
A Review of Natural and Synthetic Chalcones as Anticancer Agents Targeting Topoisomerase Enzymes


Abstract
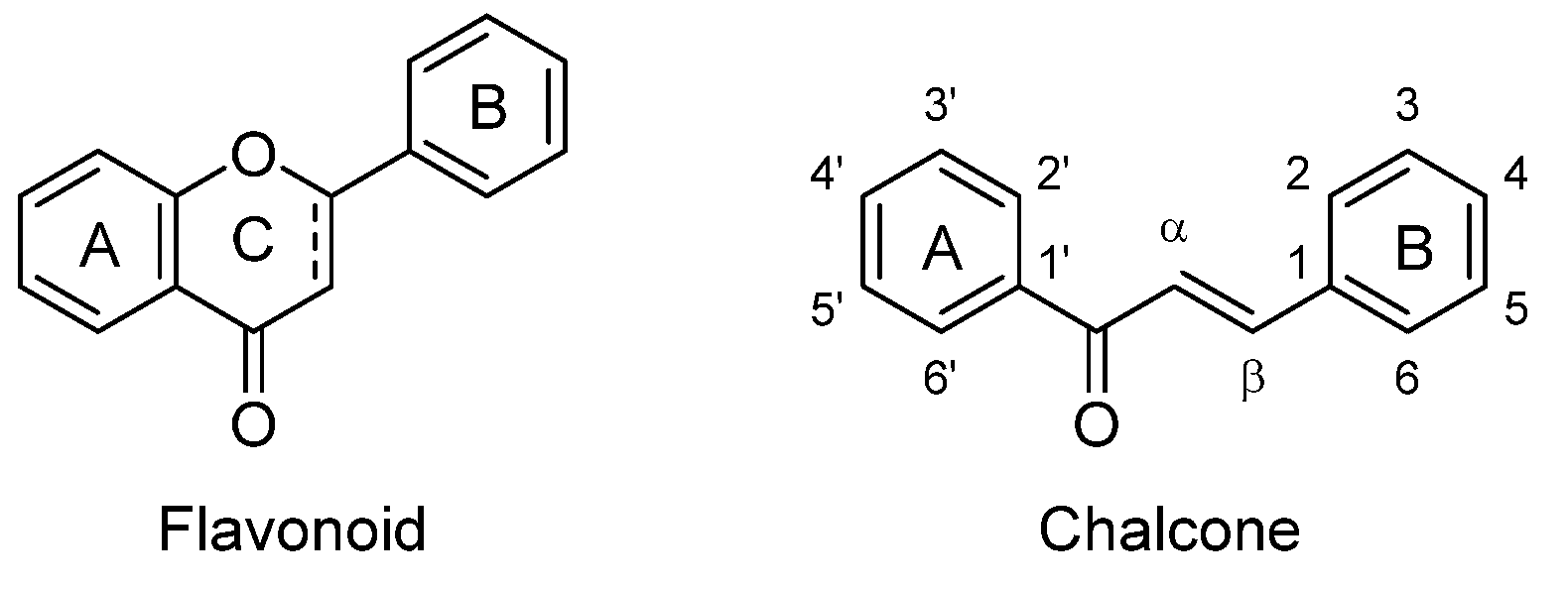
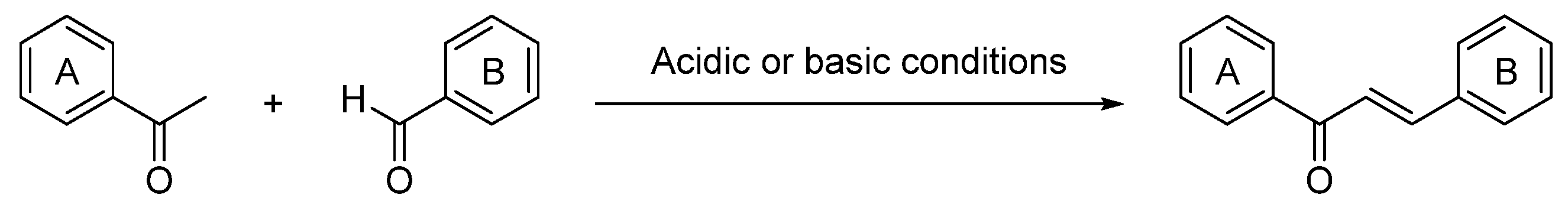
1. Introduction
- toward the electron density on the ring and consequently the electrophilicity of the α,β-unsaturated ketone system, affecting the binding ability and the biological activity of chalcones;
- on the capacity of chalcones to establish hydrogen bonds with pertinent residues (amino acids) of proteins or active sites of enzymes.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C. No (Name) | Structure | Activity on Topo: IC50 (µM) or % Inhibition | Activity on Cancer Cells IC50 (µM) | Ref. |
|---|---|---|---|---|
| 1 (camptothecin) | 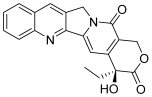 | Topo I: 5.7 µM 72% at 100 µM 31% at 20 µM | HCT15: 7.1 BT474: 7.0 T47D: 4.1 | [5,15] |
| 2 (topotecan) | 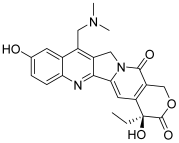 | Topo I: 3.2 µM Topo IIB: 99.1 µM | HCT116: 12.2 SR: 13.4 | [6,16] |
| 3 (doxorubicin) | 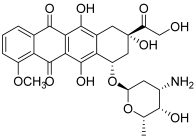 | Topo II: 0.9 µM | Caco-2: 0.7 Hep-2: 0.5 HepG2: 0.6 | [7] |
| 4 (etoposide) |  | Topo II: 34.5 µM 78% at 100 µM 39% at 20 µM | HCT15: 6.9 BT474: 6.6 T47D: 6.4 | [5,15] |
2. Natural Chalcones as Topoisomerase Inhibitors
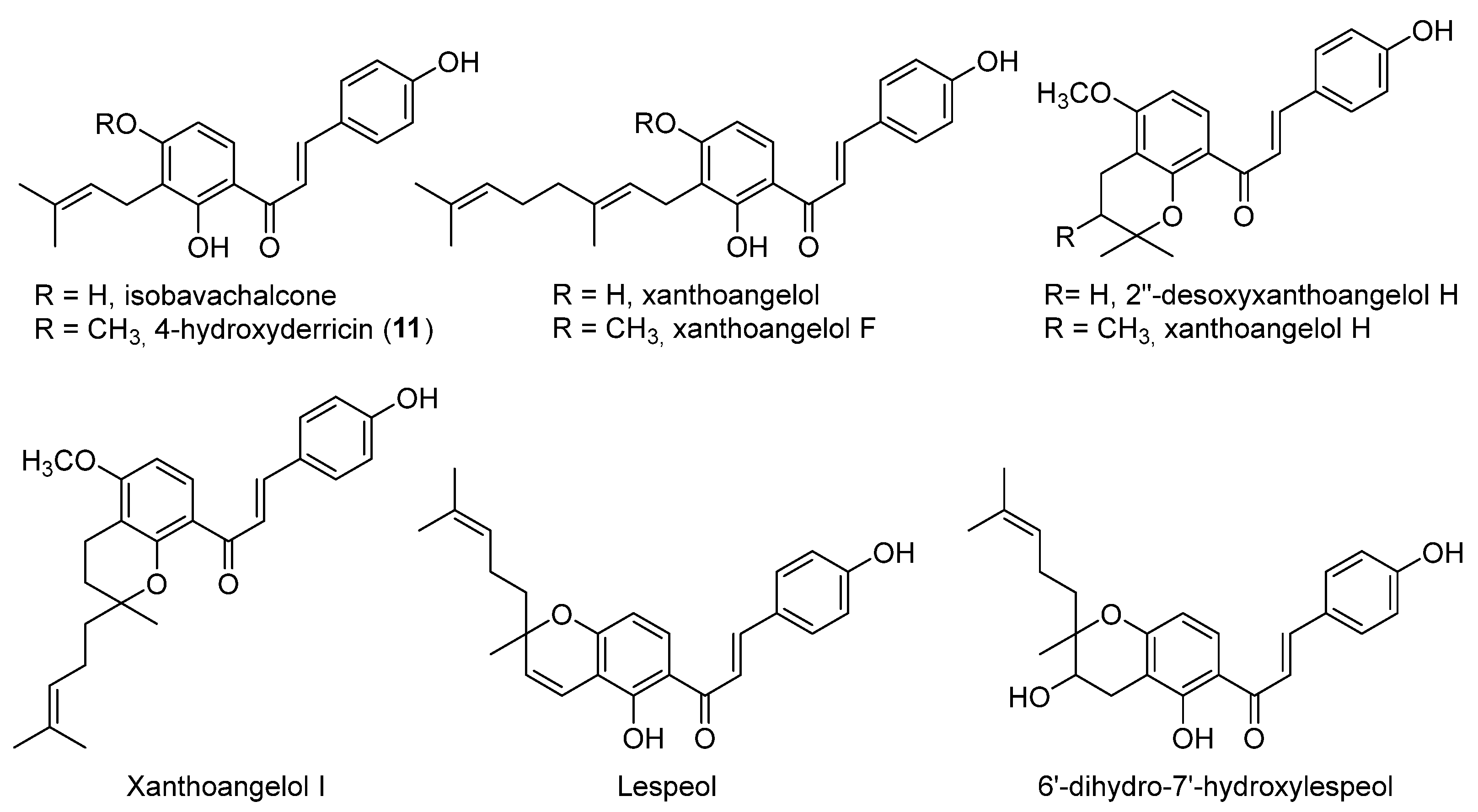
2.1. Natural Monomeric Chalcones
- xanthoangelol F bearing a geranyl moiety was inactive while 4-hydroxyderricine, with a prenyl substituent, was able to significantly inhibit topo II;
- derivatives of xanthoangelol H, which were inactive against topoisomerases, are characterized by a 3,4-dihydrobenzopyran ring and a 4-hydroxy group, compared to millepachine, a topo II inhibitor, which presents a benzopyran and a 4-methoxy substituent.
