
Synthesis, Characterisation, Biological Evaluation and In Silico Studies of Quinoline–1,2,3-Triazole–Anilines as Potential Antitubercular and Anti-HIV Agents


 and
and
Abstract

1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. In Vitro Biological Activities
2.3. In Silico Studies
2.3.1. Molecular Docking
2.3.2. Density Functional Theory Studies
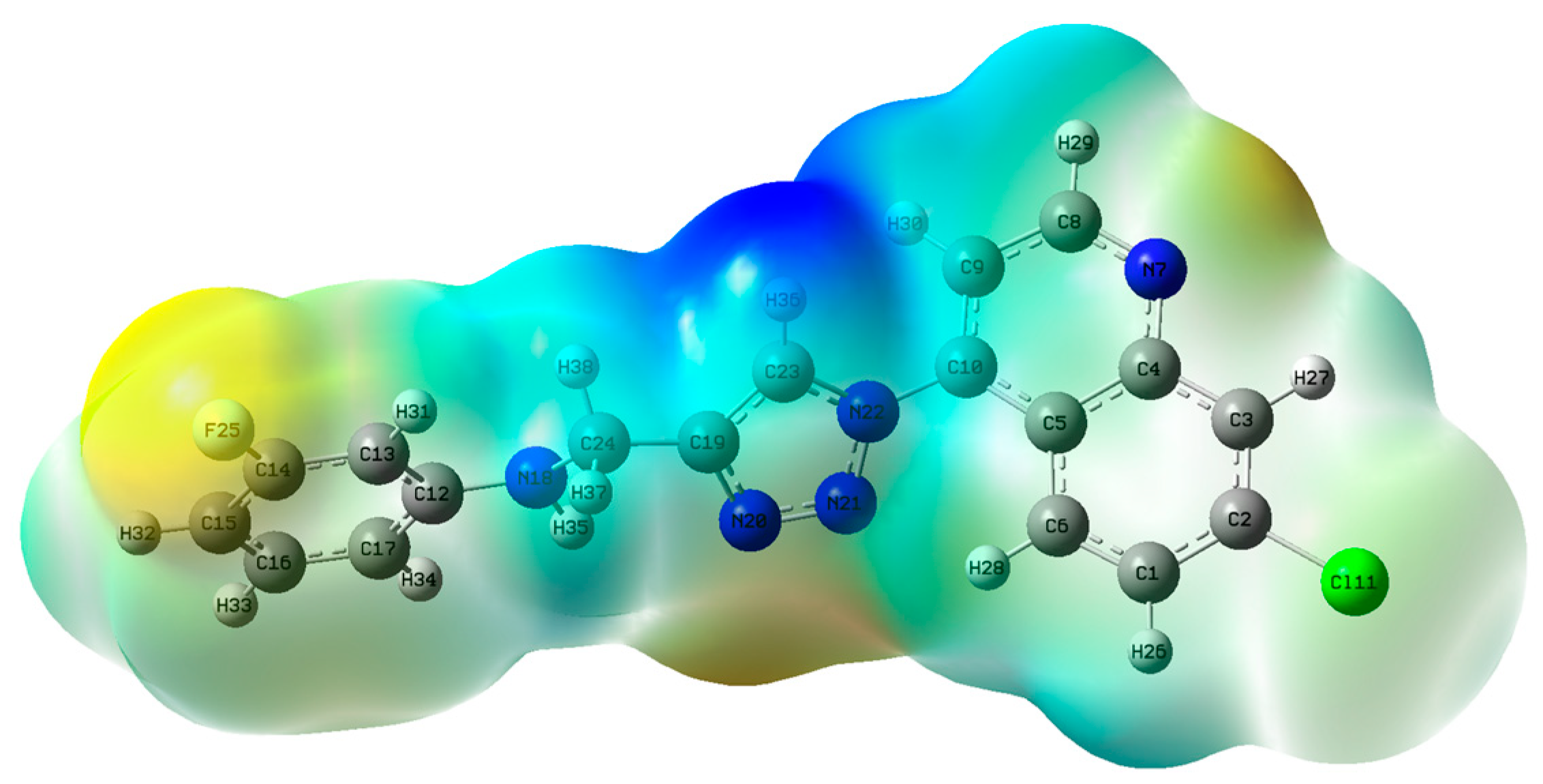
- HOMO, LUMO and ESP surface maps of 11h
2.3.3. ADMET Predictions
3. Materials and Methods
3.1. Chemistry
- Preparation of 4-azido-7-chloroquinoline (7)
- Preparation of the propargylated (alkyne) compounds (10a–j). Compound 10a is chosen as representative.
- N-2-propyl-1-yl-benzanamine (10a) (reference) as a brown liquid, 132 mg (43%) yield, 1H-NMR (DMSO-d6, 600 MHz, ppm) δH 3.0 (1H, t, J = 2.4 Hz, H-6), 3.8 (2H, dd, J1 = 6.1; J2 = 2.40 Hz, H-1), 5.9 (1H, t, J = 6.1 Hz, H-2), 6.6 (1H, t, J = 7.04 Hz, H-5), 6.63 (2H, d, J = 8.14 Hz, H-3), 7.1 (2H, t, J = 8.4 Hz, H-4). Compound 10a is used as a representative compound below.
- Preparation of N-[1-(7-chloroquinolin-4-yl)-1H-1,2,3-triazol-4-yl]anilines (11a–j).
- 1-(7-Chloro-4-quinolinyl)-1H-1,2,3,-triazole-4-methanamine (11a): As a cream-white solid, yield 228.4 mg (88%), mp 15–152 °C, IR (cm−1) C-H 2900–3000, 1H-NMR (DMSO-d6, 600 MHz, ppm) δH 4.47 (2H, d, J = 5.7 Hz, H-1), 6.21 (1H, t, J = 5.7 Hz, H-2), 6.58 (1H, t, J = 7.5 Hz, H-5), 6.72 (2H, d, J = 7.9 Hz, H-3 and H-3′), 7.11 (2H, dd, J1 = 8.3 Hz, J2 = 7.4 Hz, H-4 and H-4′), 7.78 (1H, dd, J1 = 9.0 Hz, J2 = 2.0 Hz, H-10), 7.8 (1H, d, J = 4.5 Hz, H-7), 8.02 (1H, d, J = 9.1 Hz, H-11), 8.28 (1H, d, J = 2.0 Hz, H-9), 8.74 (1H, s, H-6), 9.14 (1H, d, J = 4.5 Hz, H-8). 13C-NMR (DMSO-d6, 150 MHz, ppm) δC 39.9 (C-1), 112.9 (C-3), 116.7 (C-5), 117.3 (C-7), 120.7 (C-15a), 125.7 (C-11), 125.9 (C-6), 128.6 (C-9), 129.4 (C-4,10), 135.8 (C-16), 140.9 (C-14), 147.1 (C-13), 148.7 (C-12), 149.9 (C-15b), 152.8 (C-8). TOFF MS ES−: (m/z) 306.0968 (100%) [(M − H) − N2]− (Calculated for C18H13ClN3− (306.0803)].
