


Agapanthussaponin A from the Underground Parts of Agapanthus africanus Induces Apoptosis and Ferroptosis in Human Small-Cell Lung Cancer Cells


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
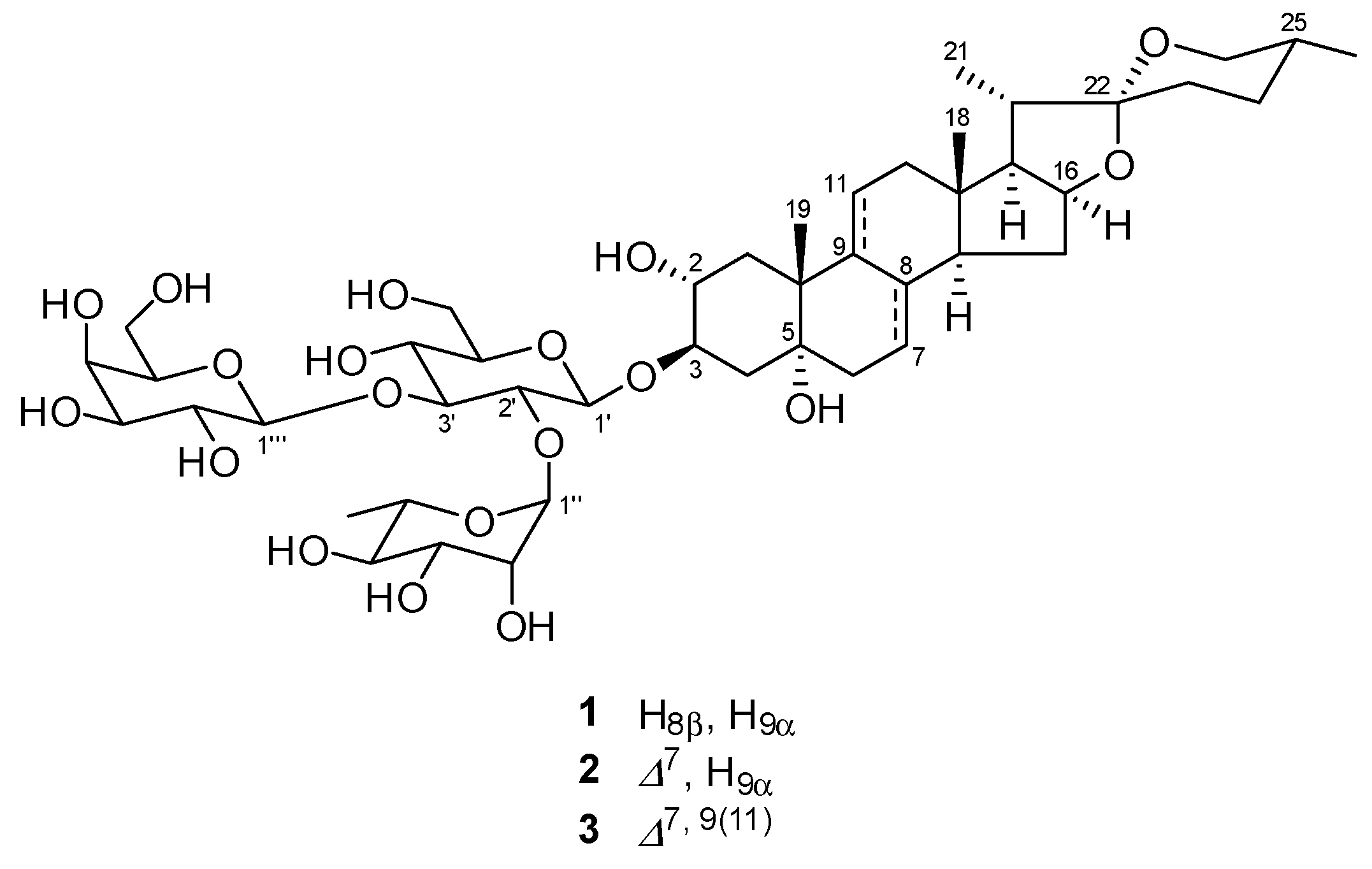
2.1. Structure Identification
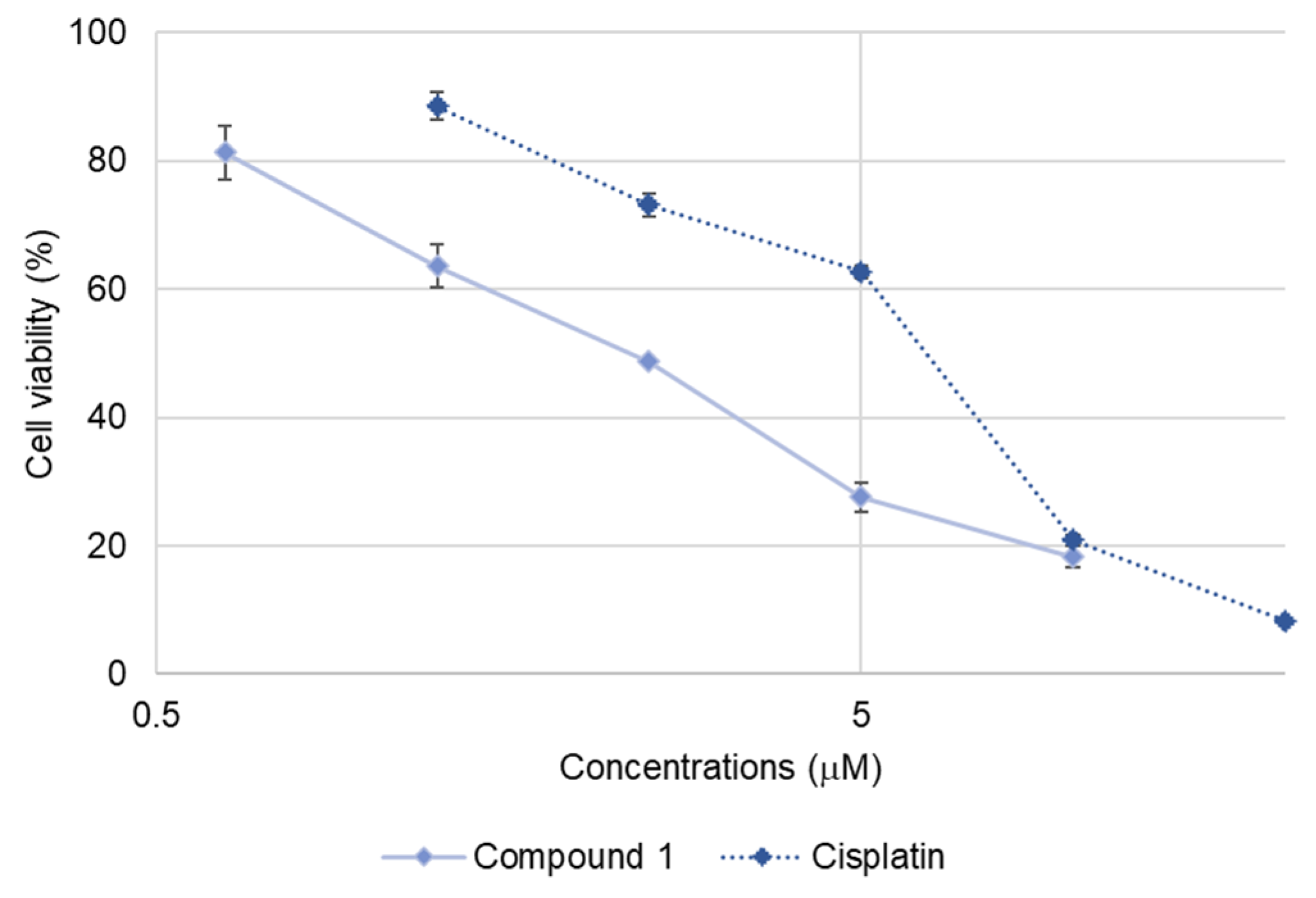
2.2. Cytotoxicity of 1–3
2.3. Apoptosis-Inducing Activity of 1
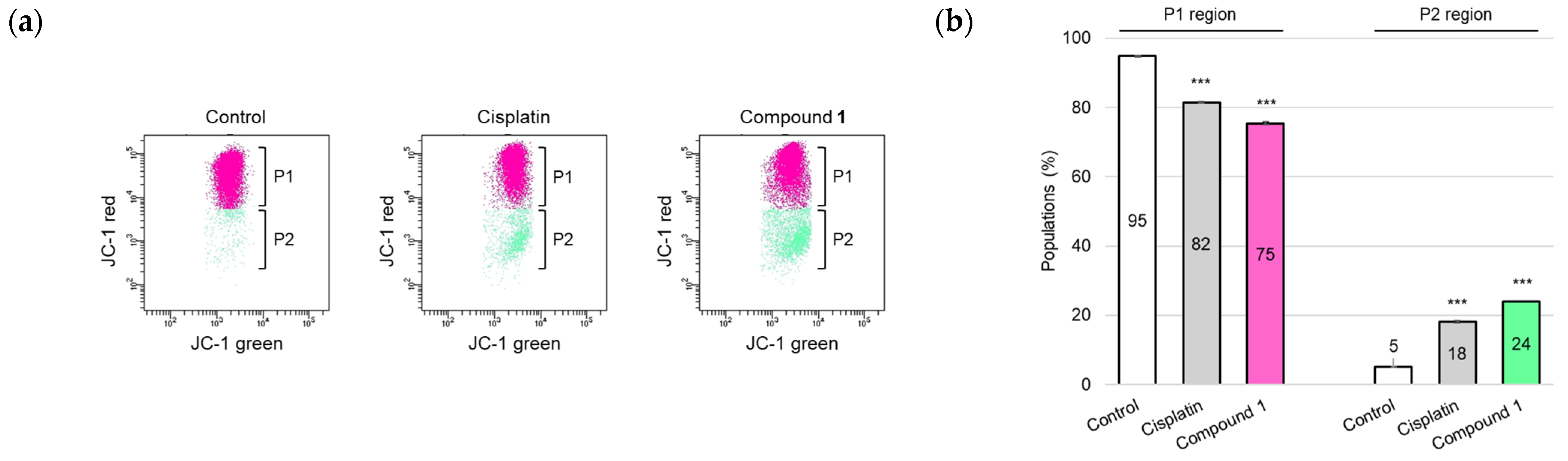
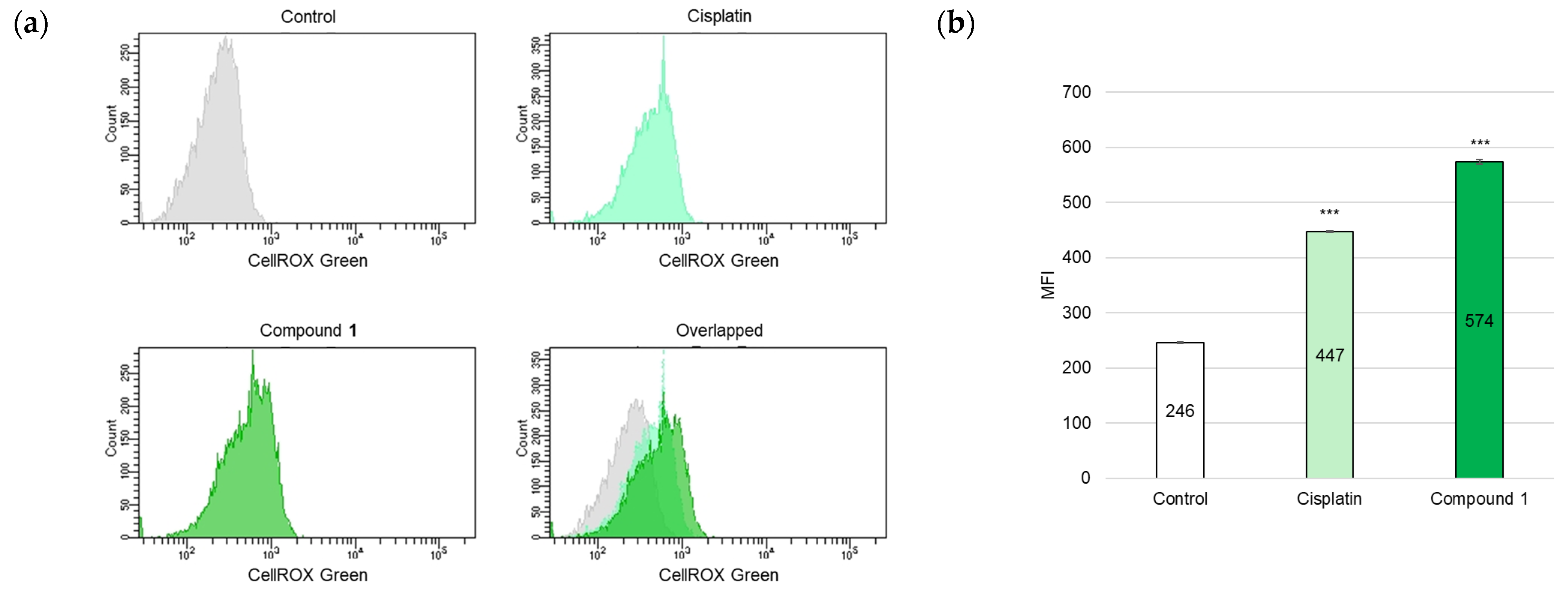
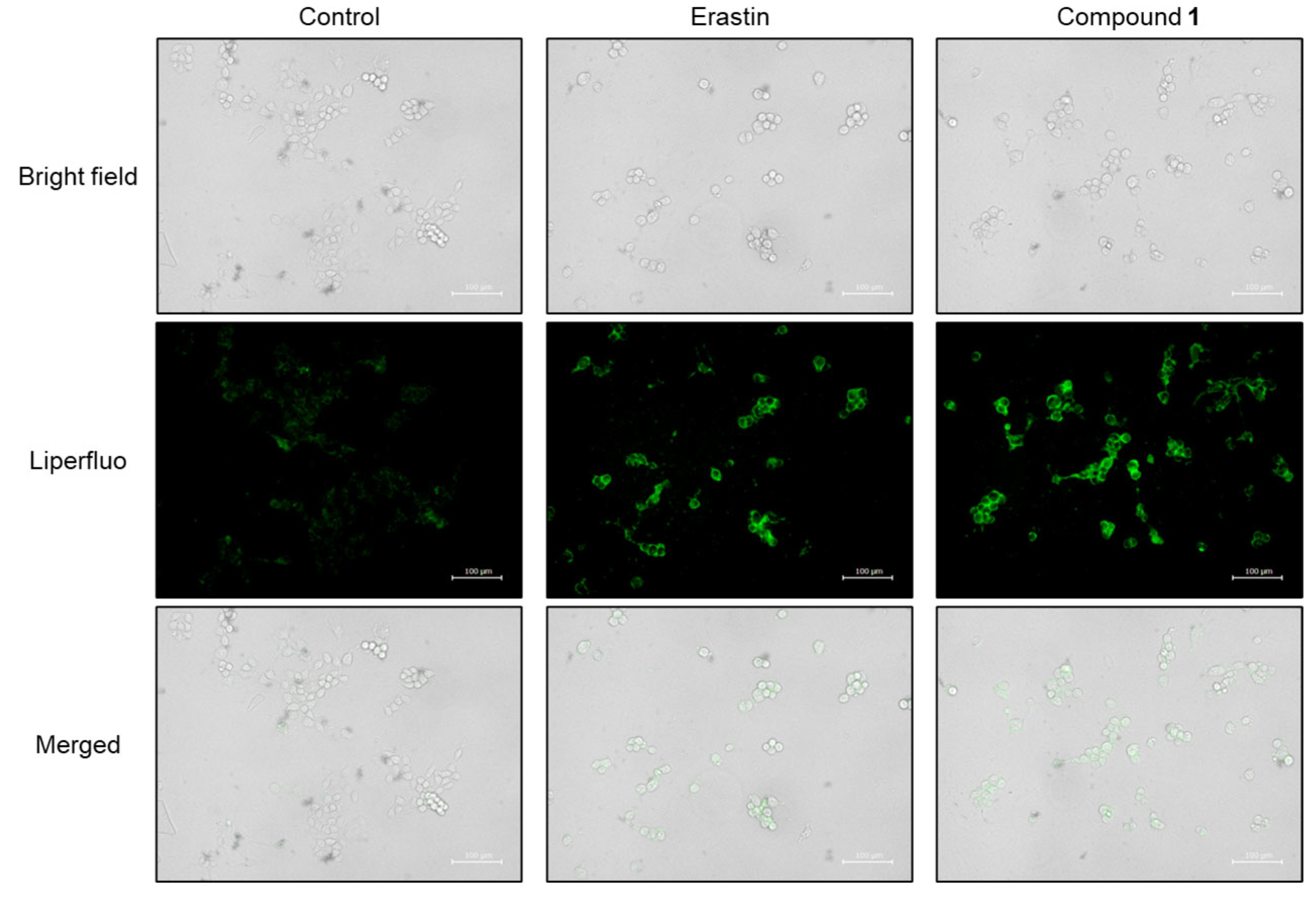
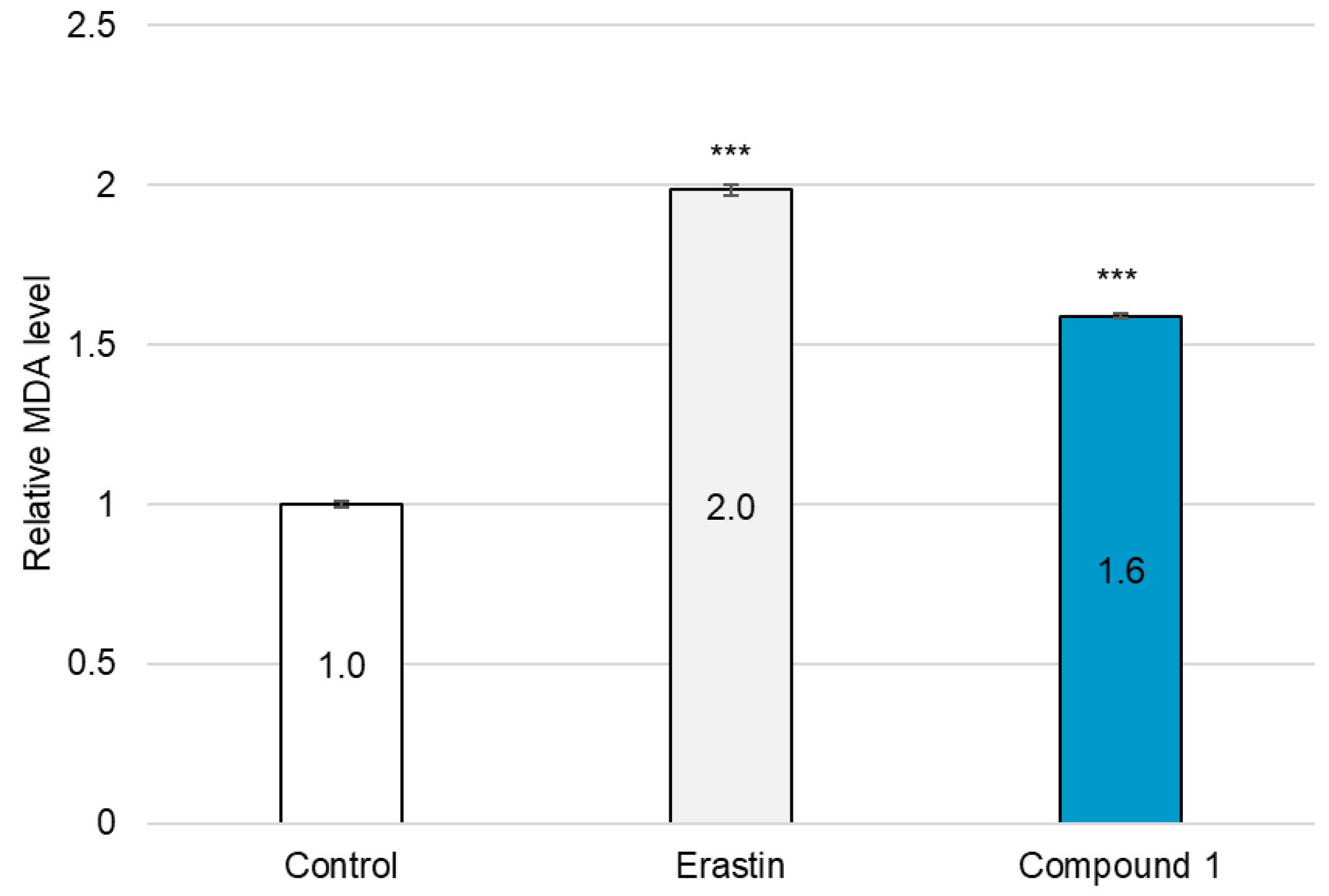
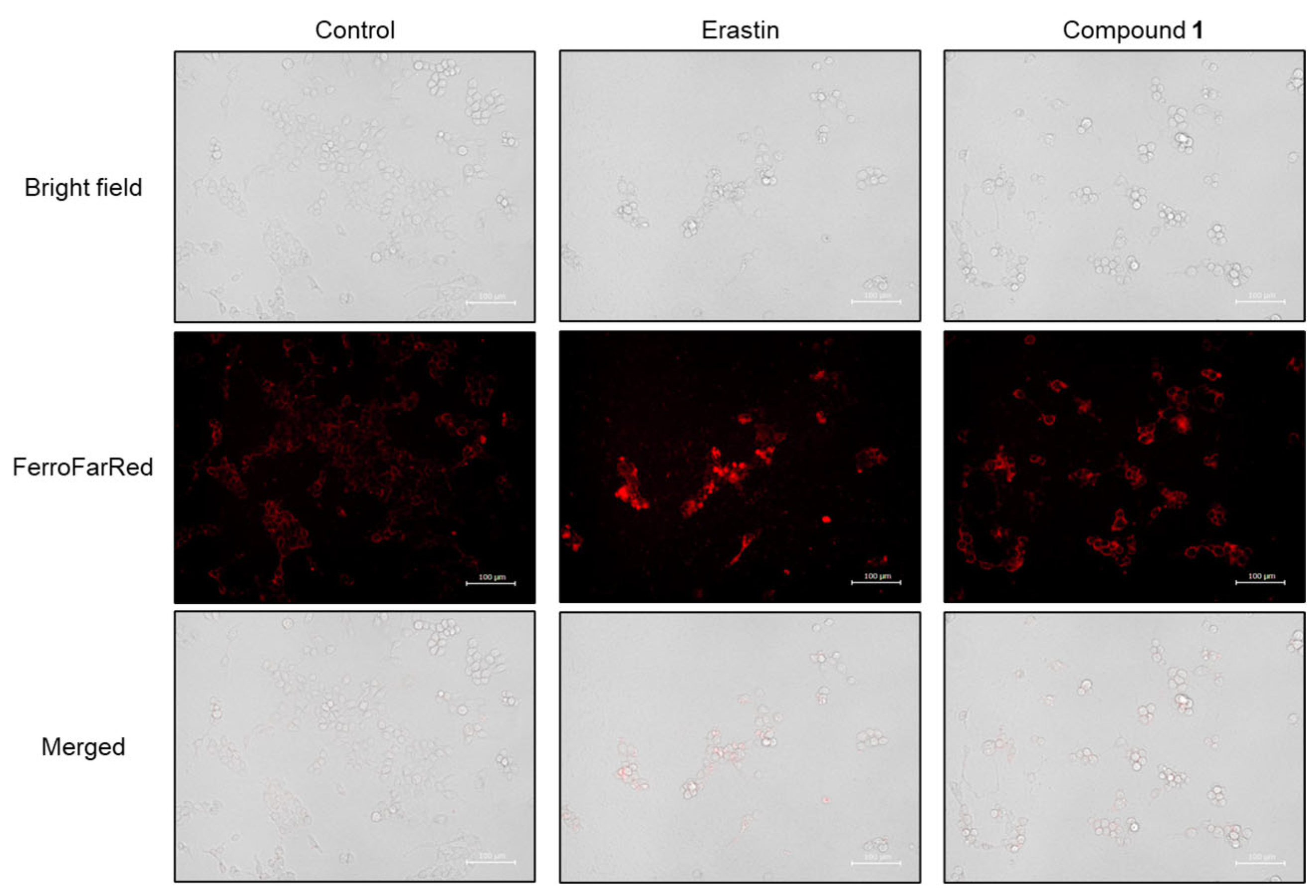
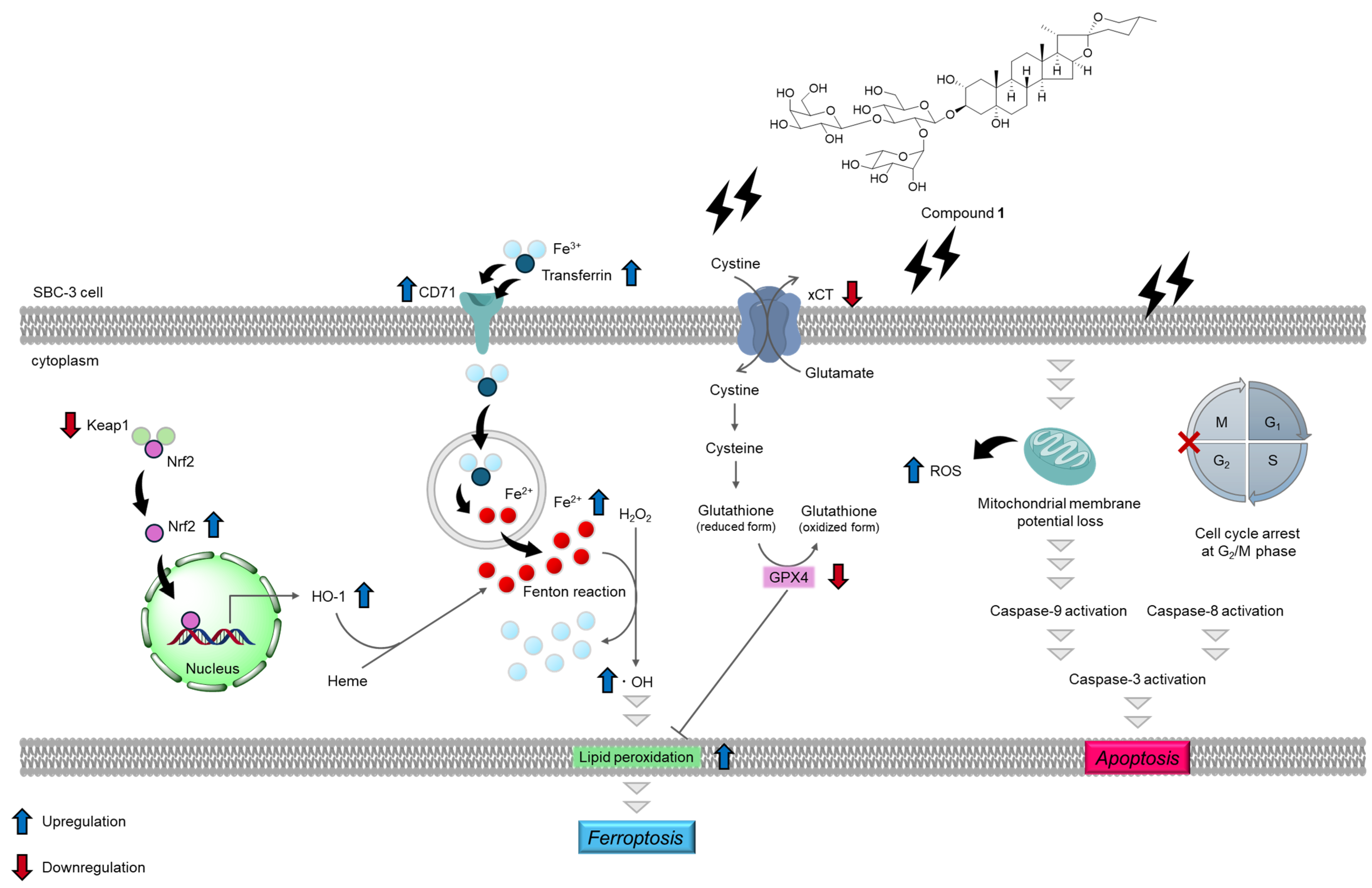
2.4. Ferroptosis-Inducing Activity of 1
3. Materials and Methods
3.1. General
3.2. Plant Material
3.3. Extraction and Isolation
3.4. Cell Culture and Cytotoxic Activity Assay
3.5. DAPI Staining
3.6. Apoptosis Detection by Annexin V-FITC and PI Double Staining
3.7. Analysis of Cell Cycle
3.8. Detection of Activated Caspases
3.9. Detection of Mitochondrial Membrane Potential
3.10. Detection of ROS Generation Levels
3.11. Detection of Hydroxyl Radicals
3.12. Detection of Lipid Peroxides
3.13. Quantification of Malondialhyde
3.14. Detection of Cellular Fe2+
3.15. Detection of Ferroptosis-Associated Proteins
3.16. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemel, A. Cancer Statistics. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar]