2.2. Natural Oligomeric Chalcones
| C. No (Name) | Structure | Activity on Topo: IC50 (µM) or % Inhibition | Activity on Cancer Cells IC50 (µM) | Ref. |
|---|---|---|---|---|
| 5 (licochalcone A) | 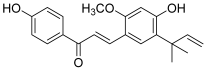 | Topo I: dose-dependent inhibition 18.5 to 295.5 µM | A549: 14.3 HCT-15: 10.1 SK-OV-3: 13.5 SK-MEL-2: 7.9 | [17] |
| 6 (licochalcone E) | 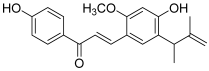 | A549: 17.3 HCT-15: 10.1 SK-OV-3: 15.5 SK-MEL-2: 8.5 | [17] | |
| 7 (isoliquiritigenin) | 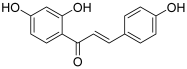 | Topo I: 178 µM | SNU475: 243 U87: 6.3 | [19,20] |
| 8 (echinatin) |  | Topo I: 45 µM | A549: 36.0 HCT116: 64.0 MCF-7: 46.0 | [21] |
| 9 (millepachine) | 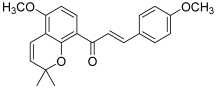 | Topo II: <100 µM dose-dependent inhibition 2 to 8 µM | A2780CP: 4.0 A2780S: 2.5 SK-OV-3: 4.0 | [24,25] |
| 10 (xanthohumol) | 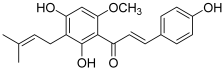 | Topo I: inhibition of activity with a concentration of 140 µM | A549: 12.1 SK-MEL-2: 14.4 HCT115: 10.2 SK-OV-3: 16.1 | [28] |
| 11 (4-hydroxyderricin) | 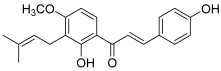 | Topo I: No activity Topo II: 21.9 µM | A549: 10.2 AZ521: 4.2 CRL1579: 4.6 HL-60: 5.5 | [5] |
| 12 (flavokawain B) | 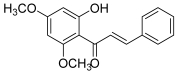 | Topo IIA | A549: 21.7 HepG2: 20.7 HuCCA-1: 19.6 MOLT-3: 10.0 | [29] |
| 13 (pauferrol A) | 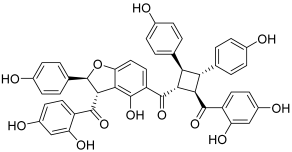 | Topo IIA: 2.1 µM | HL-60: 5.2 | [30] |
| 14–15 (pauferrol B and C) | 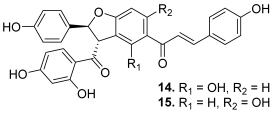 | Topo IIA: 14. 15.3 µM 15. 14.5 µM | HL-60: 14. 11.6 15. 12.1 | [31] |
| 16 (tomoroside A) | 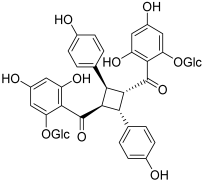 | Topo IIA: decreased mRNA expression in cancer cells | NCI-H460: 44.4 NCI-H460/R: 199 | [32] |
| 17 (carthorquinoside B) |  | Topo I: inhibition at 100 µM (no lower dose tests) | HeLa, HepG-2, A549, K562, and HCT116: between 3.13 and 200 | [34] |
3. Synthetic Chalcones as Topoisomerase Inhibitors
3.1. Synthetic Chalcones
| C. No (Name) | Structure | Activity on Topo: IC50 (µM) or % Inhibition | Activity on Cancer Cells IC50 (µM) | Ref. |
|---|---|---|---|---|
| 18 | 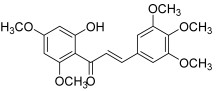 | Topo IIA ATPase: 7.5 nM | HeLa: 3.2 HT-1376: 10.8 MCF-7: 21.1 | [37] |
| 19 (salvicine) |  | Topo IIA ATPase: 326.5 nM | HeLa: 70.1 HT-1376: 106.5 MCF-7: >200 | [37,38] |
| 20 |  | Topo IIA: decreased mRNA expression at 5 µM | DLD-1: 0.3 HCT116: 0.3 HT-29: 0.7 LS174T: 0.4 | [39] |
| 21 | 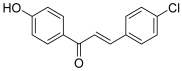 | Topo I: 76% at 3.9 mM | Jurkat: 9.3 | [41] |
| 22 | 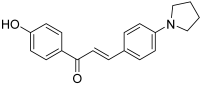 | Topo I: 75% at 100 µM 60% at 20 µM Topo II: 97% at 100 µM 19% at 20 µM | SNU638: 0.6 T47D: 1.4 | [42] |
| 23 |  | Topo I: 0% at 100 µM Topo II: 61% at 100 µM 21% at 20 µM | DU145: 1.1 HCT15: 0.2 K562: 7.6 MCF-7: 0.5 | [44] |
| 24 | 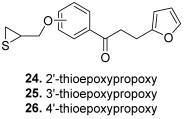 | Topo I: 61% at 100 µM 6% at 20 µM Topo II: 95% at 100 µM 31% at 20 µM | MDA-MB-231: 32.2 MDA-MB-468: 8.3 T47D: 6.6 | [47] |
| 25 | Topo I: 75% at 100 µM 1% at 20 µM Topo II: 90% at 100 µM 30% at 20 µM | MDA-MB-231: 28.9 MDA-MB-468: 7.2 T47D: 10.4 | ||
| 26 | Topo I: 64% at 100 µM 0% at 20 µM Topo II: 95% at 100 µM 25% at 20 µM | MDA-MB-231: 18.0 MDA-MB-468: 4.3 T47D: 9.4 | ||
| 27 |  | Topo I: 37% at 100 µM 24% at 20 µM Topo II: 82% at 100 µM 0% at 20 µM | MDA-MB-231: 28.7 MDA-MB-468: 48.3 T47D: 19.4 | [47] |
| 28 |  | Topo IIA ATPase | DU145: 6.9 | [48] |
| 29 |  | Topo IIA: decreased mRNA expression in cancer cells at 40 µM | DH82: 38.2 | [49] |
| 30 | 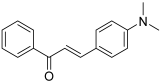 | Topo I: inhibition at 60 µM | - | [50] |
| 31–32 | 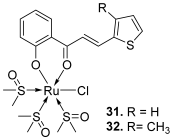 | Topo II: 31. 18 µM 32. 13 µM | - | [51] |
| 33 | 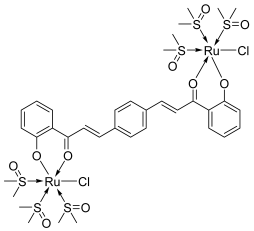 | Topo I: 87 µM | - | [54] |
| 34–35 | 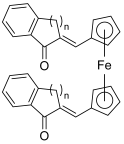 | Topo I: partial inhibition at 30 µM Topo II: partial inhibition at 5 µM | - | [55] |
3.2. Chalcone Hybrids as Topoisomerases Inhibitors
3.2.1. Fluoroquinolone Hybrids
3.2.2. Chalcone Hybrids Including Natural Compounds
3.2.3. Nitrogen Heterocycle-Based Hybrids
| C. No (Name) | Structure | Activity on Topo: IC50 (µM) or % Inhibition | Activity on Cancer Cells IC50 (µM) | Ref. |
|---|---|---|---|---|
| 36 (ciprofloxacin CP) | 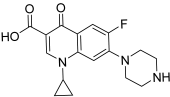 | Topo II: 104 µM | Hela: 300 MG63: 480 K562: >150 NCI-H460: 60.0 | [59] |
| 37 | 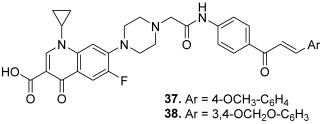 | Topo I: 68% at 100 µM 25% at 20 µM Topo II: 85% at 100 µM 50% at 20 µM | - | [15] |