- [1-(7-Chloro-4-quinolinyl)-1H-1,2,3-triazole-4-methyl]-4-bromoaniline (11b): As a light brown solid, yield 278.2 mg (87%), mp 187–190 °C, IR (cm−1) C-H 2900–3000; 1H-NMR (DMSO-d6, 600 MHz, ppm) δH 4.48 (2H, d, J = 5.2 Hz, H-1), 6.4 (1H, s, H-2), 6.71 (2H, d, J = 8.7 Hz, H-4), 7.25 (2H, d, J = 8.7 Hz, H-3), 7.76 (1H, dd, J1 = 9.0, J2 = 1.5 Hz, H-10), 7.81 (1H, d, J = 4.6 Hz, H-7), 8.01 (1H, d, J = 9.0 Hz, H-11), 8.26 (1H, d, J = 1.5 Hz, H-9), 8.74 (1H, s, H-6), 9.14 (1H, d, J = 4.5 Hz, H-8). 13C-NMR (DMSO-d6, 150 MHz, ppm) δC 38.0 (C-1)114.0 (C-4), 131.0 (C-3), 117.3 (C-7), 120.7 (C-15a), 125.6 (C-6), 125.8 (C-11), 128.6 (C-9), 129.4 (C-10), 135.8 (C-16), 140.9 (C-14), 148.0 (C-12), 147.0 (C-13), 149.8 (C-15b), 152.0 (C-8), 107.0 (C-5). TOF MSMS ES−: (m/z) 451.4776 [(M− + HCl]− [Calculated for C18H19BrCl2N5 (451.1490)].
- [1-(7-Chloro-4-quinolinyl)-1H-1,2,3-triazole-4-methyl]-4-iodoaniline (11c): As a brown solid, yield 328.1 mg (92%), mp 195–199 °C, IR (cm−1) C-H 2900–3000; 1H-NMR (DMSO-d6, 600 MHz, ppm) δH 4.45 (2H, d, J = 5.5 Hz, H-1), 6.47 (1H, t, J = 5.5 Hz, H-2), 6.58 (2H, d, J = 8.7 Hz, H-4), 7.37 (2H, d, J = 8.7 Hz, H-3), 7.78 (1H, dd, J1 = 9.0, J2 = 1.9 Hz, H-10), 7.81 (1H, d, J = 4.5 Hz, H-7), 7.99 (1H, d, J = 8.8 Hz, H-11), 8.28 (1H, s, H-9), 8.72 (1H, s, H-6), 9.15 (1H, s, H-8). 13C-NMR (DMSO-d6, 150 MHz, ppm) δC 39.0 (C-1), 137.6 (C-3),117.3 (C-7), 77.4 (C-5), 120.7 (C-15a), 125.6 (C-6), 125.9 (C-11), 128.5 (C-9), 129.3 (C-10), 115.8 (C-4), 135.7 (C-16), 140.9 (C-14), 137.9 (C-12), 147.1 (C-13), 149.8 (C-15b), 152.8 (C-8). TOFF MS ES−: (m/z) [(M + Cl]− 495.9821 (100%) (Calculated for C18H13Cl2N5− (495.9593)].
- [1-(7-Chloro-4-quinolinyl)-1H-1,2,3-triazole-4-methyl]-4-flouroaniline (11d): As a light grey solid, yield 216.0 mg (79%), mp-145–148, IR (cm−1) C-H 2900–3000, 1H-NMR (DMSO-d6, 600 MHz, ppm) δH 4.44 (2H, d, J = 5.2 Hz, H-1), 6.15 (1H, t, J = 5.6 Hz, H-2), 6.71 (2H, dd, J1 = 9.1 Hz, 2J(H-F) = 4.4 Hz, H-4), 6.96 (2H, t, J = 9.1 Hz, H-3), 7.79 (1H, dd, J1 = 9.0 Hz, 3J(H-F) = 1.5 Hz, H-10), 7.82 (1H, d, J = 4.5 Hz, H-7), 8.01 (1H, d, J = 9.0, H-11), 8.29 (1H, d, J = 1.5 Hz, H-9), 8.73 (1H, s, H-6), 9.15 (1H, d, J = 4.5 Hz, H-8). 13C-NMR (DMSO-d6, 150 MHz, ppm) δC 39.0 (C-1), 113.7 (d, 3J(C-F) = 7.02 Hz, C-3), 115,8 (d, 2J(C-F) = 21.9 Hz, C-4), 117.3 (C-7), 120.7 (C-15a), 125.6 (C-6), 125.8 (C-11), 128.6 (C-9), 129.4 (C-10), 135.8 (C-16), 140.9 (C-14), 145.4 (C-12), 147.0 (C-13), 149.8 (C-15b), 152 (C-8), 154.3/155.8 (d, 1J(C-F) = 230.04 Hz, C-5). TOFF MS ES−: (m/z) [(M − H) − N2]− 324.0866 (100%) (Calculated for C18H12ClFN3− (324.0709)].