- Veronika, P.; Dorothea, P.M.; Clemens, A.; Joachim, W.; Kristiina, B.; Zsolt, M.; Balazs, D. Prophylactic cranial irradiation for small cell lung cancer in the era of immunotherapy and molecular subtypes. Curr. Opin. Oncol. 2025, 37, 27–34. [Google Scholar]
- Wang, Q.; Gumus, Z.H.; Colarossi, C.; Memeo, L.; Wang, X.; Kong, C.Y.; Boffetta, P. SCLC: Epidemiology, risk factors, genetic susceptibility, molecular pathology, screening, and early detection. J. Thorac. Oncol. 2023, 18, 31–46. [Google Scholar] [CrossRef]
- Bertaglia, V.; Petrelli, F.; Dottorini, L.; Carnio, S.; Morelli, A.M.; Nepote, A.; Maccioni, A.; Scartozzi, M.; Solinas, C.; Novello, S. Chemotherapy plus immunotherapy as first line combination in order patients with extensive stage small cell lung cancer: A systematic review and meta-analysis. Semin. Oncol. 2025, 52, 14–18. [Google Scholar] [CrossRef]
- Qiang, M.; Liu, H.; Yang, L.; Wang, H.; Guo, R. Immunotherapy for small cell lung cancer: The current state and future trajectories. Discov. Oncol. 2024, 15, e355. [Google Scholar] [CrossRef]
- Takahashi, N.; Iguchi, T.; Kuroda, M.; Mishima, M.; Mimaki, Y. Novel oleanane-type triterpene glycosides from the Saponaria officinalis L. seeds and apoptosis-inducing activity via mitochondria. Int. J. Mol. Sci. 2022, 23, 2047. [Google Scholar] [CrossRef] [PubMed]
- Iguchi, T.; Kuroda, M.; Morita, H.; Mimaki, Y. Phenolic compounds from the underground parts of Eremuru robustus and their cytotoxicity. Jpn. J. Pharmacog. 2022, 76, 45–46. [Google Scholar]