| 38 | Topo I: 91% at 100 µM 26% at 20 µM Topo II: 86% at 100 µM 44% at 20 µM | - | ||
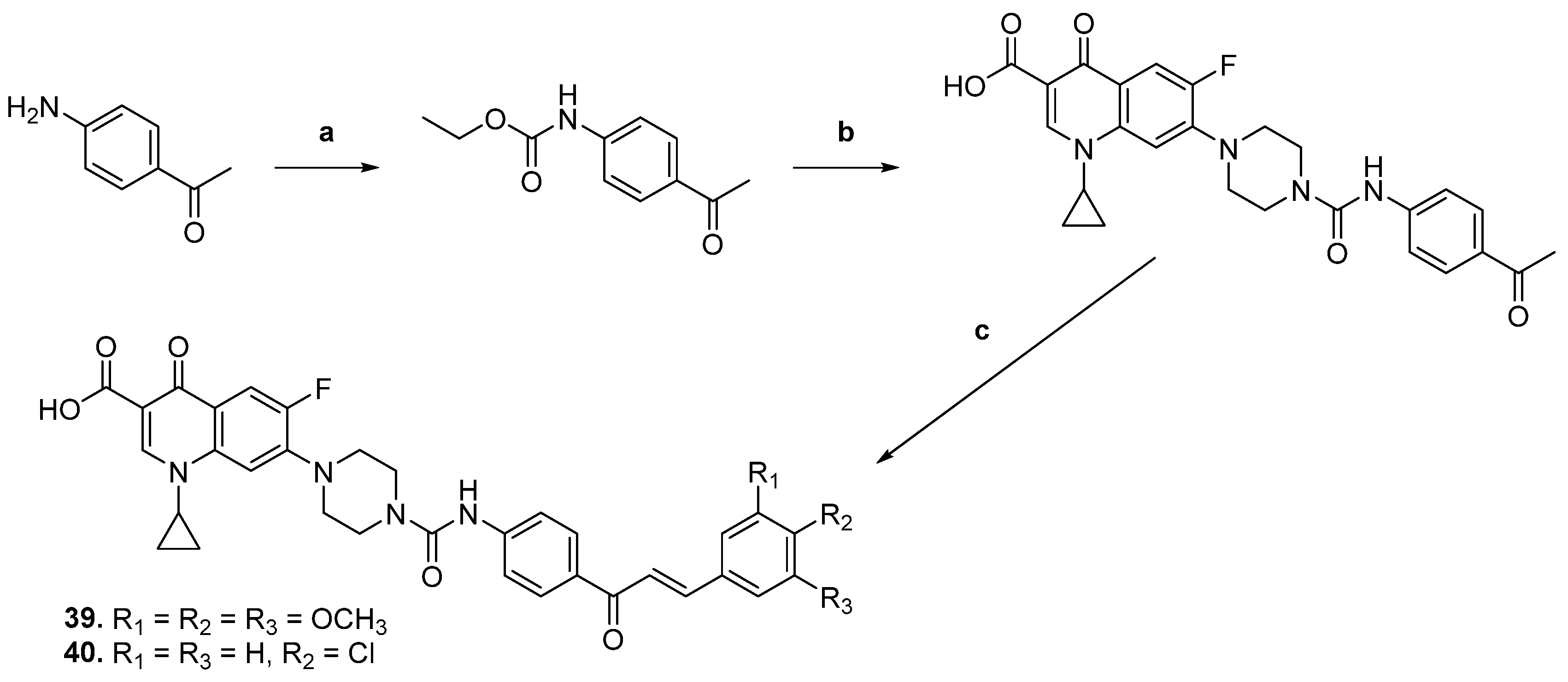
| 39 | 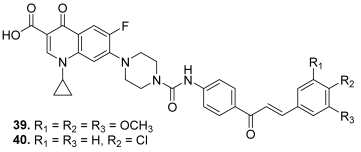 | Topo I: 16.0 µM Topo IIA: 137 µM | HCT116: 2.0 SR: 0.6 | [16] |
| 40 | Topo I: 18.0 µM Topo IIB: 107 µM | HCT116: 2.5 SR: 0.7 | ||
| Topo I: 37.5 µM Topo II: 19.9 µM | HCT116: 5.0 LOX IMVI: 1.3 | [60] | ||
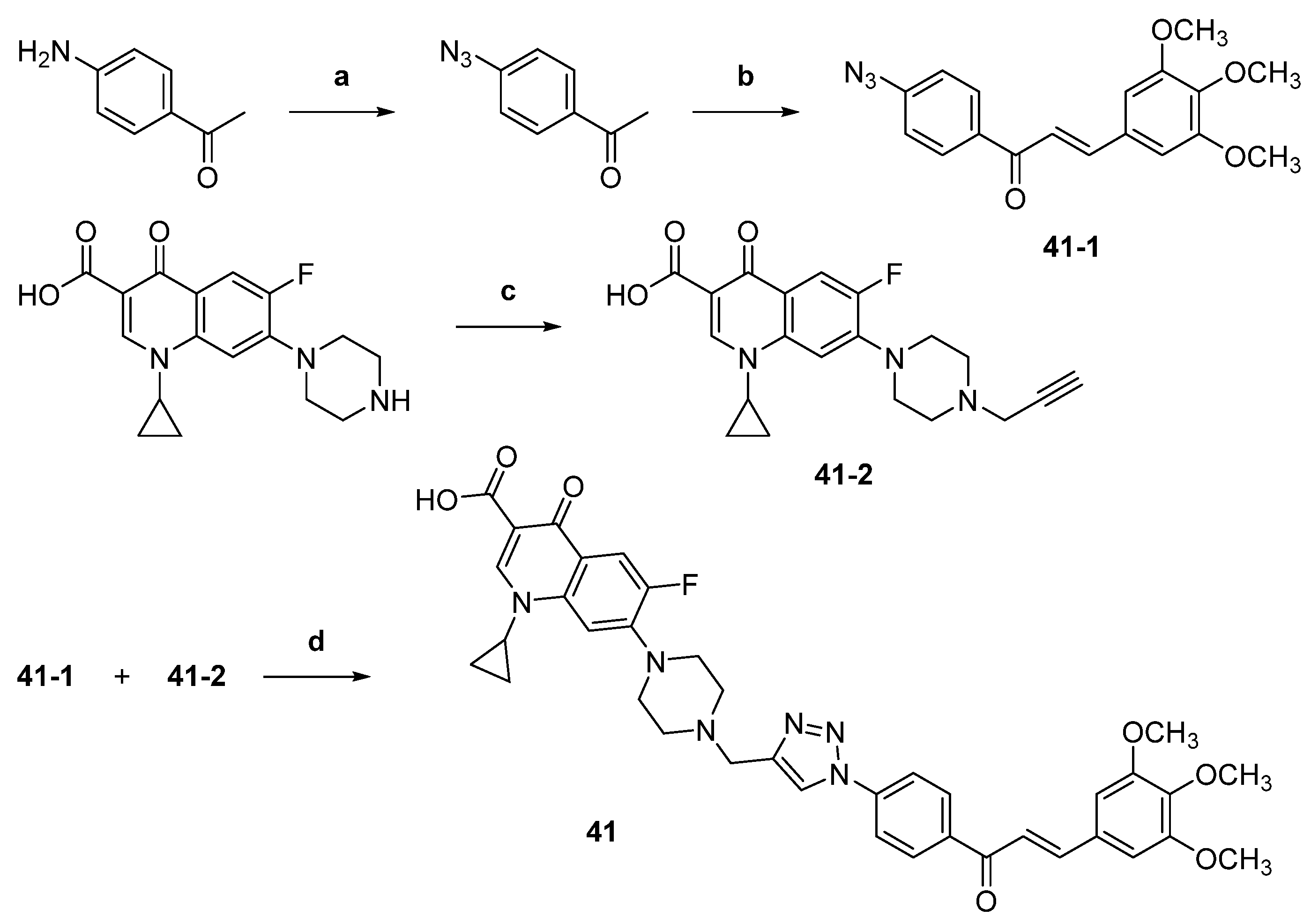
| 41 | 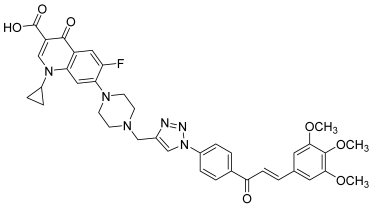 | Topo I: 25% at 2.5 µM Topo IIB: 100% at 100 µM 95% at 10 µM | Caco-2: 7.1 HCT116: 2.5 HT-29: 13.2 RPMI-8226: 0.4 µM | [61] |
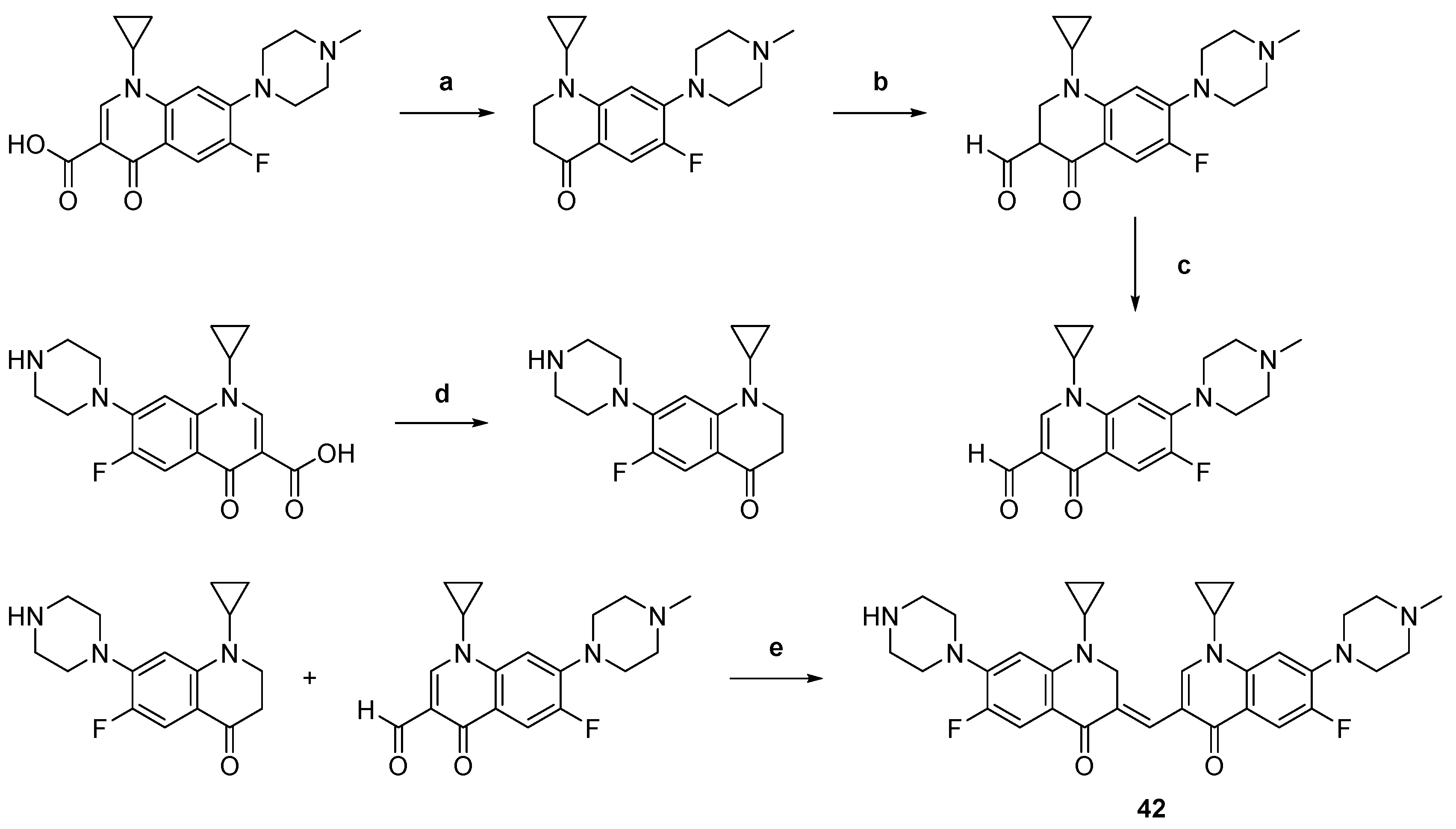
| 42 | 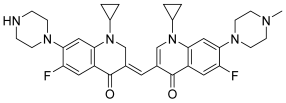 | Topo II: Partial inhibition at 1.6 μM, with Virtually complete inhibition at 3.2 μM | BGC-823: 3.8 Capan-1: 2.9 HGC-27: 5.6 Panc-1: 3.5 DU145: 3.2 T24: 3.7 | [63] |
| 43 | 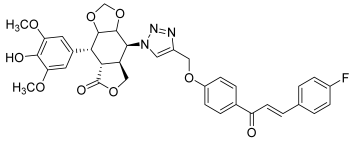 | Topo II | Colo-205: 12.5 DU145: 12.0 HCT-15: 12.4 HeLa: 4.5 SKN-SH: 0.4 SW-620: 0.4 | [65] |
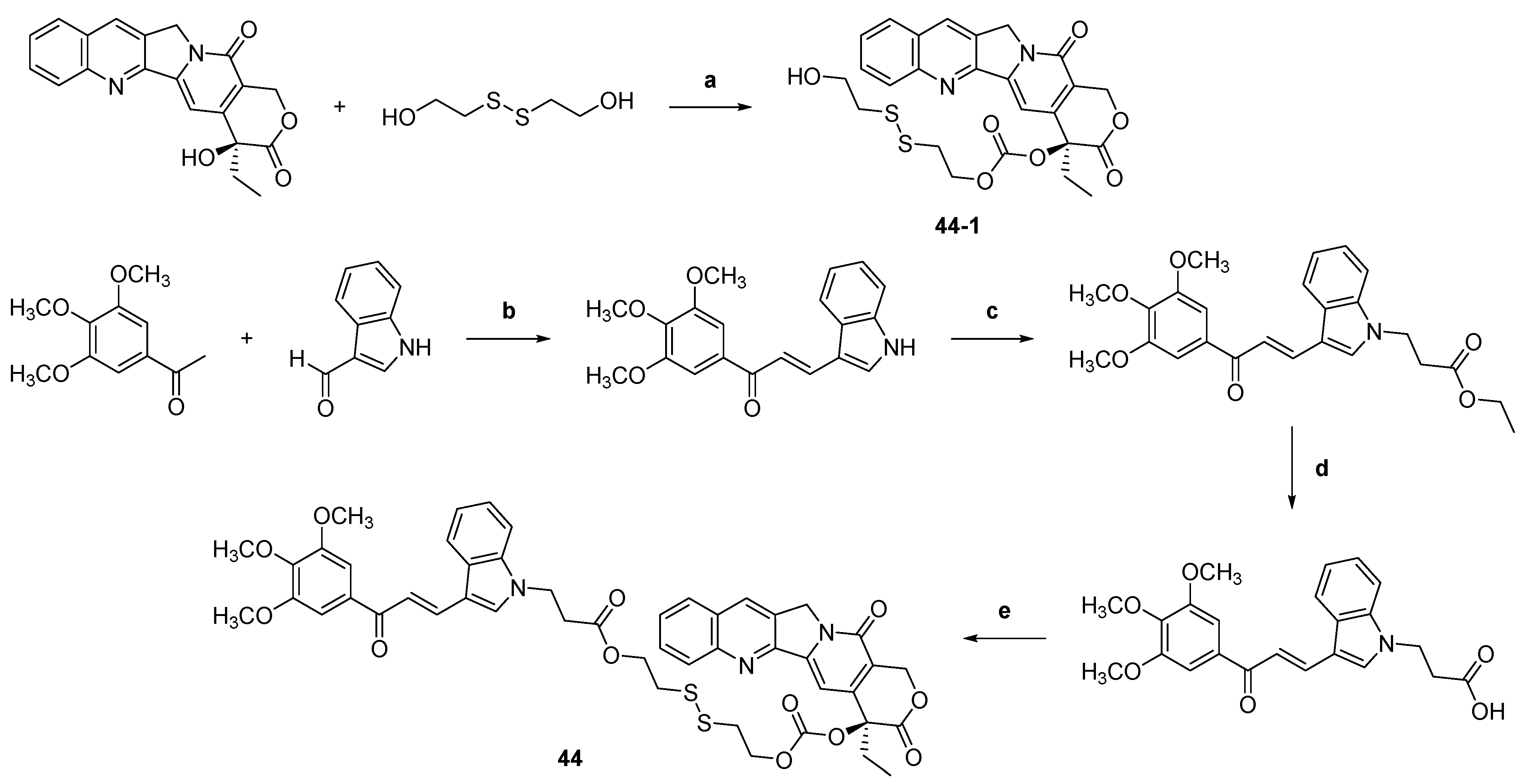
| 44 | 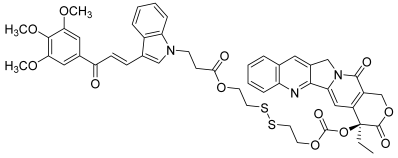 | Topo I: | A549: 0.4 A549/PTX: 0.6 HCT116: 0.2 HCT116/PTX: 0.3 | [66] |
| ||||
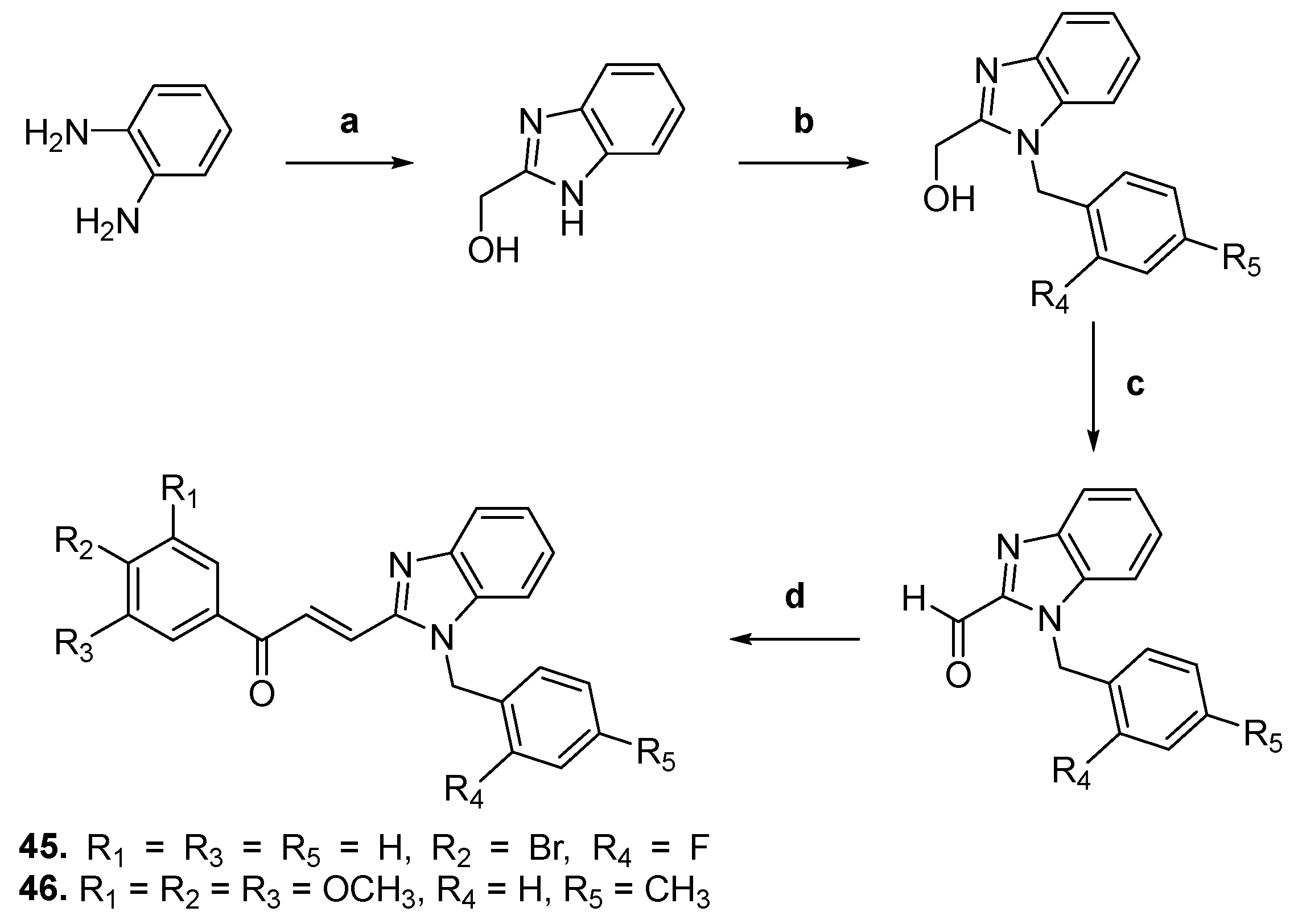
| 45 | 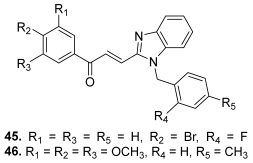 | Topo II: 87% at 20 µM | A549: 3.6 HePG2: 4.5 MG63: 4.7 LNCaP: 5.4 | [68] |
| 46 | Topo II: 95% at 20 µM | A549: 3.8 HePG2: 4.6 MG63: 4.1 LNCaP: 3.6 | ||
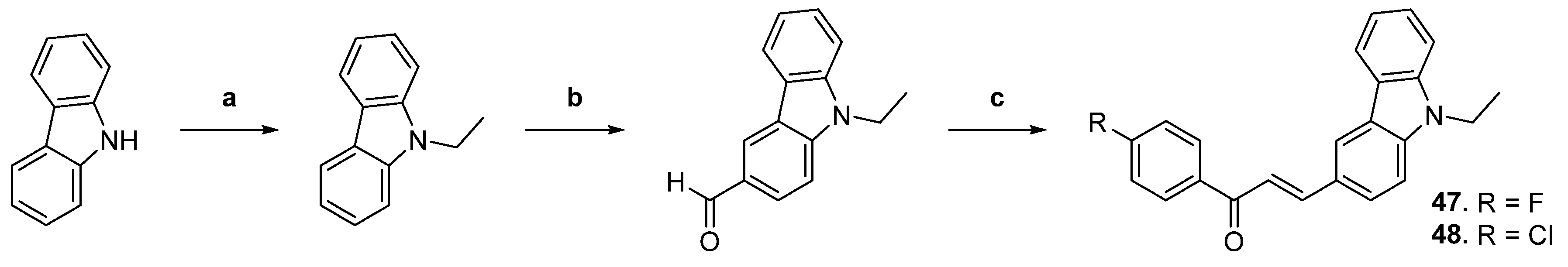
| 47 |  | Topo II: inhibition at 20 µM. | A549: 16.7 HeLa: >50 HL-60: 12.9 PC-3: 32.3 | [71] |
| 48 | Topo II: inhibition at 20 µM. | A549: 9.6 HeLa: 5.5 HL-60: 2.9 PC-3: 7.1 | ||
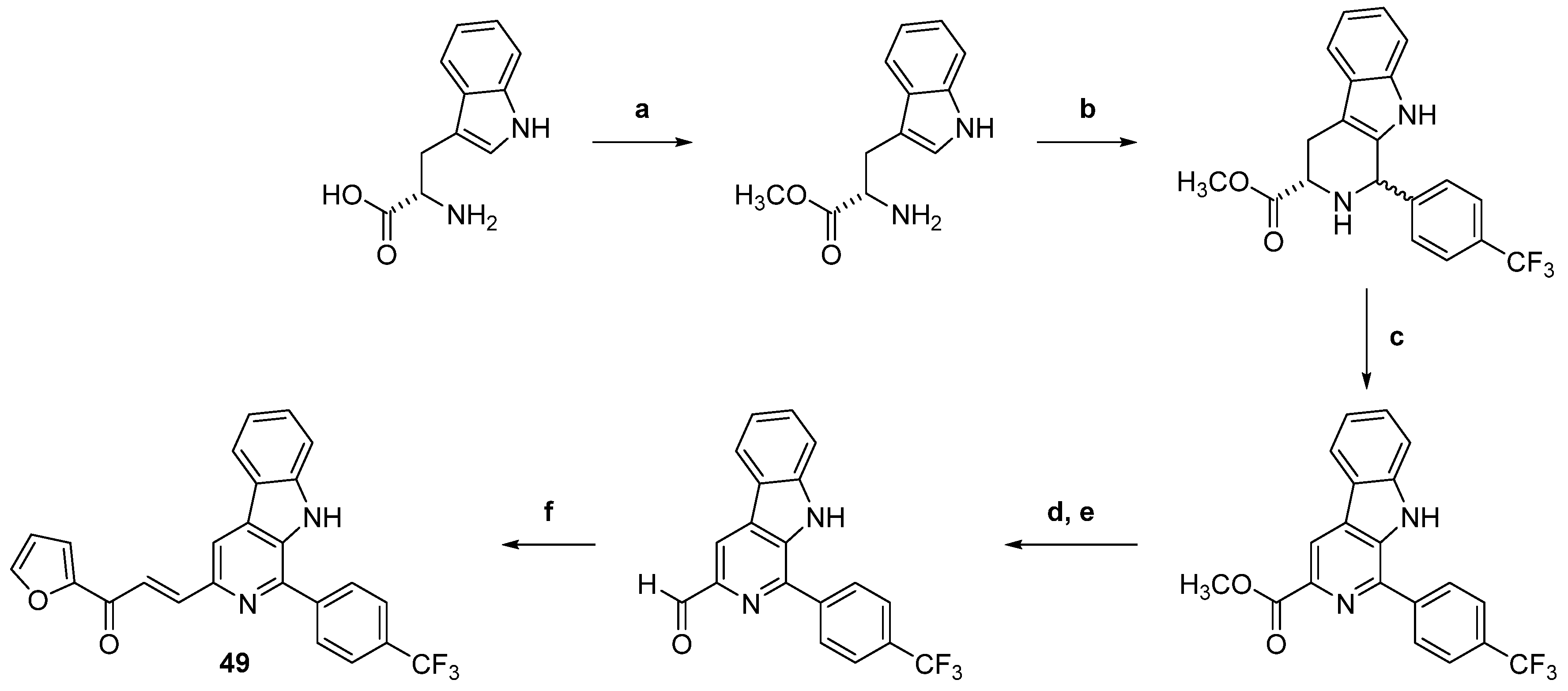
| 49 | 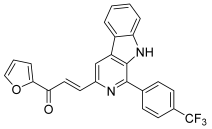 | Topo I: inhibition at 100 µM | A549: 2.6 ACHN: 2.13 DU145: 3.3 MCF-7: 1.9 HeLa: 2.8 | [73] |
| 50 (harmine) | 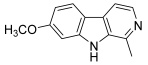 | Topo I: inhibition at 150 µM | - | [73,75] |
| 51 | 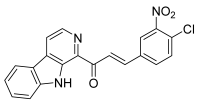 | Topo I: dose-dependent inhibition 25 to 100 µM | A549: 2.0 HepG2: 1.6 HT29: 0.6 MCF-7: 0.3 MDA-MB-231: 1.0 PC-3: 1.2 | [76] |
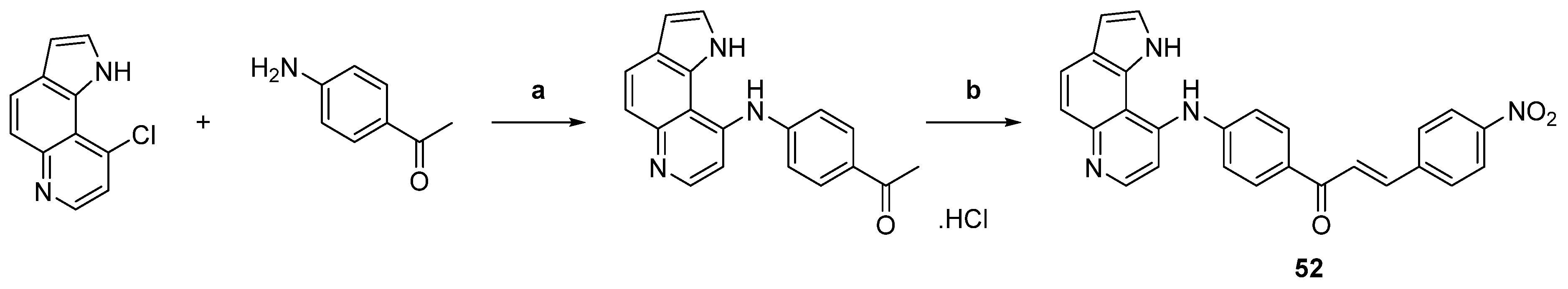
| 52 |  | Topo II: inhibition at 10 µM. | HeLa: 10.4 HL-60: 4.4 JR8: 1.2 | [77] |
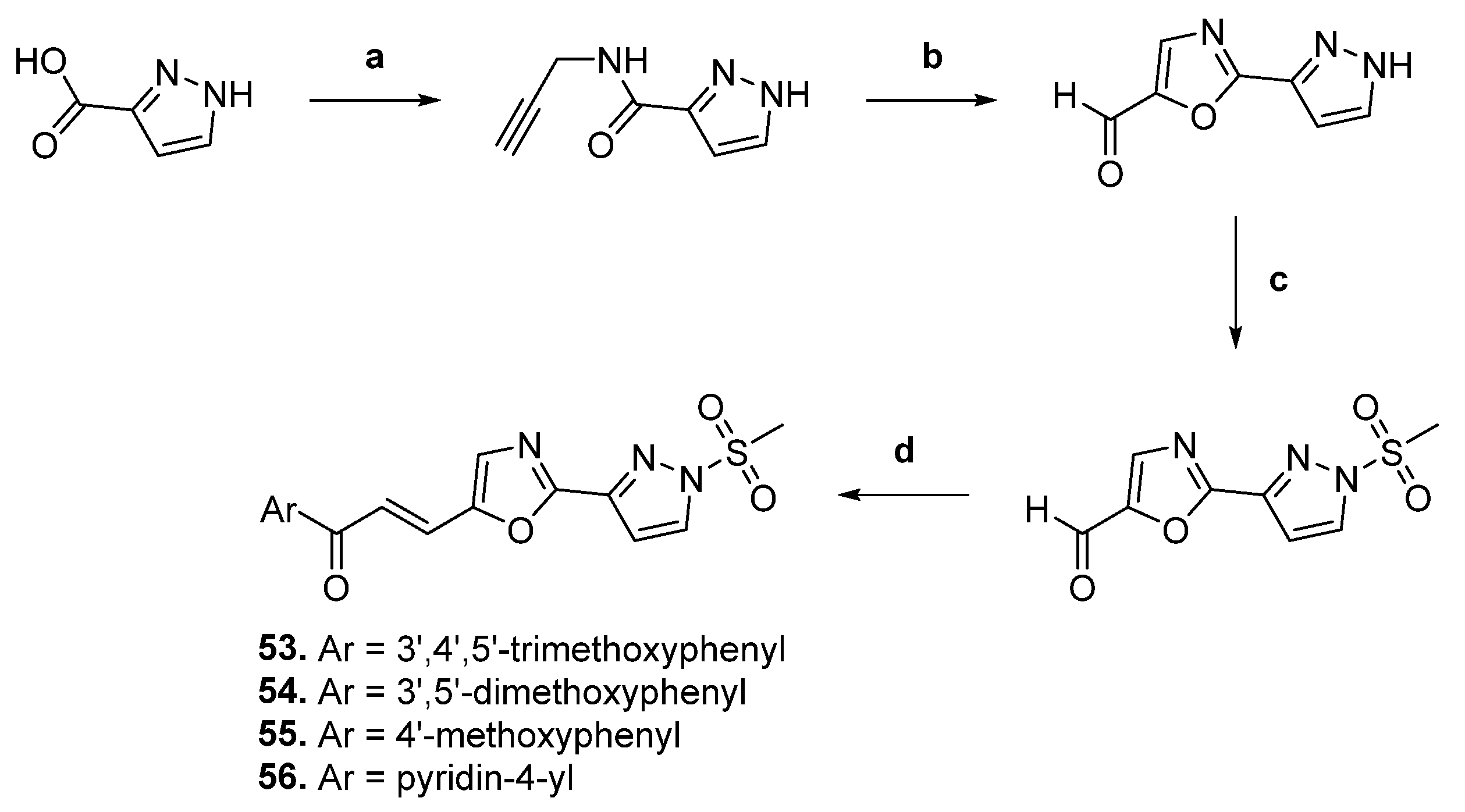
| 53 |  | Topo II | A549: 0.1 Colo-205: 0.1 MCF-7: 0.1 SiHa: 0.2 | [79] |
| 54 | A549: 0.9 Colo-205: 1.4 MCF-7: 1.2 SiHa: 0.9 | |||
| 55 | A549: 1.7 Colo-205: 2.0 MCF-7: 1.8 SiHa: 1.7 | |||
| 56 | A549: 0.01 Colo-205: 0.04 MCF-7: 0.07 SiHa: 0.1 |
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CPT | Camptothecin |
| CuAAC | Copper-Catalyzed Azide-Alkyne Cycloaddition |
| CP | Ciprofloxacin |
| DC | Deoxycytidine |
| DCM | Dichloromethane |
| DG | Deoxyguanosine |
| DIPEA | N,N-diisopropylethylamine |
| DMAP | 4-ddimethylaminopyridine |
| DMF | Dimethylformamide |
| DNA | Deoxyribonucleic acid |
| DOX | Doxorubicin |
| EDCI | 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide |
| GSH | Glutathione |
| HATU | Hexafluorophosphate azabenzotriazole tetramethyl uronium |
| HOBt | Hydroxybenzotriazole |
| IARC | International Agency for Research on Cancer |
| IC50 | Half maximal inhibitory concentration |
| ISL | Isoliquiritigenin |
| MDAs | Microtubule destabilizing agents |
| MDR | Multidrug resistance |
| MTDs | Multi-targeted drugs |
| topo | Topoisomerase |
| Cancer Cell Lines | |
| The following cancer cell lines are used in this manuscript: | |
| A2780 | Ovarian endometrioid adenocarcinoma |
| A2780CP | Cisplatin-resistant A2780 |
| A2780S | Cisplatin-sensitive A2780 |
| A549 | Lung adenocarcinoma |
| A549/PTX | Paclitaxel-resistant A549 |
| ACHN | Papillary renal cell carcinoma |
| AZ521 | Gastric carcinoma |
| BGC-823 | Human papillomavirus-related endocervical adenocarcinoma |
| BT474 | Invasive breast carcinoma of no special type |
| Caco-2 | Colon adenocarcinoma |
| Capan-1 | Pancreatic ductal adenocarcinoma |
| Colo-205 | Colon adenocarcinoma |
| CRL-1579 | Amelanotic melanoma |
| DH82 | Canine histiocytic sarcoma |
| DLD-1 | Colon adenocarcinoma |
| DU145 | Prostate carcinoma |
| HCT116 | Colon carcinoma |
| HCT116/PTX | Paclitaxel-resistant HCT116 |
| HCT-15 | Colon adenocarcinoma |
| Hep-2 | Human papillomavirus-related endocervical adenocarcinoma |
| HepG2 | Hepatoblastoma |
| HGC-27 | Hepatoblastoma |
| HL-60 | Adult acute myeloid leukemia |
| HT-1376 | Bladder carcinoma |
| HT-29 | Colon adenocarcinoma |
| HuCCA-1 | Cholangiocarcinoma |
| JR8 | Melanoma |