- [1-(7-Chloro-4-quinolinyl)-1H-1,2,3-triazole-4-methyl]-3-chloroaniline (11e): As a yellow solid, yield 197.2 mg (69%), mp 169–172 °C, IR (cm−1) C-H 2900–3000, 1H-NMR (DMSO-d6, 600 MHz, ppm) δH 4.48 (2H, d, J = 5.6 Hz, H-1), 6.53 (1H, t, J = 9.2 Hz, H-2), 6.66 (H, t, J = 8.0 Hz, H-5), 6.74 (1H, s, H-3′), 6.74 (1H, dd, J1 = 3 Hz, J2 = 2.4 Hz H-4′), 7.10 (1H, dd, J1 = 9.2 Hz, J2 = 1.9 Hz, H-3), 7.12 (1H, s, H-3), 7.80–7.79 (2H, m, H-7 and H-10), 8.01 (1H, d, J = 9.0 Hz, H-11), 8.27 (1H, d, J = 1.6 Hz, H-9), 8.72 (1H, s, H-6), 9.13 (1H, d, J = 4.5 Hz, H-8). 13C-NMR (DMSO-d6, 150 MHz, ppm) δC 38.6 (C-1), 112.1 (C-3), 111.5(C-3′), 117.3 (C-7), 130.8 (C-5), 120.7 (C-15a), 125.7 (C-6), 125.8 (C-11), 128.6 (C-9), 129.3 (C-10), 124.7 (C-4′), 135.8 (C-16), 134.1 (C-4), 140.9 (C-14), 150.2 (C-12), 147.0 (C-13), 149.8 (C-15b), 152.8 (C-8). TOFF MS ES−: (m/z) [(M − H) − N2]− 340.0580 (100%) (Calculated for C18H12Cl2N3− (340.0414)].
- [1-(7-Chloro-4-quinolinyl)-1H-1,2,3-triazole-4-methyl]-2-methoxyaniline (11f): As a brown liquid, yield 121.6 mg (43%), IR (cm−1) C-H 2900–3000 1H-NMR (DMSO-d6, 600 MHz, ppm) δH 3.79 (3H, s, H-3″), 4.52 (2H, d, J = 6 Hz, H-1), 5.46 (1H, t, J = 6 Hz, H-2), 6.59 (1H, td, J1 = 7.7 Hz, J2 = 1.4 Hz, H-5), 6.72 (1H, dd, J1 = 7.7 Hz, J2 = 1.4, H-3′), 6.78 (1H, d, J = 8.0 Hz, H-4), 6.83 (1H, td, J1 = 7.7 Hz, J2 = 0.6 Hz, H-4′), 7.76 (1H, dd, J1 = 9.1 Hz, J2 = 2.1 Hz, H-10), 7.81 (1H, d, J = 4.6 Hz, H-7), 8.01 (1H, d, J = 9.1 Hz, H-11), 8.26 (1H, d, J = 2.0 Hz, H-9), 8.69 (1H, s, H-6), 9.12 (1H, d, J = 4.6 Hz, H-8). 13C-NMR (DMSO-d6, 150 MHz, ppm) δC 39.0 (C-1), 110.2 (C-3′), 138.0 (C-3), 117.3 (C-7), 116.7 (C-5), 120.7 (C-15a), 125.6 (C-6), 125.9 (C-11), 128.5 (C-9), 129.3 (C-10), 110.3 (C-4), 135.7 (C-16), 121.4 (C-4′), 140.9 (C-14), 137.9 (C-12), 147.1 (C-13), 149.8 (C-15b), 152.8 (C-8), 55.7 (C-3″). TOF MSMS ES+: (m/z) 388.1096 (M + Na)+ [Calculated for C19H16ClN5NaO (388.0941)].
- [1-(7-Chloro-4-quinolinyl)-1H-1,2,3-triazole-4-methyl]-2-triflouromethylaniline (11g): As a yellow liquid, yield 115.3 mg (48%), IR (cm−1) C-H 2900–3000 1H-NMR(DMSO-d6, 600 MHz, ppm) δH 4.65 (2H, d, J = 5.7 Hz, H-1), 6.11 (1H, s, H-2), 6.73 (1H, t, J = 7.5 Hz, H-5), 7.01 (1H, d, J = 8.4 Hz, H-3′), 7.43 (1H, m, H-4,4′), 7.76 (1H, dd, J1 = 9.0 Hz, J2 = 2.0 Hz, H-10), 7.80 (1H, d, J = 4.6 Hz, H-7), 7.96 (1H, d, J = 9.0 Hz, H-11), 8.25 (1H, d, J = 2.0, H-9), 8.65 (1H, s, H-6), 9.11 (1H, d, J = 4.6 Hz, H-8). 13C-NMR (DMSO-d6, 150 MHz, ppm) δC 39.0 (C-1), 145.4 (C-3), 112.9 (C-3″), 117.4 (C-7), 116.2 (C-5), 120.7 (C-15a), 126.1 (C-6), 125.8 (C-11), 128.5 (C-9), 129.3 (C-10), 126.7 (C-4′), 135.8 (C-16), 134.0 (C-4), 140.9 (C-14), 146.5 (C-12), 146.5 (C-13), 149.8 (C-15b), 152.8 (C-8), 126 (C-3′). LCMS (ACN with 0.1% formic acid) (m/z) (M + 1) 404.
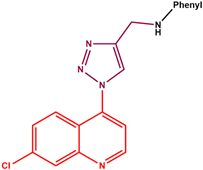
- [1-(7-Chloro-4-quinolinyl)-1H-1,2,3-triazole-4-methyl]-3-flouroaniline (11h): As a cream-white solid, yield 149.7 mg (85%), mp 145–148 °C, IR (cm−1) C-H 2900 1H-NMR (DMSO-d6, 600 MHz, ppm) δH 4.47 (2H, d, J = 5.7 Hz, H-1), 5.40 (1H, s, H-2), 6.35 (1H, td, J1 = 8.6, J2 = 2 Hz, H-4), 6.49–6.55 (3H, m, H-3, H-3′ and H5), 7.76 (1H, dd, J1 = 9.0 Hz, J2 = 1.9 Hz, H-10), 7.80 (1H, d, J = 4.7 Hz, H-7), 8.00 (1H, d, J = 9.2 Hz, H-11), 8.28 (1H, s, Hz, H-9), 8.72 1(H, s, H-6), 9.13 (1H, d, J = 4.6 Hz, H-8). 13C-NMR (DMSO-d6, 150 MHz, ppm) δC 38.7 (C-1), 99.2 (d, 2JC-F = 33.2 Hz, C-3), 109.2 (C-3′), 117.3 (C-7), 130.7 (d, 2JC-F = 30.1 Hz, C-5), 143.5 (d, 3JC-F = 4.1 Hz, C-4′) 120.7 (C-15a), 125.6 (C-6), 125.9 (C-11), 128.5 (C-9), 129.4 (C-10), 135.8 (C-16), 165.1(C-4), 140.9 (C-14), 153.5 (C-12), 146.6 (C-13), 149.8 (C-15b), 152.8 (C-8). TOFF MS ES−: (m/z) [(M − H) − N2]− 324.0868 (100%) (Calculated for C18H12ClFN3− (324.0709)].