- Shimazaki, T.; Iguchi, T.; Takahashi, Y.; Yamamoto, K.; Takahashi, N.; Mimaki, Y. Determination of structure and cytotoxicity of ten undescribed steroidal glycosides from Allium cristophii×A. macleanii ‘Globemaster’. Molecules 2023, 28, 6248. [Google Scholar] [CrossRef]
- Shimazaki, T.; Iguchi, T.; Kanda, A.; Yamamoto, K.; Takahashi, N.; Mimaki, Y. Six unprecedented steroidal glycosides from Allium atropurpureum bulbs and their cytotoxicities against SBC-3 human small-cell lung cancer cells. Phytochem. Lett. 2023, 57, 200–209. [Google Scholar] [CrossRef]
- Shimazaki, T.; Iguchi, T.; Takahashi, N.; Sano, Y.; Nakamura, K.; Mimaki, Y. Steroidal glycosides from Ornithogalum thyrsoides bulbs and their cytotoxicity toward HL-60 human promyelocytic leukemia cells and SBC-3 human small-cell lung cancer cells. Phytochemistry 2024, 219, e113985. [Google Scholar] [CrossRef]
- Yoshizawa, Y.; Yokosuka, A.; Inomata, M.; Iguchi, T.; Mimaki, Y. Steroidal constituents in the whole plants of Helleborus niger and their cytotoxic activity in vitro. Phytochemistry 2025, 229, e114272. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Iguchi, T.; Nagamine, A.; Shirai, R.; Nagata, A.; Yamauchi, J.; Mimaki, Y. Structure elucidation of 16 undescribed steroidal glycosides from the underground parts of Agapanthus africanus and apoptosis-inducing activity in small-cell lung cancer cell. ACS Omega 2023, 8, 2808–2830. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Zhou, Y.; Chen, W.; Feng, S.; Liu, S.; Chen, C.; Yao, B.; Shen, X.L. Ochratoxin A-induced mitochondrial pathway apoptosis and ferroptosis by promoting glycolysis. Apoptosis 2025, 30, 1440–1452. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Yu, T.; Zhu, R.; Lu, J.; Ouyang, X.; Zhang, Z.; Chen, Q.; Li, J.; Cui, J.; Jiang, F.; et al. Timosaponin AIII promotes non-small-cell lung cancer ferroptosis through targeting and facilitating HSP90 mediated GPX4 ubiquitination and degradation. Int. J. Biol. Sci. 2023, 19, 1471–1489. [Google Scholar] [CrossRef]
- Wei, G.; Sun, J.; Hou, Z.; Luan, W.; Wang, S.; Cui, S.; Cheng, M.; Liu, Y. Novel antitumor compound optimized from natural saponin albiziabioside A induced caspase-dependent apoptosis and ferroptosis as a p53 activator through the mitochondrial pathway. Eur. J. Med. Chem. 2018, 157, 759–772. [Google Scholar] [CrossRef]
- Zhou, Y.; Yang, J.; Chen, C.; Li, Z.; Chen, Y.; Zhang, X.; Wang, L.; Zhou, J. Polyphyllin III-induced ferroptosis in MDA-MB-231 triple-negative breast cancer cells can be protected against by KLF4-mediated upregulation of xCT. Front. Pharmacol. 2021, 12, e670224. [Google Scholar]
- Xie, Y.; Chen, G. Dioscin induces ferroptosis and synergistic cytotoxicity with chemotherapeutics in melanoma cells. Biochem. Biophys. Res. Commun. 2021, 557, 213–220. [Google Scholar] [CrossRef]