| Jurkat | Childhood T acute lymphoblastic leukemia |
| K562 | Chronic myeloid leukemia |
| LNCaP | Prostate carcinoma |
| LOX-IMVI | Amelanotic melanoma |
| LS174T | Colon adenocarcinoma |
| MCF-7 | Invasive breast carcinoma of no special type |
| MDA-MB-231 | Breast adenocarcinoma |
| MDA-MB-468 | Breast adenocarcinoma |
| MG63 | Osteosarcoma |
| MOLT-3 | Adult T acute lymphoblastic leukemia |
| NCI-H460 | Lung large cell carcinoma |
| NCI-H460/R | Doxorubicin-resistance NCI-H460 |
| Panc-1 | Pancreatic ductal adenocarcinoma |
| PC-3 | Prostate carcinoma |
| RPMI-8226 | Multiple myeloma |
| SiHa | Human papillomavirus-related cervical squamous cell carcinoma |
| SK-MEL-2 | Melanoma |
| SKN-SH | Neuroblastoma |
| SK-OV-3 | Ovarian serous cystadenocarcinoma |
| SR | Anaplastic large cell lymphoma |
| SNU475 | Adult hepatocellular carcinoma |
| SNU638 | Gastric adenocarcinoma |
| SW-620 | Colon adenocarcinoma |
| T24 | Bladder carcinoma |
| T47D | Invasive breast carcinoma of no special type |
| U87 | Glioblastoma |
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global Cancer Statistics 2022: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- Buzun, K.; Bielawska, A.; Bielawski, K.; Gornowicz, A. DNA Topoisomerases as Molecular Targets for Anticancer Drugs. J. Enzym. Inhib. Med. Chem. 2020, 35, 1781–1799. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; Sun, Y.; Huang, S.-Y.N.; Nitiss, J.L. Roles of Eukaryotic Topoisomerases in Transcription, Replication and Genomic Stability. Nat. Rev. Mol. Cell Biol. 2016, 17, 703–721. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Wu, Q.; Luan, S.; Yin, Z.; He, C.; Yin, L.; Zou, Y.; Yuan, Z.; Li, L.; Song, X.; et al. A Comprehensive Review of Topoisomerase Inhibitors as Anticancer Agents in the Past Decade. Eur. J. Med. Chem. 2019, 171, 129–168. [Google Scholar] [CrossRef]
- Akihisa, T.; Kikuchi, T.; Nagai, H.; Ishii, K.; Tabata, K.; Suzuki, T. 4-Hydroxyderricin from Angelica keiskei Roots Induces Caspase-Dependent Apoptotic Cell Death in HL60 Human Leukemia Cells. J. Oleo Sci. 2011, 60, 71–77. [Google Scholar] [CrossRef]
- Kingsbury, W.D.; Boehm, J.C.; Jakas, D.R.; Holden, K.G.; Hecht, S.M.; Gallagher, G.; Caranfa, M.J.; McCabe, F.L.; Faucette, L.F.; Johnson, R.K.; et al. Synthesis of Water-Soluble (Aminoalkyl) Camptothecin Analogs: Inhibition of Topoisomerase I and Antitumor Activity. J. Med. Chem. 1991, 34, 98–107. [Google Scholar] [CrossRef]
- Ibrahim, M.K.; Taghour, M.S.; Metwaly, A.M.; Belal, A.; Mehany, A.B.M.; Elhendawy, M.A.; Radwan, M.M.; Yassin, A.M.; El-Deeb, N.M.; Hafez, E.E.; et al. Design, Synthesis, Molecular Modeling and Anti-Proliferative Evaluation of Novel Quinoxaline Derivatives as Potential DNA Intercalators and Topoisomerase II Inhibitors. Eur. J. Med. Chem. 2018, 155, 117–134. [Google Scholar] [CrossRef]
- Yadav, A.; Sharma, V.; Singh, G. Anti-Inflammatory Potential of Chalcone Related Compounds: An Updated Review. ChemistrySelect 2024, 9, e202401321. [Google Scholar] [CrossRef]
- Xu, M.; Wu, P.; Shen, F.; Ji, J.; Rakesh, K.P. Chalcone Derivatives and Their Antibacterial Activities: Current Development. Bioorg. Chem. 2019, 91, 103133. [Google Scholar] [CrossRef]
- Elkhalifa, D.; Al-Hashimi, I.; Al Moustafa, A.E.; Khalil, A. A Comprehensive Review on the Antiviral Activities of Chalcones. J. Drug Target. 2021, 29, 403–419. [Google Scholar] [CrossRef]
- Mittal, A.; Vashistha, V.K.; Das, D.K. Recent Advances in the Antioxidant Activity and Mechanisms of Chalcone Derivatives: A Computational Review. Free Radic. Res. 2022, 56, 378–397. [Google Scholar] [CrossRef] [PubMed]
- Yadav, C.S.; Azad, I.; Khan, A.R.; Ahmad, N.; Gupta, S.K.; Verma, V.K.; Hansda, D.; Lohani, M.B. Exploring the Therapeutic Potential of Chalcones in Oncology: A Comprehensive Review. Curr. Bioact. Compd. 2024, 20, 12–47. [Google Scholar] [CrossRef]
- Chen, S.; Wang, X.; Cheng, Y.; Gao, H.; Chen, X. A Review of Classification, Biosynthesis, Biological Activities and Potential Applications of Flavonoids. Molecules 2023, 28, 4982. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, M.F.A.; Abuo-Rahma, G.E.-D.A. Molecular Targets and Anticancer Activity of Quinoline-Chalcone Hybrids: Literature Review. RSC Adv. 2020, 10, 31139–31155. [Google Scholar] [CrossRef]
- Abdel-Aziz, M.; Park, S.-E.; Abuo-Rahma, G.E.-D.A.A.; Sayed, M.A.; Kwon, Y. Novel N-4-Piperazinyl-Ciprofloxacin-Chalcone Hybrids: Synthesis, Physicochemical Properties, Anticancer and Topoisomerase I and II Inhibitory Activity. Eur. J. Med. Chem. 2013, 69, 427–438. [Google Scholar] [CrossRef]