- [1-(7-Chloro-4-quinolinyl)-1H-1,2,3-triazole-4-methyl]-3-nitroaniline (11i): As an orange solid, yield 232.4 mg (79%), mp 192–197 °C, IR (cm−1) C-H 2900–3000. 1H-NMR(DMSO-d6, 600 MHz, ppm) δH 4.58 (2H, d, J = 5.7 Hz, H-1), 7.00 (H, s, H-2), 7.15 (1H, d, J = 7.74 Hz, H-3′), 7.37 (2H, m, H-4′ and H-5), 7.52 (1H, d, J = 7.7 Hz, H-3), 7.77 (1H, t, J = 2.0 Hz, H-10), 7.78 (1H, dd, J1 = 8.7 Hz, J2 = 2, H-7), 7.81 (H, dd, J1 = 9.0 Hz, J2 = 1.5 Hz, H-11), 8.29 (1H, d, J = 2 Hz, H-9), 8.77 (1H, s, H-6), 9.14 (1H, d, J = 4.5 Hz, H-8). 13C-NMR (DMSO-d6, 150 MHz, ppm) δC 39.1 (C-1), 130 (C-4′), 106 (C-3′), 117.4 (C-7), 119.1 (C-3) 120.8 (C-15a), 125.8 (C-6), 125.8 (C-11), 128.6 (C-9), 129.4 (C-10), 135.8 (C-16), 140.9 (C-14), 145.4 (C-12), 146.2 (C-13), 149.81 (C-4), 149.8 (C-15b), 152 (C-8), 111.0 (C-5). TOF MSMS ES+: (m/z) 381.2533 (M + 1)− [Calculated for C18H14ClN6O2 (381.7920)].
- [1-(7-Chloro-4-quinolinyl)-1H-1,2,3-triazole-4-methyl]-4-methoxyaniline (11j): As a brown solid, yield 243.1 mg (86%), mp 169–172, IR (cm−1) C-H 2900–3000. 1H-NMR(DMSO-d6, 600 MHz, ppm) δH 3.64 (3H, s, H-5′), 4.42 (2H, d, J = 5.8 Hz, H-1), 5.76 (1H, t, J = 5.8 Hz, H-2), 6.68 (2H, d, J = 8.8 Hz, H-4), 6.74 (2H, d, J = 8.8 Hz, H-3), 7.75 (1H, dd, J1 = 9.0 Hz, J2 = 1.8 Hz, H-10), 7.79 (1H, d, J = 4.6 Hz, H-7), 8.01 (1H, d, J = 9.0 Hz, H-11), 8.25 (1H, d, J = 1.7 Hz, H-9), 8.68 (1H, s, H-6), 9.11 (1H, d, J = 4.6 Hz, H-8). 13C-NMR (DMSO-d6, 150 MHz, ppm) δC 40.4 (C-1), 115.07 (C-3), 117.2 (C-7), 151.6 (C-5), 55.7 (C-5′), 120.7 (C-15a), 125.6 (C-6), 125.9 (C-11), 128.5 (C-9), 129.3 (C-10), 135.7 (C-16), 114.1 (C-4), 140.9 (C-14), 142.9 (C-12), 147.1 (C-13), 149.8 (C-15b), 152.8 (C-8). TOFF MS ES−: (m/z) [(M − H) − N2]− 336.1097 (100%) [Calculated for C19H15ClN3O− (336.0909)].
3.2. Biology
3.2.1. Antimycobacterial Evaluation
3.2.2. MTT Cytotoxicity Evaluation
3.2.3. Luciferase-Based Antiviral Assay Evaluating Human Immunodeficiency Virus
- Maintenance of cell lines
- Antiviralassay
3.3. In Silico Studies
3.3.1. Molecular Docking
3.3.2. DFT Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Global Tuberculosis Report 2024. World Health Organisation: Geneva, Switzerland, 2024. Available online: https://iris.who.int/bitstream/handle/10665/379339/9789240101531-eng.pdf?sequence=1 (accessed on 17 January 2025).
- Quimque, M.T.G.; Go, A.D.; Lim, J.A.K.; Vidar, W.S.; Macabeo, A.P.G. Mycobacterium tuberculosis Inhibitors Based on arylated quinoline carboxylic acid backbones with anti-Mtb gyrase activity. Int. J. Mol. Sci. 2023, 24, 11632. [Google Scholar] [CrossRef] [PubMed]
- Martinez, R.M. Bacillus subtilis. In Brenner’s Encyclopedia of Genetics, 2nd ed.; Maloy, S., Hughes, K., Eds.; Academic Press: San Diego, CA, USA, 2013; pp. 246–248. [Google Scholar]
- Smith, T.; Wolff, K.A.; Nguyen, L. Molecular biology of drug resistance in Mycobacterium tuberculosis. Curr. Top. Microbiol. Immunol. 2013, 374, 53–80. [Google Scholar] [CrossRef]
- Sawyer, E.B.; Grabowska, A.D.; Cortes, T. Translational regulation in mycobacteria and its implications for pathogenicity. Nucleic Acids Res. 2018, 46, 6950–6961. [Google Scholar] [CrossRef]
- Bhat, Z.S.; Rather, M.A.; Maqbool, M.; Ahmad, Z. Drug targets exploited in Mycobacterium tuberculosis: Pitfalls and promises on the horizon. Biomed. Pharmacother. 2018, 103, 1733–1747. [Google Scholar] [CrossRef]
- Amusengeri, A.; Khan, A.; Tastan Bishop, O. The structural basis of Mycobacterium tuberculosis RpoB drug-Resistant clinical mutations on rifampicin drug binding. Molecules 2022, 27, 885. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, D.E.; Siliciano, R.F.; Jacobs, W.R. Outwitting evolution: Fighting drug-resistant TB, malaria, and HIV. Cell 2012, 148, 1271–1283. [Google Scholar] [CrossRef]
- Aguilar Diaz, J.M.; Abulfathu, A.A.; te Brake, L.H.M.; van Ingen, J.; Kuipers, S.; Magis-Escurra, C.; Raiijmaker, J.; Svensson, E.M.; Boeree, M.J. New and repurposed drugs for the treatment of active tuberculosis: An Update for clinicians. Respiration 2023, 102, 83–100. [Google Scholar] [CrossRef] [PubMed]
- Rendon, A.; Tiberi, S.; Scardigli, A.; D’Ambrosio, L.; Centis, R.; Caminero, J.A.; Migloori, G.B. Classification of drugs to treat multidrug-resistant tuberculosis (MDR-TB): Evidence and perspectives. J. Thorac. Dis. 2016, 8, 2666–2671. [Google Scholar] [CrossRef]
- Karim, Q.A.; Karim, S.S.A. The evolving HIV epidemic in South Africa. Int. J. Epidemol. 2002, 31, 37–40. [Google Scholar] [CrossRef]
- The Urgency of Now: AIDS at a Crossroads; Joint United Nations Programme on HIV/AIDS: Geneva, Switzerland, 2024; Available online: https://www.unaids.org/sites/default/files/media_asset/2024-unaids-global-aids-update_en.pdf (accessed on 17 March 2025).