- Chen, H.C.; Tang, H.H.; Hsu, W.H.; Wu, S.Y.; Cheng, W.H.; Wang, B.Y.; Su, C.L. Vulnerability of triple-negative breast cancer to saponin formosanin C-induced ferroptosis. Antioxidants 2022, 11, 298. [Google Scholar] [CrossRef]
- Yan, C.; Xuan, F. Paris saponin VII promotes ferroptosis to inhibit breast cancer via Nrf2/GPX4 axis. Biochem. Biophys. Res. Commun. 2024, 697, e149524. [Google Scholar] [CrossRef]
- Nakamura, O.; Mimaki, Y.; Sashida, Y.; Nikaido, T.; Ohmoto, T. Agapanthussaponin A-D, new potent cAMP phosphodiesterase inhibitors from the underground parts of Agapanthus inapertus. Chem. Pharm. Bull. 1993, 41, 1784–1789. [Google Scholar] [CrossRef]
- Koopman, G.; Reutelingsperger, C.P.M.; Kuijten, G.A.M.; Keehnen, R.M.J.; Pals, S.T.; van Oers, M.H.J. Annexin V for flow cytometric detection of phophatidylserine expression on B cells undergoing apoptosis. Blood 1994, 84, 1415–1420. [Google Scholar] [CrossRef] [PubMed]
- McIlwain, D.R.; Berger, T.; Mak, W.T. Caspase function in cell death and disease. Cold Spring Harb. Perspect. Biol. 2013, 5, a008656. [Google Scholar] [CrossRef]
- Eeva, J.; Nuutinen, U.; Ropponen, A.; Mättö, M.; Eray, M.; Pellinen, R.; Wahlfors, J.; Pelkonen, J. The involvement of mitochondrial and the caspase-9 activation pathway in rituximab-induced apoptosis in FL cells. Apoptosis 2009, 14, 687–698. [Google Scholar] [CrossRef]