- Mohammed, H.H.H.; Abbas, S.H.; Hayallah, A.M.; Abuo-Rahma, G.E.-D.A.; Mostafa, Y.A. Novel Urea Linked Ciprofloxacin-Chalcone Hybrids Having Antiproliferative Topoisomerases I/II Inhibitory Activities and Caspases-Mediated Apoptosis. Bioorg. Chem. 2021, 106, 104422. [Google Scholar] [CrossRef]
- Yoon, G.; Kang, B.Y.; Cheon, S.H. Topoisomerase I Inhibition and Cytotoxicity of Licochalcones A and E from Glycyrrhiza inflata. Arch. Pharmacal. Res. 2007, 30, 313–316. [Google Scholar] [CrossRef]
- Wang, K.-L.; Yu, Y.-C.; Hsia, S.-M. Perspectives on the Role of Isoliquiritigenin in Cancer. Cancers 2021, 13, 115. [Google Scholar] [CrossRef]
- Zhao, S.; Chang, H.; Ma, P.; Gao, G.; Jin, C.; Zhao, X.; Zhou, W.; Jin, B. Inhibitory Effect of DNA Topoisomerase Inhibitor Isoliquiritigenin on the Growth of Glioma Cells. Int. J. Clin. Exp. Pathol. 2015, 8, 12577–12582. [Google Scholar]
- Li, Z.; Li, J.; Li, Y.; You, K.; Xu, H.; Wang, J. Novel Insights into the Apoptosis Mechanism of DNA Topoisomerase I Inhibitor Isoliquiritigenin on HCC Tumor Cell. Biochem. Biophys. Res. Commun. 2015, 464, 548–553. [Google Scholar] [CrossRef]
- Zhang, H.-L.; Zhang, Y.; Yan, X.-L.; Xiao, L.-G.; Hu, D.-X.; Yu, Q.; An, L.-K. Secondary Metabolites from Isodon ternifolius (D. Don) Kudo and Their Anticancer Activity as DNA Topoisomerase IB and Tyrosyl-DNA Phosphodiesterase 1 Inhibitors. Bioorg. Med. Chem. 2020, 28, 115527. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Fu, A.; Wu, W.; Li, Y.; Wang, G.; Tang, M.; Li, S.; He, S.; Zhong, S.; Lai, H.; et al. Cytotoxic and Apoptotic Effects of Constituents from Millettia pachycarpa Benth. Fitoterapia 2012, 83, 1402–1408. [Google Scholar] [CrossRef] [PubMed]
- Rampogu, S.; Badvel, P.; Hoon Jo, B.; Kim, Y.; Kim, S.-W.; Lee, K.W. A Review on Millepachine and Its Derivatives as Potential Multitarget Anticancer Agents. Biochem. Biophys. Res. Commun. 2023, 681, 249–270. [Google Scholar] [CrossRef]
- Wu, W.; Ma, B.; Ye, H.; Wang, T.; Wang, X.; Yang, J.; Wei, Y.; Zhu, J.; Chen, L. Millepachine, a Potential Topoisomerase II Inhibitor Induces Apoptosis via Activation of NF-κB Pathway in Ovarian Cancer. Oncotarget 2016, 7, 52281–52293. [Google Scholar] [CrossRef]
- Wu, W.; Liu, Y.; Ye, H.; Li, Z. Millepachine Showed Novel Antitumor Effects in Cisplatin-Resistant Human Ovarian Cancer through Inhibiting Drug Efflux Function of ATP-Binding Cassette Transporters. Phytother. Res. 2018, 32, 2428–2435. [Google Scholar] [CrossRef]
- Liu, M.; Hansen, P.E.; Wang, G.; Qiu, L.; Dong, J.; Yin, H.; Qian, Z.; Yang, M.; Miao, J. Pharmacological Profile of Xanthohumol, a Prenylated Flavonoid from Hops (Humulus lupulus). Molecules 2015, 20, 754–779. [Google Scholar] [CrossRef]
- Harish, V.; Haque, E.; Śmiech, M.; Taniguchi, H.; Jamieson, S.; Tewari, D.; Bishayee, A. Xanthohumol for Human Malignancies: Chemistry, Pharmacokinetics and Molecular Targets. Int. J. Mol. Sci. 2021, 22, 4478. [Google Scholar] [CrossRef]
- Lee, S.H.; Kim, H.J.; Lee, J.S.; Lee, I.-S.; Kang, B.Y. Inhibition of Topoisomerase I Activity and Efflux Drug Transporters’ Expression by Xanthohumol from Hops. Arch. Pharmacal. Res. 2007, 30, 1435–1439. [Google Scholar] [CrossRef]
- Savaspun, K.; Boonchaisri, S.; Chaisuriya, P.; Srisomsap, C.; Svasti, J.; Ardkhean, R.; Phanumartwiwath, A.; Sam-ang, P. Cytotoxicity and Molecular Docking to DNA Topoisomerase II of Chalcone FLavokawain B Isolated from Kaempferia elegans Rhizomes. ScienceAsia 2023, 49, 421–427. [Google Scholar] [CrossRef]
- Nozaki, H.; Hayashi, K.; Kido, M.; Kakumoto, K.; Ikeda, S.; Matsuura, N.; Tani, H.; Takaoka, D.; Iinuma, M.; Akao, Y. Pauferrol A, a Novel Chalcone Trimer with a Cyclobutane Ring from Caesalpinia ferrea Mart Exhibiting DNA Topoisomerase II Inhibition and Apoptosis-Inducing Activity. Tetrahedron Lett. 2007, 48, 8290–8292. [Google Scholar] [CrossRef]
- Ohira, S.; Takaya, K.; Mitsui, T.; Kido, M.; Kakumoto, K.; Hayashi, K.; Kuboki, A.; Tani, H.; Ikeda, S.; Iinuma, M.; et al. New Chalcone Dimers from Caesalpinia ferrea Mart Act as Potent Inhibitors of DNA Topoisomerase II. Tetrahedron Lett. 2013, 54, 5052–5055. [Google Scholar] [CrossRef]
- Aljančić, I.S.; Vučković, I.; Jadranin, M.; Pešić, M.; Đorđević, I.; Podolski-Renić, A.; Stojković, S.; Menković, N.; Vajs, V.E.; Milosavljević, S.M. Two Structurally Distinct Chalcone Dimers from Helichrysum zivojinii and Their Activities in Cancer Cell Lines. Phytochemistry 2014, 98, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Yue, S.; Tang, Y.; Li, S.; Duan, J.-A. Chemical and Biological Properties of Quinochalcone C-Glycosides from the Florets of Carthamus tinctorius. Molecules 2013, 18, 15220–15254. [Google Scholar] [CrossRef] [PubMed]
