). The organic layer was separated, dried over MgSO4, and concentrated under reduced pressure. The crude product was purified by flash chromatography, eluting with hexane. Spectra of all the synthesized compounds are available in the Supplementary Materials (Figures S1–S28).
Spectral Data of Synthesized Products (3a–3j, 5a–5c, 7a, 9a)
- (Z)-1-methoxy-4-styrylbenzene 3a [44]. Yield: 83% (0.035 g), colorless liquid. RMN 1H (CDCl3, 500 MHz): δ = 7.27–7.17 (m, 7H); 6.75 (d, J = 8.7 Hz, 2H); 6.54–6.48 (m, 2H); 3.77 (s, 3H). RMN 13C (CDCl3, 125 MHz): δ = 158.60; 135.56; 137.57; 130.16; 129.78; 129.67; 128.83; 128.77; 128.25; 126.9; 55.20.
- (Z)-1-methil-4-styrylbenzene 3b [45]. Yield: 87% (0.034 g), colorless liquid. RMN 1H (CDCl3, 500 MHz): δ = 7.46 (d, J = 7.9 Hz, 3H); 7.25–7.12 (m, 5H); 6.54 (s, 1H); 2.37 (s, 3H). RMN 13C (CDCl3, 125 MHz): δ = 138.49; 136.91; 130.41; 129.64; 129.11; 129.05; 128.99; 128.37; 128.39; 127.02; 21.25.
- (Z)-1,2-diphenylethene 3c [44]. Yield: 72% (0.026 g), colorless liquid. RMN 1H (CDCl3, 500 MHz): δ = 7.65 (d, J = 7.2 Hz, 1H); 7.50 (t, J = 7,7 Hz, 1H) 7.43–7.39 (m, 1H); 7.31–7.23 (m, 7H); 6.66 (s, 2H). RMN 13C (CDCl3, 125 MHz): δ = 137.29, 130.29, 128.92, 128.25, 127.13.
- (Z)-1-fluoro-4-styrylbenzene 3d [44]. Yield: 63% (0.024 g), colorless liquid. RMN 1H (CDCl3, 500 MHz): δ = 7.49–7.46 (m, 1H); 7.22–7.17 (m, 5H); 7.12–7,09 (m, 1H); 6,91–6.87 (m, 2H); 6,56 (q, J = 12.1 Hz, 2H). RMN 13C (CDCl3, 125 MHz): δ = 161,97 (d, J = 246.6 Hz); 137.24; 1336.37 (d, J = 3.4 HZ); 130.73 (d, 7.9 Hz); 130.46; 129.28, 129.03; 128.51; 127.40; 115.45 (d, J = 21.4 Hz).
- (Z)-1-methoxy-4-(4-methylstyryl)benzene 3f [45]. Yield: 87% (0.0386 g), colorless liquid. RMN 1H (CDCl3, 500 MHz): δ = 7.21–7.14 (m, 4H); 7.03 (d, J = 8.0 Hz, 2H); 6.75 (d, J = 8.7 Hz, 2H); 6.47 (s, 2H); 2.31 (s, 3H); 3.79 (s, 3H); 2.33 (s, 3H). RMN 13C (CDCl3, 125 MHz): δ = 158.60; 136.65; 134.63; 130.28; 130.12; 129.12; 128.94; 128.76; 128.76; 113.58; 55.20; 21.25.
- (Z)-1-methoxy-4-(4-methylstyryl)benzene 3g [44]. Yield: 69% (0.031 g), colorless liquid. RMN 1H (CDCl3, 500 MHz): δ = 7.22–7.17 (m, 4H); 7.05 (d, J = 8.0 Hz, 2H); 6.77 (d, J = 8.7 Hz, 2H); 6.49 (s, 2H); 2.31 (s, 3H); 3.79 (s, 3H); 2.33 (s, 3H). RMN 13C (CDCl3, 125 MHz): δ = 158.68; 136.81; 134.80; 130.29; 130.12; 129.28; 129.11; 128.93; 128.90; 113.75; 55.35; 21.42.