- Zorova, D.L.; Popkov, A.V.; Plotnikov, Y.E.; Silachev, N.D.; Pevzner, B.I.; Jankauskas, S.S.; Babenko, A.V.; Zorov, D.S.; Balakireva, V.A.; Juhaszova, M.; et al. Mitochondrial membrane potential. Anal. Biochem. 2018, 552, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Elefantova, K.; Lakatos, B.; Kubickova, J.; Sulova, Z.; Breier, A. Detection of the mitochondrial membrane potential by the cationic dye JC-1 in L1210 cells with massive overexpression of the plasma membrane ABCB1 drug transporter. Int. J. Mol. Sci. 2018, 19, 1985. [Google Scholar] [CrossRef]
- Fleury, C.; Mignotte, B.; Vayssière, J.L. Mitochondrial reactive oxygen species in cell death signaling. Biochimie 2014, 84, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Endale, T.H.; Tesfaya, W.; Mengstie, A.T. ROS induced lipid peroxidation and their role in ferroptosis. Front. Cell Dev. Biol. 2023, 11, e1226044. [Google Scholar] [CrossRef]
- Wang, L.L.; Mai, Y.Z.; Zheng, M.H.; Wu, X.; Jin, J.Y. Fluorescent probe disclosing hydroxyl radical generation in mitochondria and nucleoli of cells during ferroptosis. Sens. Actuators B: Chem. 2022, 373, e132707. [Google Scholar] [CrossRef]
- Ma, W.; Hu, N.; Xu, W.; Zhao, L.; Tian, C. Ferroptosis inducers: A new frontier in cancer therapy. Bioorg. Chem. 2024, 146, e107331. [Google Scholar] [CrossRef]
- Mortensen, S.M.; Ruiz, J.; Watts, L.J. Polyunsaturated fatty acids drive lipid peroxidation during ferroptosis. Cell 2023, 12, 804. [Google Scholar] [CrossRef]