- Yue, S.-J.; Qu, C.; Zhang, P.-X.; Tang, Y.-P.; Jin, Y.; Jiang, J.-S.; Yang, Y.-N.; Zhang, P.-C.; Duan, J.-A. Carthorquinosides A and B, Quinochalcone C-Glycosides with Diverse Dimeric Skeletons from Carthamus tinctorius. J. Nat. Prod. 2016, 79, 2644–2651. [Google Scholar] [CrossRef]
- Kazuma, K.; Takahashi, T.; Sato, K.; Takeuchi, H.; Matsumoto, T.; Okuno, T. Quinochalcones and Flavonoids from Fresh Florets in Different Cultivars of Carthamus tinctorius L. Biosci. Biotechnol. Biochem. 2000, 64, 1588–1599. [Google Scholar] [CrossRef]
- Tang, Y.; Duan, J.; Le, S. Quinoid Chalcone C-Glycoside Dimer Compound Having Anti-Tumor Activity and Anti-Inflammatory Activity and Method for Preparing Same. Patent Number WO2017219510 A1, 28 December 2017. [Google Scholar]
- Sangpheak, K.; Mueller, M.; Darai, N.; Wolschann, P.; Suwattanasophon, C.; Ruga, R.; Chavasiri, W.; Seetaha, S.; Choowongkomon, K.; Kungwan, N.; et al. Computational Screening of Chalcones Acting against Topoisomerase IIα and Their Cytotoxicity towards Cancer Cell Lines. J. Enzym. Inhib. Med. Chem. 2019, 34, 134–143. [Google Scholar] [CrossRef]
- Hu, C.-X.; Zuo, Z.-L.; Xiong, B.; Ma, J.-G.; Geng, M.-Y.; Lin, L.-P.; Jiang, H.-L.; Ding, J. Salvicine Functions as Novel Topoisomerase II Poison by Binding to ATP Pocket. Mol. Pharmacol. 2006, 70, 1593–1601. [Google Scholar] [CrossRef]
- Chen, X.; Lv, X.; Gao, L.; Liu, J.; Wang, W.; Guo, L.; Frasinyuk, M.S.; Zhang, W.; Watt, D.S.; Liu, C.; et al. Chalcone Derivative CX258 Suppresses Colorectal Cancer via Inhibiting the TOP2A/Wnt/β-Catenin Signaling. Cells 2023, 12, 1066. [Google Scholar] [CrossRef]
- Liu, C.; Watt, D.S.; Liu, X.; Frasinyuk, M.S. Substituted 2-Cinnamoylphenyl Benzoates and Related Heterocycles as Wnt Signaling Inhibitors for the Treatment of Cancer. Patent Number WO2024064827 A2, 28 March 2024. [Google Scholar]
- Gul, H.I.; Cizmecioglu, M.; Zencir, S.; Gul, M.; Canturk, P.; Atalay, M.; Topcu, Z. Cytotoxic Activity of 4′-Hydroxychalcone Derivatives against Jurkat Cells and Their Effects on Mammalian DNA Topoisomerase I. J. Enzym. Inhib. Med. Chem. 2009, 24, 804–807. [Google Scholar] [CrossRef]
- Kim, S.-H.; Lee, E.; Baek, K.H.; Kwon, H.B.; Woo, H.; Lee, E.-S.; Kwon, Y.; Na, Y. Chalcones, Inhibitors for Topoisomerase I and Cathepsin B and L, as Potential Anti-Cancer Agents. Bioorg. Med. Chem. Lett. 2013, 23, 3320–3324. [Google Scholar] [CrossRef]
- Kwon, Y.; Na, Y. Novel Chalcone Derivatives and the Use Thereof. Patent Number KR20130071218 A, 28 June 2013. [Google Scholar]
- Na, Y.; Nam, J.-M. Synthesis and Topoisomerase II Inhibitory and Cytotoxic Activity of Oxiranylmethoxy- and Thiiranylmethoxy-Chalcone Derivatives. Bioorg. Med. Chem. Lett. 2011, 21, 211–214. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.; Jung, J.; Lee, C.; Kwon, Y.; Na, Y. Synthesis of New Xanthone Analogues and Their Biological Activity Test—Cytotoxicity, Topoisomerase II Inhibition, and DNA Cross-Linking Study. Bioorg. Med. Chem. Lett. 2007, 17, 1163–1166. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.-J.; Jung, M.-J.; Kwon, Y.; Na, Y. Oxiranylmethyloxy or Thiiranylmethyloxy-Azaxanthones and -Acridone Analogues as Potential Topoisomerase I Inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 6766–6769. [Google Scholar] [CrossRef]
- Jeon, K.-H.; Yu, H.-B.; Kwak, S.Y.; Kwon, Y.; Na, Y. Synthesis and Topoisomerases Inhibitory Activity of Heteroaromatic Chalcones. Bioorg. Med. Chem. 2016, 24, 5921–5928. [Google Scholar] [CrossRef]
- Kasetti, A.B.; Singhvi, I.; Nagasuri, R.; Bhandare, R.R.; Shaik, A.B. Thiazole—Chalcone Hybrids as Prospective Antitubercular and Antiproliferative Agents: Design, Synthesis, Biological, Molecular Docking Studies and In Silico ADME Evaluation. Molecules 2021, 26, 2847. [Google Scholar] [CrossRef]
- Santos, M.B.; Pinhanelli, V.C.; Garcia, M.A.R.; Silva, G.; Baek, S.J.; França, S.C.; Fachin, A.L.; Marins, M.; Regasini, L.O. Antiproliferative and Pro-Apoptotic Activities of 2′- and 4′-Aminochalcones against Tumor Canine Cells. Eur. J. Med. Chem. 2017, 138, 884–889. [Google Scholar] [CrossRef]
- Štefanišinová, M.; Tomečková, V.; Kožurková, M.; Ostró, A.; Mareková, M. Study of DNA Interactions with Cyclic Chalcone Derivatives by Spectroscopic Techniques. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2011, 81, 666–671. [Google Scholar] [CrossRef]