- (Z)-1-choro-4-(4-methoxystyryl)benzene 3h [46]. Yield: 77% (0.038 g), colorless liquid. RMN 1H (CDCl3, 500 MHz): δ = 7.17 (d, J = 7.3 Hz, 4H); 7.15 (d, J = 8.6 Hz, 2H); 6.75 (d, J = 8.6 Hz, 2H); 6.49 (s, 2H); 6.53 (d, J = 12.1 Hz, 1H); 6.42 (d, J = 12.1 Hz, 1H); 3.79 (s, 3H). RMN 13C (CDCl3, 125 MHz): δ = 158.68; 136.81; 134.80; 130.29; 130.12; 129.28; 129.11; 128.93; 128.90; 113.75; 55.35.
- (Z)-1-methoxy-4-(4-(trifluoromethyl)ystyryl)benzene 3i [47]. Yield: 69% (0.038 g), colorless liquid. RMN 1H (CDCl3, 500 MHz): 7.46 (d, J = 8.1 Hz, 2H); 7.35 (d, J = 8.1 Hz, 2H); 7.14 (d, J = 8.6 Hz, 2H); 6.76 (d, J = 8.6 Hz, 2H); 6.63 (d, J = 12.2 Hz, 1H); 6.48 (d, J = 12.2 Hz, 1H); 3.78 (s, 3H). RMN 13C (CDCl3, 125 MHz): δ = 159.25; 141.53; 132.02; 130.38; 129.28; 129.11; 127.41; 125.37 (q, J = 3.8 Hz); 123.34; 113.99; 54.42.
- (Z)-3-(4-mehoxyphenyl)prop-2-en-1-ol 3j [44]. Yield: 43% (0.013 g), colorless liquid. RMN 1H (CDCl3, 500 MHz): 7.14 (d, J = 8.6 Hz, 2H); 6.85 (d, J = 8.6 Hz, 2H); 6.49 (d, J = 11.6 Hz, 1H); 5.78–5.73 (m, 1H); 4.41 (d, J = 6.4 Hz, 1H); 3.80 (s, 3H). RMN 13C (CDCl3, 125 MHz): δ = 158.75; 130.67; 130.11; 129.41; 127.15; 113.71; 59.78; 55.29.
- 2-(4-methoxyphenyl)pyridine5a [30]. Yield: 83% (0.031 g), colorless solid. RMN 1H (CDCl3, 500 MHz): δ = 8.63 (ddd, J = 4.8, 1.6, 1.0 Hz, 1H); 7.95–7.91 (m, 2H); 7.71–7.63 (m, 2H); 7.15 (ddd, J = 7.2, 4.8, 1.3 Hz, 1H); 7.71–7.63 (m, 2H); 3.84 (s, 3H). RMN 13C (CDCl3, 125 MHz): δ = 160.65; 157.29; 149.93; 136.85; 132.24; 128.19; 121.67; 120.00; 114.41; 55.44.
- 2-(p-tolyl)pyridine5b [48]. Yield: 73% (0.025 g), colorless solid. RMN 1H (CDCl3, 500 MHz): δ = 8.66 (ddd, J = 4.8, 1.6, 1.0 Hz, 1H); 7.87 (d, J = 8.2 Hz, 1H); 7.73–7.67 (m, 2H); 7.26 (d, J = 8.2 Hz, 1H); 7.18 (ddd, J = 6.7, 4.8, 1.6, Hz, 1H); 3.84 (s, 3H). RMN 13C (CDCl3, 125 MHz): δ = 157.70; 149.68; 139.19; 136.91; 129.69; 126.88; 122.01; 120.37; 115.36; 21.47.
- 2-(4-chorophenyl)pyridine5c [48]. Yield: 61% (0.023 g), colorless solid. RMN 1H (CDCl3, 500 MHz): δ = 8.71 (ddd, J = 4.8, 1.6, 1.0 Hz, 1H); 7.96 (d, J = 7.9 Hz, 2H); 7.77 (td, J = 7.7, 1.7 Hz, 2H); 7.72 (d, J = 8.0 Hz, 2H); 7.47 (d, J = 7.9 Hz, 2H); 7.28–7.25 (m, 1H). RMN 13C (CDCl3, 125 MHz): δ = 156.42; 150.00; 138.01; 137.13; 129.16; 128.37; 122.56; 120.40.