- Tsikas, D. Assessment of lipid peroxidation by measuring malondialdehyde (MDA) and relatives in biological samples: Analytical and biological challenges. Anal. Biochem. 2017, 524, 13–30. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Zhao, B.; Zhou, L.; Zhang, Z.; Shen, Y.; Lv, H.; AlQudsy, L.H.H.; Shang, P. Ferroptosis, a novel pharmacological mechanism of anti-cancer drugs. Cancer Lett. 2020, 483, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Schorpp, K.; Jin, J.; Yozwiak, E.C.; Hoffstrom, G.B.; Decker, M.A.; Rajbhandari, P.; Stokes, E.M.; Bender, G.H.; Csuka, M.J.; et al. Transferrin receptor is a specific ferroptosis marker. Cell rep. 2020, 30, 3411–3423. [Google Scholar] [CrossRef]
- Bellezza, I.; Giambanco, I.; Minelli, A.; Donato, R. Nrf2-Keap1 signaling in oxidative and reductive stress. BBA-Molecular Cell Research 2018, 1865, 721–733. [Google Scholar] [CrossRef]
- Jayasuriya, R.; Dhamodharan, U.; Ali, D.; Ganesan, K.; Xu, B.; Ramkumar, M.K. Targeting Nrf2/Keap1 signaling pathway by bioactive natural agents: Possible therapeutic strategy to combat liver disease. Phytomedicine 2021, 92, e153755. [Google Scholar] [CrossRef]
- Chiang, S.K.; Chen, S.E.; Chang, L.C. A dual role of heme oxygenase-1 in cancer cells. Int. J. Mol. Sci. 2019, 20, 39. [Google Scholar] [CrossRef] [PubMed]