- Gaur, R.; Mishra, L. Synthesis and Characterization of Ru(II)–DMSO–Cl–Chalcone Complexes: DNA Binding, Nuclease, and Topoisomerase II Inhibitory Activity. Inorg. Chem. 2012, 51, 3059–3070. [Google Scholar] [CrossRef]
- Clarke, M.J. Ruthenium Metallopharmaceuticals. Coord. Chem. Rev. 2003, 236, 209–233. [Google Scholar] [CrossRef]
- Brindell, M.; Kuliś, E.; Elmroth, S.K.C.; Urbańska, K.; Stochel, G. Light-Induced Anticancer Activity of [RuCl2(DMSO)4] Complexes. J. Med. Chem. 2005, 48, 7298–7304. [Google Scholar] [CrossRef]
- Gaur, R.; Mishra, L. Bi-Nuclear Ru(II) Complexes of Bis-Chalcone and Bis-Flavonol: Synthesis, Characterization, Photo Cleavage of DNA and Topoisomerase I Inhibition. RSC Adv. 2013, 3, 12210–12219. [Google Scholar] [CrossRef]
- Konkoľová, E.; Janočková, J.; Perjési, P.; Vašková, J.; Kožurková, M. Selected Ferrocenyl Chalcones as DNA/BSA-Interacting Agents and Inhibitors of DNA Topoisomerase I and II Activity. J. Organomet. Chem. 2018, 861, 1–9. [Google Scholar] [CrossRef]
- Vashisht Gopal, Y.N.; Jayaraju, D.; Kondapi, A.K. Topoisomerase II Poisoning and Antineoplastic Action by DNA-Nonbinding Diacetyl and Dicarboxaldoxime Derivatives of Ferrocene. Arch. Biochem. Biophys. 2000, 376, 229–235. [Google Scholar] [CrossRef]
- Doostmohammadi, A.; Jooya, H.; Ghorbanian, K.; Gohari, S.; Dadashpour, M. Potentials and Future Perspectives of Multi-Target Drugs in Cancer Treatment: The next Generation Anti-Cancer Agents. Cell Commun. Signal. 2024, 22, 228. [Google Scholar] [CrossRef] [PubMed]
- Morphy, R.; Rankovic, Z. Designed Multiple Ligands. Emerg. Drug Discov. Paradigm. J. Med. Chem. 2005, 48, 6523–6543. [Google Scholar] [CrossRef]
- Azéma, J.; Guidetti, B.; Dewelle, J.; Le Calve, B.; Mijatovic, T.; Korolyov, A.; Vaysse, J.; Malet-Martino, M.; Martino, R.; Kiss, R. 7-((4-Substituted)Piperazin-1-Yl) Derivatives of Ciprofloxacin: Synthesis and in Vitro Biological Evaluation as Potential Antitumor Agents. Bioorg. Med. Chem. 2009, 17, 5396–5407. [Google Scholar] [CrossRef]
- Ali, D.M.E.; Aziz, H.A.; Bräse, S.; Al Bahir, A.; Alkhammash, A.; Abuo-Rahma, G.E.-D.A.; Elshamsy, A.M.; Hashem, H.; Abdelmagid, W.M. Unveiling the Anticancer Potential of a New Ciprofloxacin-Chalcone Hybrid as an Inhibitor of Topoisomerases I & II and Apoptotic Inducer. Molecules 2024, 29, 5382. [Google Scholar] [CrossRef]
- Mohammed, H.H.H.; Abd El-Hafeez, A.A.; Ebeid, K.; Mekkawy, A.I.; Abourehab, M.A.S.; Wafa, E.I.; Alhaj-Suliman, S.O.; Salem, A.K.; Ghosh, P.; Abuo-Rahma, G.E.-D.A.; et al. New 1,2,3-Triazole Linked Ciprofloxacin-Chalcones Induce DNA Damage by Inhibiting Human Topoisomerase I & II and Tubulin Polymerization. J. Enzym. Inhib. Med. Chem. 2022, 37, 1346–1363. [Google Scholar] [CrossRef]
- Ducki, S.; Rennison, D.; Woo, M.; Kendall, A.; Chabert, J.F.D.; McGown, A.T.; Lawrence, N.J. Combretastatin-like Chalcones as Inhibitors of Microtubule Polymerization. Part 1: Synthesis and Biological Evaluation of Antivascular Activity. Bioorg. Med. Chem. 2009, 17, 7698–7710. [Google Scholar] [CrossRef]
- Ma, Y.-C.; Wang, Z.-X.; Jin, S.-J.; Zhang, Y.-X.; Hu, G.-Q.; Cui, D.-T.; Wang, J.-S.; Wang, M.; Wang, F.-Q.; Zhao, Z.-J. Dual Inhibition of Topoisomerase II and Tyrosine Kinases by the Novel Bis-Fluoroquinolone Chalcone-Like Derivative HMNE3 in Human Pancreatic Cancer Cells. PLoS ONE 2016, 11, e0162821. [Google Scholar] [CrossRef]
- Liu, Y.; Ding, Y.; Zhao, H.; Wang, R.; Gao, L.; Li, T.; Xie, Y.; Yan, Q.; Wu, S.; Ni, L.; et al. 3,3′-Methylene-Bisfluoroquinolone Derivative Containing Cyclopropylquinoline Ring as Well as Preparation Method and Application of 3,3′-Methylene-Bisfluoroquinolone Derivative. Patent Number CN104370812 A, 25 February 2015. [Google Scholar]
- Banday, A.H.; Kulkarni, V.V.; Hruby, V.J. Design, Synthesis, and Biological and Docking Studies of Novel Epipodophyllotoxin–Chalcone Hybrids as Potential Anticancer Agents. MedChemComm 2015, 6, 94–104. [Google Scholar] [CrossRef]
- Wang, H.; Nie, C.; Luo, M.; Bai, Q.; Yao, Z.; Lv, H.; Chen, B.; Wang, J.; Xu, W.; Wang, S.; et al. Novel GSH-Responsive Prodrugs Derived from Indole-Chalcone and Camptothecin Trigger Apoptosis and Autophagy in Colon Cancer. Bioorg. Chem. 2024, 143, 107056. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Chen, J.; Zhang, S.; Hu, J.; Huang, L.; Li, X. Synthesis, Evaluation, and Mechanism Study of Novel Indole-Chalcone Derivatives Exerting Effective Antitumor Activity Through Microtubule Destabilization in Vitro and in Vivo. J. Med. Chem. 2016, 59, 5264–5283. [Google Scholar] [CrossRef]