- 1-methoxy-4-(phenylethynyl)benzene7a [30]. Yield: 35% (0.015 g), colorless solid. RMN 1H (CDCl3, 500 MHz): δ = 7.51–7.49 (m, 2H); 7.45 (d, J = 8.6 Hz, 2H); 7.34–7.29 (m, 3H); 6.86. (d, J = 8.6 Hz, 2H); 3.81 (s, 3H). RMN 13C (CDCl3, 125 MHz): δ = 159.23; 133.20; 131.66; 128.61; 128.09; 123.66; 115.49; 114.13; 89.57; 88.23; 55.51.
- 4-methoxy-1,1′-biphenyl9a [30]. Yield: 61% (0.022 g), white solid. RMN 1H (CDCl3, 500 MHz): δ = 7.55–7.51 (m, 4H); 7.42–7.39 (m, 2H); 7.31–7.28 (m, 1H); 6.97 (d, J = 8.6 Hz, 2H); 3.84 (s, 3H). RMN 13C (CDCl3, 125 MHz): δ = 159.34; 141.03; 135.44; 133.92; 128.92; 128.36; 126.94; 114.29; 55.55.
4. Conclusions
In summary, we have developed a practical and efficient cobalt-catalyzed cross-coupling of organoselenides with Grignard reagents under mild conditions. Under the optimized conditions, the reaction operates without any ligands or co-solvents, and good yields were achieved in just 3 h using only 5 mol% of the cobalt catalyst. This method significantly expands the scope of metal-catalyzed cross-coupling reactions involving freshly prepared Grignard reagent, accommodating not only vinyl selenides but also aryl, heteroaryl, and alkynyl derivatives. Given the facile preparation of organoselenium compounds and their inherent chemo-, regio-, and stereoselective reactivity, this transformation offers a versatile and powerful strategy for C–C bond formation.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules30214232/s1, NMR Spectra (Figures S1–S28).
Author Contributions
Conceptualization: J.R. and A.L.S.; Methodology: T.V.d.S.P., K.D.V.P.S., M.E.C.d.S., S.A.d.O., A.P.V.d.J. and T.A.N.R.; Validation: T.V.d.S.P., K.D.V.P.S., M.E.C.d.S., S.A.d.O., A.P.V.d.J. and T.A.N.R.; Formal analysis: T.V.d.S.P., K.D.V.P.S., M.E.C.d.S., S.A.d.O., A.P.V.d.J., T.A.N.R., L.G.d.V., P.T.d.S.J. and S.S.; Investigation: T.V.d.S.P., K.D.V.P.S., M.E.C.d.S., S.A.d.O., A.P.V.d.J. and T.A.N.R.; Resources: J.R. and A.L.S.; Data curation: T.V.d.S.P., K.D.V.P.S., M.E.C.d.S., S.A.d.O., A.P.V.d.J. and T.A.N.R.; Writing—Original draft: A.L.S.; Writing—Review & Editing: S.S., J.R. and A.L.S.; Visualization: S.S., J.R. and A.L.S.; Supervision: J.R. and A.L.S.; Project administration: J.R. and A.L.S.; Funding acquisition: S.S., J.R. and A.L.S. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES, Finance Code 001), UFMS, FUDECT, UFMT, UFG, and FAPEMAT for financial support and fellowships. This study was financed in part by the Universidade Federal de Mato Grosso do Sul- Brasil (UFMS)—Finance Code 001. A.L.S. acknowledge CNPq for Universal 420048/2023-5. S.S., and J.R. would like to acknowledge CNPq (401355/2025-0, 316687/2023-5, 309975/2022-0, 404172/2023-7, and 405655/2023-1). S.S. also acknowledges the following FAPEG public calls: Chamada Pública FAPEG/SES No. 18/2025 (ARB2025191000003), and Chamada Pública FAPEG No. 05/2025 (PVE2025041000055).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions presented in this study are included in the article and Supplementary Materials. Further inquiries can be directed to the cor-responding authors.