- Koppula, P.; Zhuang, L.; Gan, B. Cystine transporter SLC7A11/xCT in cancer: Ferroptosis, nutrient dependency, and cancer therapy. Protein Cell 2021, 12, 599–620. [Google Scholar] [CrossRef]
- Lu, B.; Chen, B.X.; Ying, D.M.; He, J.Q.; Cao, J.; Yang, B. The role of ferroptosis in cancer development and treatment response. Front. Pharmacol. 2018, 8, e992. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iguchi, T.; Shimazaki, T.; Mimaki, Y. Agapanthussaponin A from the Underground Parts of Agapanthus africanus Induces Apoptosis and Ferroptosis in Human Small-Cell Lung Cancer Cells. Molecules 2025, 30, 3189. https://doi.org/10.3390/molecules30153189
Iguchi T, Shimazaki T, Mimaki Y. Agapanthussaponin A from the Underground Parts of Agapanthus africanus Induces Apoptosis and Ferroptosis in Human Small-Cell Lung Cancer Cells. Molecules. 2025; 30(15):3189. https://doi.org/10.3390/molecules30153189
Chicago/Turabian StyleIguchi, Tomoki, Tamami Shimazaki, and Yoshihiro Mimaki. 2025. "Agapanthussaponin A from the Underground Parts of Agapanthus africanus Induces Apoptosis and Ferroptosis in Human Small-Cell Lung Cancer Cells" Molecules 30, no. 15: 3189. https://doi.org/10.3390/molecules30153189
APA StyleIguchi, T., Shimazaki, T., & Mimaki, Y. (2025). Agapanthussaponin A from the Underground Parts of Agapanthus africanus Induces Apoptosis and Ferroptosis in Human Small-Cell Lung Cancer Cells. Molecules, 30(15), 3189. https://doi.org/10.3390/molecules30153189