- Maeda, K.; Das, D.; Kobayakawa, T.; Tamamura, H.; Takeuchi, H. Discovery and development of anti-HIV therapeutic agents: Progress towards improved HIV medication. Curr. Top. Med. Chem. 2019, 19, 1621–1649. [Google Scholar] [CrossRef]
- Phanuphak, N.; Gulick, R.M. HIV treatment and prevention 2019: Current standards of care. Curr. Opin. HIV AIDS 2020, 15, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Arts, E.J.; Hazuda, D.J. HIV-1 antiretroviral drug therapy. Cold Spring Harb. Perspect. Med. 2012, 2, a007161. [Google Scholar] [CrossRef]
- Holec, A.D.; Mandal, S.; Prathipati, P.K.; Destache, C.J. Nucleotide reverse transcriptase inhibitors: A thorough review, present status and tuture perspective as HIV therapeutics. Curr. HIV Res. 2017, 15, 411–421. [Google Scholar] [CrossRef]
- Koren, D.E. 225 Classes of antiretrovirals. In Fundamentals of HIV Medicine; Oxford University Press: New York, NY, USA, 2020; pp. 225–238. [Google Scholar]
- Di, L.; Kerns, E.H. Drug-Like Properties: Concepts, Structure Design and Methods from ADME to Toxicity Optimization; Academic Press: San Jose, CA, USA, 2012. [Google Scholar]
- Sluis-Cremer, N.; Tachedjian, G. Mechanisms of inhibition of HIV replication by non-nucleoside reverse transcriptase inhibitors. Virus Res. 2008, 134, 147–156. [Google Scholar] [CrossRef]
- Ramprasad, J.; Sthalam, V.K.; Thampunuri, R.L.M.; Bhukya, S.; Ummanni, R.; Balasubramanian, S.; Pabbaraja, S. Synthesis and evaluation of a novel quinoline-triazole analogs for antitubercular properties via molecular hybridization approach. Bioorg. Med. Chem. Lett. 2019, 29, 126671. [Google Scholar] [CrossRef]
- Thomas, K.D.; Adhikari, A.V.; Chowdhury, I.H.; Sumesh, E.; Pal, N.K. New quinolin-4-yl-1,2,3-triazoles carrying amides, sulphonamides and amidopiperazines as potential antitubercular agents. Eur. J. Med. Chem. 2011, 46, 2503–2512. [Google Scholar] [CrossRef] [PubMed]
- Nyoni, N.T.P.; Ncube, N.B.; Kubheka, M.X.; Mkhwanazi, N.P.; Senzani, S.; Tukulula, M. Synthesis, characterization, in vitro antimycobacterial and cytotoxicity evaluation, DFT calculations, molecular docking and ADME studies of new isomeric benzimidazole-1,2,3-triazole-quinoline hybrid mixtures. Bioorg. Chem. 2023, 141, 106904. [Google Scholar] [CrossRef]
- Reddyrajula, R.; Dalimba, U. Quinoline-1,2,3-triazole hybrids: Design and synthesis through Click Reaction, evaluation of antitubercular activity, molecular docking and in silico ADME studies. ChemistrySelect 2019, 4, 2685–2693. [Google Scholar] [CrossRef]
- Ganesan, M.S.; Raja, K.K.; Murugesan, S.; Karankumar, B.; Faheem, F.; Thirunavukkarasu, S.; Shetye, G.; Ma, R.; Franzablau, S.G.; Wan, B.; et al. Quinoline-proline, triazole hybrids: Design, synthesis, antituberculosis, molecular docking, and ADMET studies. J. Heterocycl. Chem. 2021, 58, 952–968. [Google Scholar] [CrossRef]
- Yadav, J.; Kaushik, C.P. Quinoline-1,2,3-triazole hybrids: Design, synthesis, antimalarial and antimicrobial evaluation. J. Mol. Struct. 2024, 1316, 138882. [Google Scholar] [CrossRef]
- Thakare, P.P.; Shinde, A.D.; Chavan, A.P.; Nyayanit, N.V.; Bobade, V.D.; Mhaske, P.C. Synthesis and biological evaluation of new 1,2,3-triazolyl-pyrazolyl-quinoline derivatives as potential antimicrobial agents. ChemistrySelect 2020, 5, 4722–4727. [Google Scholar] [CrossRef]
- Jamshidi, H.; Naimi-Jamal, M.R.; Safavi, M.; Sanati, K.R.; Azerang, P.; Tahhighi, A. Synthesis and biological activity profile of novel triazole/quinoline hybrids. Chem. Biol. Drug Des. 2022, 100, 935–946. [Google Scholar] [CrossRef]
- Bhoye, M.R.; Shinde, A.; Shaik, A.L.N.; Shisode, V.; Chavan, A.; Maliwal, D.; Pisssurlenkar, R.R.S. New thiazolyl-isoxazole derivatives as potential anti-infective agents: Design, synthesis, in vitro and in silico antimicrobial efficacy. J. Biomol. Struct. Dynamic. 2024, 23, 1–15. [Google Scholar] [CrossRef]
- Costa, C.C.P.; Boechat, N.; Bastos, M.M.; da Silva, F.-d.C.; Marttorelli, A.; Souza, T.M.L.; Baptista, M.S.; Hoelz, L.V.B.; Caffarena, E.R. New efavirenz derivatives and 1,2,3-triazolyl-phosphonates as inhibitors of reverse transcriptase of HIV-1. Curr. Top. Med. Chem. 2018, 18, 1494–1505. [Google Scholar] [CrossRef]