Acknowledgments
The authors thank the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Fundação Universidade Federal de Mato Grosso do Sul (UFMS), Universidade Federal de Mato Grosso (UFMT), and Universidade Federal de Goiás (UFG) for the support offered in this research. S.S. would like to acknowledge Programa futuras Cientistas (Edital CETENE No. 04/2025).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Korch, K.M.; Watson, D.A. Cross-Coupling of Heteroatomic Electrophiles. Chem. Rev. 2019, 119, 8192–8228. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.-F.; Anbarasan, P.; Neumann, H.; Beller, M. Palladium-Catalyzed Cross-Coupling: A Historical Contextual Perspective to the 2010 Nobel Prize. Angew. Chem. Int. Ed. 2010, 49, 9047–9050. [Google Scholar] [CrossRef] [PubMed]
- Beletskaya, I.P.; Alonso, F.; Tyurin, V. The Suzuki-Miyaura reaction after the Nobel prize. Coord. Chem. Rev. 2019, 385, 137–173. [Google Scholar] [CrossRef]
- Miyaura, N.; Yamada, K.; Suzuki, A. A new stereospecific cross-coupling by the palladium-catalyzed reaction of 1-alkenylboranes with 1-alkenyl or 1-alkynyl halides. Tetrahedron Lett. 1979, 36, 3437. [Google Scholar] [CrossRef]
- Tamao, K.; Sumitani, K.; Kumada, M. Selective carbon-carbon bond formation by cross-coupling of Grignard reagents with organic halides. Catalysis by nickel-phosphine complexes. J. Am. Chem. Soc. 1972, 94, 4374. [Google Scholar] [CrossRef]
- Sonogashira, K.; Tohda, Y.; Hasihara, N. A convenient synthesis of acetylenes: Catalytic substitutions of acetylenic hydrogen with bromoalkenes, iodoarenes and bromopyridines. Tetrahedron Lett. 1975, 50, 4467. [Google Scholar] [CrossRef]
- Milstein, D.; Stille, K. A general, selective, and facile method for ketone synthesis from acid chlorides and organotin compounds catalyzed by palladium. J. Am. Chem. Soc. 1978, 100, 3636. [Google Scholar] [CrossRef]
- King, A.O.; Okukado, N.; Negishi, E. Highly general stereo-, regio-, and chemo-selective synthesis of terminal and internal conjugated enynes by the Pd-catalysed reaction of alkynylzinc reagents with alkenyl halides. J. Chem. Soc. Chem. Commun. 1977, 683–684. [Google Scholar] [CrossRef]
- Fujita, M.; Hiyama, T. Fluoride ion-catalyzed reduction of aldehydes and ketones with hydrosilanes. Synthetic and mechanistic aspects and an application to the threo-directed reduction of .alpha.-substituted alkanones. J. Org. Chem. 1988, 53, 5405–5415. [Google Scholar] [CrossRef]
- Gu, D.-Y.; Li, B.-J.; Shi, Z.-J. Exploration of New C–O Electrophiles in Cross-Coupling Reactions. Acc. Chem. Res. 2010, 43, 1486–1495. [Google Scholar]
- Oliveira, J.A.; Dhawa, U.; Ackermann, L. Insights into the Mechanism of Low-Valent Cobalt-Catalyzed C–H Activation. ACS Catal. 2021, 11, 1505–1515. [Google Scholar] [CrossRef]
- Cattani, S.; Pandit, N.K.; Buccio, M.; Balestri, D.; Ackermann, L.; Cera, G. Iron-Catalyzed C–H Alkylation/Ring Opening with Vinylbenzofurans Enabled by Triazoles. Angew. Chem. Int. Ed. 2024, 63, e202404319. [Google Scholar] [CrossRef] [PubMed]