- Zhou, W.; Zhang, W.; Peng, Y.; Jiang, Z.-H.; Zhang, L.; Du, Z. Design, Synthesis and Anti-Tumor Activity of Novel Benzimidazole-Chalcone Hybrids as Non-Intercalative Topoisomerase II Catalytic Inhibitors. Molecules 2020, 25, 3180. [Google Scholar] [CrossRef]
- Li, P.; Zhang, W.; Jiang, H.; Li, Y.; Dong, C.; Chen, H.; Zhang, K.; Du, Z. Design, Synthesis and Biological Evaluation of Benzimidazole-Rhodanine Conjugates as Potent Topoisomerase II Inhibitors. MedChemComm 2018, 9, 1194–1205. [Google Scholar] [CrossRef]
- Wang, W.; Sun, X.; Sun, D.; Li, S.; Yu, Y.; Yang, T.; Yao, J.; Chen, Z.; Duan, L. Carbazole Aminoalcohols Induce Antiproliferation and Apoptosis of Human Tumor Cells by Inhibiting Topoisomerase I. ChemMedChem 2016, 11, 2675–2681. [Google Scholar] [CrossRef]
- Li, P.-H.; Jiang, H.; Zhang, W.-J.; Li, Y.-L.; Zhao, M.-C.; Zhou, W.; Zhang, L.-Y.; Tang, Y.-D.; Dong, C.-Z.; Huang, Z.-S.; et al. Synthesis of Carbazole Derivatives Containing Chalcone Analogs as Non-Intercalative Topoisomerase II Catalytic Inhibitors and Apoptosis Inducers. Eur. J. Med. Chem. 2018, 145, 498–510. [Google Scholar] [CrossRef]
- Luo, B.; Song, X. A Comprehensive Overview of β-Carbolines and Its Derivatives as Anticancer Agents. Eur. J. Med. Chem. 2021, 224, 113688. [Google Scholar] [CrossRef]
- Kamal, A.; Srinivasulu, V.; Nayak, V.L.; Sathish, M.; Shankaraiah, N.; Bagul, C.; Reddy, N.V.S.; Rangaraj, N.; Nagesh, N. Design and Synthesis of C3-Pyrazole/Chalcone-Linked Beta-Carboline Hybrids: Antitopoisomerase I, DNA-Interactive, and Apoptosis-Inducing Anticancer Agents. ChemMedChem 2014, 9, 2084–2098. [Google Scholar] [CrossRef]
- Chen, Z.; Cao, R.; Shi, B.; Guo, L.; Sun, J.; Ma, Q.; Fan, W.; Song, H. Synthesis and Biological Evaluation of 1,9-Disubstituted β-Carbolines as Potent DNA Intercalating and Cytotoxic Agents. Eur. J. Med. Chem. 2011, 46, 5127–5137. [Google Scholar] [CrossRef]
- Timbilla, A.A.; Vrabec, R.; Havelek, R.; Rezacova, M.; Chlebek, J.; Blunden, G.; Cahlikova, L. The Anticancer Properties of Harmine and Its Derivatives. Phytochem. Rev. 2024, 24, 1535–1564. [Google Scholar] [CrossRef]
- Guo, Y.-L.; Yu, J.-W.; Cao, Y.; Cheng, K.-X.; Dong-Zhi, S.-N.-M.; Zhang, Y.-F.; Ren, Q.-J.; Yin, Y.; Li, C.-L. Design, Synthesis, and Biological Evaluation of Harmine Derivatives as Topoisomerase I Inhibitors for Cancer Treatment. Eur. J. Med. Chem. 2024, 265, 116061. [Google Scholar] [CrossRef] [PubMed]
- Via, L.D.; Gia, O.; Chiarelotto, G.; Ferlin, M.G. DNA-Targeting Pyrroloquinoline-Linked Butenone and Chalcones: Synthesis and Biological Evaluation. Eur. J. Med. Chem. 2009, 44, 2854–2861. [Google Scholar] [CrossRef]
- Ferlin, M.G.; Dalla Via, L.; Gia, O.M. Synthesis and Antiproliferative Activity of Some New DNA-Targeted Alkylating Pyrroloquinolines. Bioorg. Med. Chem. 2004, 12, 771–777. [Google Scholar] [CrossRef] [PubMed]
- Rangaswamy, S.; Sreenivasulu, R.; Bhuvan Tej, M.; Ramesh Babu, V.; Kumar Kapavarapu, R.; Kumar Abbaraju, V.D.N. Design and Synthesis of Oxazole-Linked Pyrazole Chalcone Derivatives: In Vitro Anticancer Evaluation and In Silico Molecular Docking Studies. ChemistrySelect 2023, 8, e202302395. [Google Scholar] [CrossRef]
- Kulkarni, S.; Kaur, K.; Jaitak, V. Recent Developments in Oxazole Derivatives as Anticancer Agents: Review on Synthetic Strategies, Mechanism of Action and SAR Studies. Anti-Cancer Agents Med. Chem. 2022, 22, 1859–1882. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toublet, F.-X.; Laurent, A.; Pouget, C. A Review of Natural and Synthetic Chalcones as Anticancer Agents Targeting Topoisomerase Enzymes. Molecules 2025, 30, 2498. https://doi.org/10.3390/molecules30122498
Toublet F-X, Laurent A, Pouget C. A Review of Natural and Synthetic Chalcones as Anticancer Agents Targeting Topoisomerase Enzymes. Molecules. 2025; 30(12):2498. https://doi.org/10.3390/molecules30122498
Chicago/Turabian StyleToublet, François-Xavier, Aurélie Laurent, and Christelle Pouget. 2025. "A Review of Natural and Synthetic Chalcones as Anticancer Agents Targeting Topoisomerase Enzymes" Molecules 30, no. 12: 2498. https://doi.org/10.3390/molecules30122498
APA StyleToublet, F.-X., Laurent, A., & Pouget, C. (2025). A Review of Natural and Synthetic Chalcones as Anticancer Agents Targeting Topoisomerase Enzymes. Molecules, 30(12), 2498. https://doi.org/10.3390/molecules30122498