- Feng, L.S.; Zheng, M.-J.; Zhao, F.; Liu, D. 1,2,3-Triazole hybrids with anti-HIV-1 activity. Arch. Pharm. 2021, 354, e2000163. [Google Scholar] [CrossRef]
- Tian, Y.; Liu, Z.; Huang, B.; Kang, D.; Zhang, H.; De Clercq, E.; Daelemans, D.; Pannecouque, C.; Leé, K.-H.; Chen, C.-H.; et al. Targeting the entrance channel of NNIBP: Discovery of diarylnicotinamide 1,4-disubstituted 1,2,3-triazoles as novel HIV-1 NNRTIs with high potency against wild-type and E138K mutant virus. Eur. J. Med. Chem. 2018, 151, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Liu, T.; Wu, G.; Kang, D.; Fu, Z.; Wang, Z.; De Clercq, E.; Pannecouque, C.; Zhan, P.; Liu, X. Targeting the hydrophobic channel of NNIBP: Discovery of novel 1,2,3-triazole-derived diarylpyrimidines as novel HIV-1 NNRTIs with high potency against wild-type and K103N mutant virus. Org. Biomol. Chem. 2019, 17, 3202–3217. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Akthar, J.; Ahmad, M.; Khan, M.I.; Wasim, R.; Islam, A.; Singh, A. Bedaquiline: An Insight Into its clinical use in multidrug-resistant pulmonary tuberculosis. Drug. Res. 2024, 74, 269–279. [Google Scholar] [CrossRef]
- Sutherland, H.S.; Tong, A.S.T.; Choi, P.J.; Blaser, A.; Conobe, D.; Franzblau, S.G.; Lotlikar, M.U.; Cooper, C.B.; Upton, A.M.; Denny, W.A.; et al. 3,5-Dialkoxypyridine analogues of bedaquiline are potent antituberculosis agents with minimal inhibition of the hERG channel. Bioorg. Med. Chem. 2019, 27, 1292–1307. [Google Scholar] [CrossRef]
- Emu, B.; Fessel, J.; Schrader, S.; Kumar, P.; Richmond, G.; Win, S.; Weinheimer, S.; Marsolais, C.; Lewis, S. Phase 3 study of ibalizumab for multidrug-resistant HIV-1. N. Engl. J. Med. 2018, 379, 645–654. [Google Scholar] [CrossRef]
- Xu, G.G.; Guo, J.; Wu, Y. Chemokine Receptor CCR5 antagonist Maraviroc: Medicinal chemistry and clinical applications. Curr. Top. Med. Chem. 2014, 14, 1504–1514. [Google Scholar] [CrossRef]
- Price, D.A.; Armour, D.; de Groot, M.; Leishman, D.; Napier, C.; Perros, M.; Stammen, B.L.; Wood, A. Overcoming hERG affinity in the discovery of maraviroc; a CCR5 antagonist for the treatment of HIV. Bioorg. Med. Chem. Lett. 2006, 16, 4633–4637. [Google Scholar] [CrossRef] [PubMed]
- Manosuthi, W.; Wiboonchutikul, S.; Sungkanuparph, S. Integrated therapy for HIV and tuberculosis. AIDS Res. Ther. 2016, 13, 22. [Google Scholar] [CrossRef]
- Khan, S.A.; Akthar, M.J.; Gogoi, U.; Meenakshi, D.U.; Das, A. An overview of 1,2,3-triazole-containing hybrids and their potential anticholinesterase activities. Pharmaceuticals 2023, 16, 179. [Google Scholar] [CrossRef]
- Eswaran, S.; Adhikari, A.V.; Shetty, N.S. Synthesis and antimicrobial activities of novel quinoline derivatives carrying 1,2,4-triazole moiety. Eur. J. Med. Chem. 2009, 44, 4637–4647. [Google Scholar] [CrossRef]
- Chen, Y.; Dubrovskiy, A.; Larock, R.C. Synthesis of quinolines by electrophilic cyclization of N-(2-alkynyl)anilines: 3-Iodo-4-phenylquinoline. Org. Synth. 2012, 89, 294–306. [Google Scholar] [CrossRef]
- Jiang, Y.-B.; Zhang, W.-S.; Cheng, H.-L.; Liu, Y.-Q.; Yang, R. One-pot synthesis of N-aryl propaylamine from aromatic boronic acid, aqueous ammonia, and propargyl bromide under microwave-assisted conditions. Chin. Chem. Lett. 2014, 25, 779–782. [Google Scholar] [CrossRef]
- Mao, L.-F.; Xu, G.-Q.; Sun, B.; Jiang, Y.-Q.; Dong, W.-P.; Zhang, S.-T.; Shen, J.-X.; He, X. Design, synthesis and antibacterial evaluation of novel 1,2,2-triazole derivatives incorporating 3′-deoxythymide. J. Chem. Res. 2017, 41, 645–649. [Google Scholar] [CrossRef]
- Rodriguez, Y.A.; Gutiérres, M.; Ramírez, D.; Alzate-Morales, J.; Bernal, C.C.; Güiza, F.M.; Bohórquez, A.R.R. Novel N-allyl/propargyl tetrahydroquinolines: Sythesis via thre-component cationic imino Diels-Alder reaction, bidning prediction, and evaluation as cholinesterae inhibitor. Chem. Biol. Drug Des. 2016, 88, 498–510. [Google Scholar] [CrossRef]
- Casano, G.; Dumétre, A.; Panneouque, C.; Hutter, S.; Azas, N.; Robin, M. Anti-HIV and antiplasmodial activity of original flavonoid derivatives. Bioorg. Med. Chem. 2010, 18, 6012–6023. [Google Scholar] [CrossRef]