- Cascella, M.; Bore, S.L.; Eisestein, O. The fellowship of the Grignard: 21st century computational tools for hundred-year-old chemistry. Chem. Sci. 2025, 16, 8196–8216. [Google Scholar] [CrossRef] [PubMed]
- Kharasch, M.S.; Field, E.K. Factors Determining the Course and Mechanisms of Grignard Reactions. IV. The Effect of Metallic Halides on the Reaction of Aryl Grignard Reagents and Organic Halides. J. Am. Chem. Soc. 1941, 63, 2316–2320. [Google Scholar] [CrossRef]
- Dai, W.; Xiao, J.; Jin, G.; Xu, J.; Cao, S. Palladium- and Nickel-Catalyzed Kumada Cross-Coupling Reactions of gem-Difluoroalkenes and Monofluoroalkenes with Grignard Reagents. J. Org. Chem. 2014, 79, 10537–10546. [Google Scholar] [CrossRef]
- Gärtner, D.; Stein, A.L.; Grupe, S.; Arp, J.; Wangelin, A.J. Iron-Catalyzed Cross-Coupling of Alkenyl Acetates. Angew. Chem. Int. Ed. 2015, 54, 10545–10549. [Google Scholar] [CrossRef]
- Nogueira, C.W.; Rocha, J.B.T. Toxicology and pharmacology of selenium: Emphasis on synthetic organoselenium compounds. Arch. Toxicol. 2011, 85, 1313–1359. [Google Scholar] [CrossRef]
- Vendruscolo, S.J.; de Oliveira, A.J.; de Sousa, J.R.; Targanski, S.; Stein, A.L.; de Vasconcelos, L.G.; Ferreira, P.A.; Soares, M.A. Nematicidal and ovicidal activity of environmentally-friendly selenol ester derivatives against Meloidogyne incognita. J. Pest Sci. 2024, 97, 2257–2272. [Google Scholar] [CrossRef]
- Braga, A.L.; Rafique, J. Synthesis of biologically relevant small molecules containing selenium. Part B. Anti-infective and anticancer Compounds. In The Chemistry of Organic Selenium and Tellurium Compounds; Rappoport, Z., Ed.; John Wiley & Sons, Ltd.: Chichester, UK, 2013; Volume 4, pp. 1053–1117. [Google Scholar]
- Organoselenium Chemistry. In Topics in Current Chemistry 208; Wirth, T., Ed.; Springer: Heidelberg, Germany, 2000. [Google Scholar]
- Krief, A. Comprehensive Organometallic Chemistry II; Abel, E.V., Stone, F.G.A., Wilkinson, G., Eds.; Pergamon Press: New York, NY, USA, 1995; Volume 11, Chapter 13. [Google Scholar]
- Chuai, H.; Zhang, S.-Q.; Bai, H.; Li, J.; Wang, Y.; Sun, J.; Wen, E.; Zhang, J.; Xin, M. Small molecule selenium-containing compounds: Recent development and therapeutic applications. Eur. J. Med. Chem. 2021, 223, 113621. [Google Scholar] [CrossRef]
- Ma, Y.-T.; Miao-Chang Liu, M.-C.; Zhou, Y.-B.; Wua, H.-Y. Synthesis of Organoselenium Compounds with Elemental Selenium. Adv. Synth. Catal. 2021, 363, 5386–5406. [Google Scholar] [CrossRef]
- Doerner, C.V.; Neto, J.; Cabreira, C.; Saba, S.; Sandjo, L.P.; Rafique, J.; Braga, A.L.; de Assis, F.F. Synthesis of 3-selanyl-isoflavones from 2-hydroxyphenyl enaminones using trichloroisocyanuric acid (TCCA): A sustainable approach. New J. Chem. 2023, 47, 5598–5602. [Google Scholar] [CrossRef]
- Moraes, C.A.O.; Santos, R.B.C.; Cavalcante, M.F.O.; Guilhermi, J.S.; Ali, M.A.; Botteselle, G.V.; Frizon, T.E.A.; Shah, M.I.A.; Liao, L.M.; Beatriz, A.; et al. Urea Hydrogen Peroxide and Ethyl Lactate, an Eco-Friendly Combo System in the Direct C(sp2)–H Bond Selenylation of Imidazo[2,1-b]thiazole and Related Structures. ACS Omega 2023, 8, 39535–39545. [Google Scholar] [CrossRef] [PubMed]