- Indrayanto, G.; Putra, G.S.; Suhud, F. Chapter Six—Validation of in-vitro bioassay methods: Application in herbal drug research. in Profiles of Drug Substances, Excipients and Related Methodology, A.A. Al-Majed, Editor. Profiles Drug Subst. Excip. Relat. Methodol. 2021, 46, 273–307. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Mehra, R.; Sharma, S.; Bokolia, N.P.; Raina, D.; Nargotra, A.; Singh, P.P.; Khan, I.A. Screening of antitubercular compound library identifies novel ATP synthase inhibitors of Mycobacterium tuberculosis. Tuberculosis 2018, 108, 56–63. [Google Scholar] [CrossRef] [PubMed]
- El-Zohairy, M.A.; Zlotos, D.P.; Berger, M.R.; Adwan, H.H.; Mandour, Y.M. Discovery of Novel CCR5 Ligands as anticolorectal cancer agents by sequential virtual screening. ACS Omega 2021, 6, 10921–10935. [Google Scholar] [CrossRef] [PubMed]
- Vadivelu, A.; Gopal, V.; Reddy, C.U.M. Molecular docking studies of 1, 3, 4-thiadiazoles as novel peptide deformylase inhibitors as potential antibacterial agents. Int. J. Pharm. Sci. Rev. Res. 2015, 31, 58–62. [Google Scholar]
- Shah, P.; Naik, D.; Jariwala, N.; Bhadane, D.; Kumar, S.; Kulkarni, S.; Bhutani, K.K.; Singh, I.P. Synthesis of C-2 and C-3 substituted quinolines and their evaluation as anti-HIV-1 agents. Bioorg. Chem. 2018, 80, 591–601. [Google Scholar] [CrossRef]
- Ibrahim, T.S.; Bokhtia, R.; Al-Mahmoudy, A.M.M.; Taher, E.S.; AlAwadh, M.A.; Elagawany, M.; Abdel-Aal, E.H.; Panda, S.; Gouda, A.M.; Asfour, H.Z.; et al. Design, synthesis and biological evaluation of novel 5-((substituted quinolin-3-yl/1-naphthyl) methylene)-3-substituted imidazolidin-2,4-dione as HIV-1 fusion inhibitors. Bioorg. Chem. 2020, 99, 103782. [Google Scholar] [CrossRef]
- Oyeneyin, O.E.; Ojo, N.D.; Ipinlogu, N.; James, A.C.; Agbaffa, E.B. Investigation of corrosion inhibition potentials of some aminopyridine Schiff bases using density functional theory and Monte Carlo simulation. Chem. Afr. 2022, 5, 319–322. [Google Scholar] [CrossRef]
- Zhan, C.-G.; Nichols, J.A.; Dixon, D.A. Ionization potential, electron affinity, electronegativity, dardness, and electron excitation energy: Molecular properties from density functional theory orbital energies. J. Phys. Chem. A 2003, 107, 4184–4195. [Google Scholar] [CrossRef]
- Kadi, I.; Güldeniz, S.; Bouled, H.; Zebbiche, Z.; Suat, T.; Fatümetüzzehra, K.; Gönül, Z.; Küçükbay, H.; Boumoud, T. Synthesis, cytotoxicity, antioxidant activity, DFT calculations, and docking studies of new pyridine-malonate derivatives as potential anticancer agents. Polycycl. Aromat. Compd. 2022, 44, 6615–6629. [Google Scholar] [CrossRef]
- Azzouzi, M.; Ouchaoui, A.A.; Azougagh, O.; El Hadad, S.E.; Abou-Salama, M.; Oussaid, A.; Pannecouque, C.; Rohand, T. Synthesis, crystal structure, and antiviral evaluation of new imidazopyridine-schiff base derivatives: In vitro and in silico anti-HIV studies. RSC Advances 2024, 14, 36902–36918. [Google Scholar] [CrossRef]
- Morad, R.; Akbari, M.; Maaza, M. Theoretical study of chemical reactivity descriptors of some repurposed drugs for COVID-19. MRS Adcances 2023, 8, 656–660. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.K.; Bajpai, A.K.; Kumar, A.; Pal, M.; Baboo, V.; Dwivedi, A. Isolation, identification, molecular and electronic tructure, vibrational spectroscopi investigation, and anti-HIV-1 activity of Karanjin using density functional theory. J. Theor. Chem. 2014, 680987. [Google Scholar] [CrossRef]
- Hussain, Z.; Ibrahim, M.A.; Hassanin, N.M.; Badran, A.-S. Synthetic approaches for novel annulated pyrido[2,3-d]pyrimidines: Design, structural sharacterization, Fukui functions, DFT calculations, molecular docking and anticancer efficiency. J. Mol. Struct. 2024, 1318, 139335. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Properties of FDA-approved small molecule protein kinase inhibitors. Pharmacol. Res. 2024, 200, 107059. [Google Scholar] [CrossRef] [PubMed]
- QikProp User Manual, Schrödinger Release 2025-1: QuikProp; Schrödinger, LLC: New York, NY, USA, 2025.