- Zeni, G.; Godoi, B.; Jurinic, C.K.; Belladona, A.L.; Schumacher, R.F. Transition Metal-Free Synthesis of Carbo- and Heterocycles via Reaction of Alkynes with Organylchalcogenides. Chem. Rec. 2021, 21, 2880–2895. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, A.; Arsenyan, P. Rise of diselenides: Recent advances in the synthesis of heteroarylselenides. Coord. Chem. Rev. 2018, 370, 55–68. [Google Scholar] [CrossRef]
- Ehhalt, L.E.; Beleh, O.M.; Prest, I.C.; Mouat, J.M.; Olszewski, A.K.; Ahern, B.N.; Cruz, A.R.; Chi, B.K.; Castro, A.J.; Kang, K.; et al. Cross-Electrophile Coupling: Principles, Methods, and Applications in Synthesis. Chem. Rev. 2024, 124, 13397–13569. [Google Scholar] [CrossRef]
- Jana, R.; Pathak, T.P.; Sigman, M.S. Advances in Transition Metal (Pd, Ni, Fe)-Catalyzed Cross-Coupling Reactions Using Alkyl-organometallics as Reaction Partners. Chem. Rev. 2011, 111, 1417–1492. [Google Scholar] [CrossRef]
- Stein, A.L.; Bilheri, F.N.; Zeni, G. Application of organoselenides in the Suzuki, Negishi, Sonogashira and Kumada cross-coupling reactions. Chem. Commun. 2015, 51, 15522–15525. [Google Scholar] [CrossRef]
- Silveira, C.C.; Mendes, S.R.; Wolf, L. Iron-catalyzed coupling reactions of vinylic chalcogenides with Grignard reagents. J. Braz. Chem. Soc. 2010, 11, 2138–2145. [Google Scholar] [CrossRef]
- Kumar, S.; Sapra, S.; Singh, B. Nickel-Catalyzed Cross-Coupling of Aryl Alkyl Selenides with Aryl Bromides: Cross-Coupling Between Two Electrophiles. Adv. Synth. Catal. 2024, 366, 4528–4534. [Google Scholar] [CrossRef]
- de Oliveira, A.J.; de Oliveira, S.A.; Vasconcelos, L.G.; Dall’oglio, E.L.; Vieira, L.C.C.; Stein, A.L. Synthesis of selenol esters via the reaction of acyl chlorides with diselenides in the presence of Zn dust catalyzed by CoCl2·6H2O. Tetrahedron Lett. 2021, 80, 153317. [Google Scholar] [CrossRef]
- Duarte, J.S.; de Oliveira, S.A.; da Silva, E.C.O.; de Jesus, A.P.V.; Vasconcelos, L.G.; de Sousa Júnior, P.T.; Rafique, J.; Saba, S.; Stein, A.l. Transition-Metal Free, One Pot Synthesis of Selenoesters via Reaction of Alkyl Selenides and Acyl Chlorides. Asian J. Org. Chem. 2023, 12, e202300474. [Google Scholar] [CrossRef]
- Botteselle, G.V.; Elias, W.C.; Bettanin, L.; Canto, R.F.S.; Salin, D.N.O.; Barbosa, F.A.R.; Saba, S.; Gallardo, H.; Ciancaleoni, G.; Domingo, J.B.; et al. Catalytic Antioxidant Activity of Bis-Aniline-Derived Diselenides as GPx Mimics. Molecules 2021, 26, 4446. [Google Scholar] [CrossRef] [PubMed]
- Rafique, J.; Farias, G.; Saba, S.; Zapp, E.; Bellettini, I.C.; Salla, C.A.M.; Bechtold, I.H.; Scheide, M.R.; Neto, J.S.S.; de Souza Junior, D.M. Selenylated-oxadiazoles as promising DNA intercalators: Synthesis, electronic structure, DNA interaction and cleavage. Dyes Pigm. 2020, 180, 108519. [Google Scholar] [CrossRef] [PubMed]