- Wei, X.; Decker, M.J.; Liu, H.; Zhang, Z.; Arani, R.B.; Kilny, J.M.; Saag, M.S.; Wu, X.; Shaw, G.M.; Kappes, J.C. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob. Agents Chemother. 2002, 46, 1896–1905. [Google Scholar] [CrossRef]
- Naidu, D.; Oduro-Kwateng, E.; Solima, M.E.S.; Ndlovu, S.I.; Mkhwanazi, N.P. Alternaria alternata (Fr) Keissl crude extract inhibits HIV subtypes and integrase drug-resistant strains at different stages of HIV replication. Pharmaceuticals 2025, 18, 189. [Google Scholar] [CrossRef]
- Sastry, M.G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Mohammadpourasl, S.; de Biani, F.F.; Coppola, C.C.; Parisi, M.L.; Zani, L.; Dessi, A.; Calamante, M.; Reginato, G.; Basosi, R.; Sinicropi, A. Ground-State redox potentials calculations of D-π-A and D-A-π-A organic dyes for DSSC and visible-light-driven hydrogen production. Energies 2020, 13, 2032. [Google Scholar] [CrossRef]
- Aslam, R.; Serdaoglu, G.; Zehra, S.; Verma, K.D.; Aslam, J.; Guo, L.; Verma, C.; Ebenzo, E.E.; Quraishi, M.A. Corrosion inhibition of steel using different families of organic compounds: Past and present progress. J. Mol. Liq. 2022, 348, 118373. [Google Scholar] [CrossRef]
- Ibrahim, M.A.; Roushdy, N.; Atta, A.A.; Badran, A.-S.; Farag, A.A.M. Comprehensive study on pyrano[3,2-c]quinoline-based indole: Synthesis, characterization, and potential for optoelectronic and photovoltaic applications. J. Mol. Struct. 2024, 1312, 138660. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Compound | Phenyl | Appearance | % Yield | m.p. (°C) | MS Data |
| 11a |  | cream white solid | 88 | 150–152 | 306.0968 [(M − H) − N2]− |
| 11b | 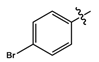 | light brown solid | 87 | 188–190 | 449.9707 [(M + HCl]− |
| 11c |  | brown solid | 92 | 198–199 | 495.9821 [(M − H) + Cl]− |
| 11d |  | light grey solid | 79 | 145–147 | 324.0866 [(M − H) − N2]− |
| 11e |  | Yellow solid | 69 | 169–171 | 340.0580 [(M − H) − N2)]− |
| 11f | 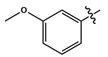 | dark brown liquid | 43 | - | 388.1096 (M + Na)+ |
| 11g |  | yellow liquid | 48 | - | 404 (M + 1) |
| 11h |  | cream-white solid | 85 | 145–147 | 324.0868 [(M − H) − N2]− |
| 11i |  | orange solid | 79 | 192–194 | 381.2533 (M + 1)− |
| 11j |  | brown solid | 86 | 169–172 | 336.1097 [(M − H) − N2]− |
| Compound | Anti-Mtb (µM) | Anti-HIV (µM) | Cytotoxicity CC50 (µM) | SI a |
|---|---|---|---|---|
| H37Rv MIC90 | HIV-1 Subtype B IC50 | TZM-bl Cell Line | (CC50/IC50) | |
| 11a | 186.52 | 3.013 | 177.1 | 58.75 |
| 11b | 1210.65 | 124.4 | 0.248 | 1.994 × 10−3 |
| 11c | 1084.6 | 23.20 | 156.9 | 6.76 |
| 11d | 176.52 | DNC * | 1320.0 | - |
| 11e | 168.81 | 713.7 | 834.6 | 1.17 |
| 11f | 171.19 | 22.75 | 3.599 | 0.158 |
| 11g | 155.05 | 0.3883 | 4414 | 11,367.49 |
| 11h | 88.72 | 0.01032 | 25.52 | 2472.87 |
| 11i | nd # | 0.167 | 0.00901 | 0.05 |
| 11j | 1369.86 | 180.4 | 4.000 | 0.02 |
| 7 | 19.09 | nd # | nd # | - |
| 10a | 3814 | nd # | nd # | - |
| Ethambutol | 9.68 | - | - | - |
| AZT | - | 0.0909 | 1122.58 | 12,349.59 |
| Compound | Docking Scores 4V1F (kcal mol−1) | Docking Scores 4MBS (kcal mol−1) |
|---|---|---|
| 11a | −2.540 | −6.990 |
| 11b | −2.291 | −6.729 |
| 11c | −2.339 | −6.899 |
| 11d | −2.528 | −7.561 |
| 11e | −2.879 | −7.371 |
| 11f | −2.035 | −4.815 |
| 11g | −2.714 | −6.427 |
| 11h | −2.606 | −7.362 |
| 11i | −2.479 | −5.825 |
| 11j | −2.570 | −5.301 |
| Compound | EHOMO (eV) | ELUMO (eV) | I (eV) | A (eV) | Eg (eV) | η (eV) | S (eV−1) | χ (eV) | ω (eV) |
|---|---|---|---|---|---|---|---|---|---|
| 11h | −5.89 | −2.64 | 5.89 | 2.64 | 3.25 | 1.63 | 0.615 | 4.27 | 5.59 |
| Compound | QPlogKhsa | QPlogS | % Human Oral Absorption | QPlogBB | CNS | #metab | Ro5 |
|---|---|---|---|---|---|---|---|
| 11a | 0.555 | −5.804 | 100.000 | −0.293 | 0 | 5 | 0 |
| 11b | 0.697 | −6.667 | 100.000 | −0.127 | 0 | 4 | 0 |
| 11c | 0.723 | −6.793 | 100.000 | −0.116 | 0 | 4 | 1 |
| 11d | 0.597 | −6.169 | 100.000 | −0.186 | 0 | 4 | 0 |
| 11e | 0.673 | −6.548 | 100.000 | −0.137 | 0 | 5 | 0 |
| 11f | 0.549 | −5.946 | 100.000 | −0.329 | 0 | 6 | 0 |
| 11g | 0.779 | −6.922 | 100.000 | −0.022 | 0 | 6 | 1 |
| 11h | 0.598 | −6.169 | 100.000 | −0.187 | 0 | 6 | 0 |
| 11i | 0.500 | −5.932 | 88.989 | −1.381 | −2 | 6 | 0 |
| 11j | 0.545 | −5.938 | 100.000 | −0.369 | 0 | 5 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Magwaza, S.S.; Naidu, D.; Oyeneyin, O.E.; Senzani, S.; Mkhwanazi, N.P.; Tukulula, M. Synthesis, Characterisation, Biological Evaluation and In Silico Studies of Quinoline–1,2,3-Triazole–Anilines as Potential Antitubercular and Anti-HIV Agents. Molecules 2025, 30, 2119. https://doi.org/10.3390/molecules30102119
Magwaza SS, Naidu D, Oyeneyin OE, Senzani S, Mkhwanazi NP, Tukulula M. Synthesis, Characterisation, Biological Evaluation and In Silico Studies of Quinoline–1,2,3-Triazole–Anilines as Potential Antitubercular and Anti-HIV Agents. Molecules. 2025; 30(10):2119. https://doi.org/10.3390/molecules30102119
Chicago/Turabian StyleMagwaza, Snethemba S., Darian Naidu, Oluwatoba E. Oyeneyin, Sibusiso Senzani, Nompumelelo P. Mkhwanazi, and Matshawandile Tukulula. 2025. "Synthesis, Characterisation, Biological Evaluation and In Silico Studies of Quinoline–1,2,3-Triazole–Anilines as Potential Antitubercular and Anti-HIV Agents" Molecules 30, no. 10: 2119. https://doi.org/10.3390/molecules30102119
APA StyleMagwaza, S. S., Naidu, D., Oyeneyin, O. E., Senzani, S., Mkhwanazi, N. P., & Tukulula, M. (2025). Synthesis, Characterisation, Biological Evaluation and In Silico Studies of Quinoline–1,2,3-Triazole–Anilines as Potential Antitubercular and Anti-HIV Agents. Molecules, 30(10), 2119. https://doi.org/10.3390/molecules30102119