- Doerner, C.V.; Scheide, M.R.; Nicoleti, C.R.; Durigon, D.C.; Idiarte, V.D.; Sousa, M.J.A.; Mendes, S.R.; Saba, S.; Neto, J.S.S.; Martins, G.M.; et al. Versatile Electrochemical Synthesis of Selenylbenzo[b]Furan Derivatives Through the Cyclization of 2-Alkynylphenols. Front. Chem. 2022, 10, 880099. [Google Scholar] [CrossRef]
- Rocha, M.S.T.; Rafique, J.; Saba, S.; Azerdo, J.B.; Back, D.; Godoi, M.; Braga, A.L. Regioselective hydrothiolation of terminal acetylene catalyzed by magnetite (Fe3O4) nanoparticles. Synth. Commun. 2017, 47, 291–298. [Google Scholar] [CrossRef]
- Tornquist, B.L.; Bueno, G.P.; Willing, J.C.M.; Oliveira, I.M.; Stefani, H.A.; Rafique, J.; Saba, S.; Iglesis, B.I.; Botteselle, G.V.; Manrin, F. Ytterbium (III) triflate/Sodium Dodecyl Sulfate: A Versatile Recyclable and Water-Tolerant Catalyst for the Synthesis of Bis(indolyl)methanes (BIMs). ChemistrySelect 2018, 3, 6358–6363. [Google Scholar] [CrossRef]
- Godoi, M.; Botteselle, G.V.; Rafique, J.; Rocha, M.S.T.; Pena, J.P.; Braga, A.L. Solvent-Free Fmoc Protection of Amines Under Microwave Irradiation. Asian J. Org. Chem. 2013, 2, 746–749. [Google Scholar] [CrossRef]
- Wakabayashi, K.; Yorimitsu, H.; Oshima, K. Cobalt-Catalyzed Tandem Radical Cyclization and Cross-Coupling Reaction: Its Application to Benzyl-Substituted Heterocycles. J. Am. Chem. Soc. 2001, 123, 5374–5375. [Google Scholar] [CrossRef]
- Ohmiya, H.; Wakabayashi, K.; Yorimitsu, H.; Oshima, K. Reductive cyclization caused by cobaloxime. I. A new method for the synthesis of .beta.-methylene-.gamma.-butyrolactones. Tetrahedron 2006, 62, 2207–2213. [Google Scholar] [CrossRef]
- Shirakawa, E.; Sato, T.; Imazaki, Y.; Kimura, T.; Hayashi, T. Cobalt-catalyzed cross-coupling of alkynyl Grignard reagents with alkenyl triflates. Chem. Commun. 2007, 4513–4515. [Google Scholar] [CrossRef]
- Chen, C.; Huang, Y.; Zhang, Z.; Xiu-Qin Dong, X.-Q.; Zhang, X. Cobalt-catalyzed (Z)-selective semihydrogenation of alkynes with molecular hydrogen. Chem. Commun. 2017, 53, 4612–4615. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Zhou, Y.; Li, Y. PVC-NHC-Pd(0): An efficient and reusable heterogeneous catalyst for highly cis-selective semihydrogenation of alkynes using formic acid as hydrogen source. Inorg. Chem. Commun. 2021, 134, 109014. [Google Scholar] [CrossRef]
- Wu, J.; Li, D.; Zhang, D. Aqueous Wittig reactions of aldehydes with in situ formed semistabilized phosphorus ylides. Synth. Commun. 2005, 35, 2543–2551. [Google Scholar] [CrossRef]
- Goraia, M.; Franzenb, J.H.; Rotering, P.; Rüffera, T.; Dielmann, F.; Teichert, J.F. Broadly Applicable Copper(I)-Catalyzed Alkyne Semihydrogenation and Hydrogenation of α,β-Unsaturated Amides Enabled by Bifunctional Iminopyridine Ligands. J. Am. Chem. Soc. 2025, 147, 14481–14490. [Google Scholar] [CrossRef]
- Yu, X.; Tang, J.; Jin, X.; Yamamoto, Y.; Bao, M. Manganese-Catalyzed C–H Cyanation of Arenes with N-Cyano-N-(4-methoxy)phenyl-p-toluenesulfonamide. Asian J. Org. Chem. 2018, 7, 550–553. [Google Scholar] [CrossRef]







































Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).