
Aminoquinoline-Based Tridentate (NNN)-Copper Catalyst for C–N Bond-Forming Reactions from Aniline and Diazo Compounds


 , , and
, , and
Abstract
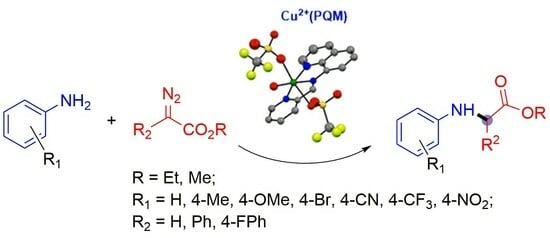
1. Introduction
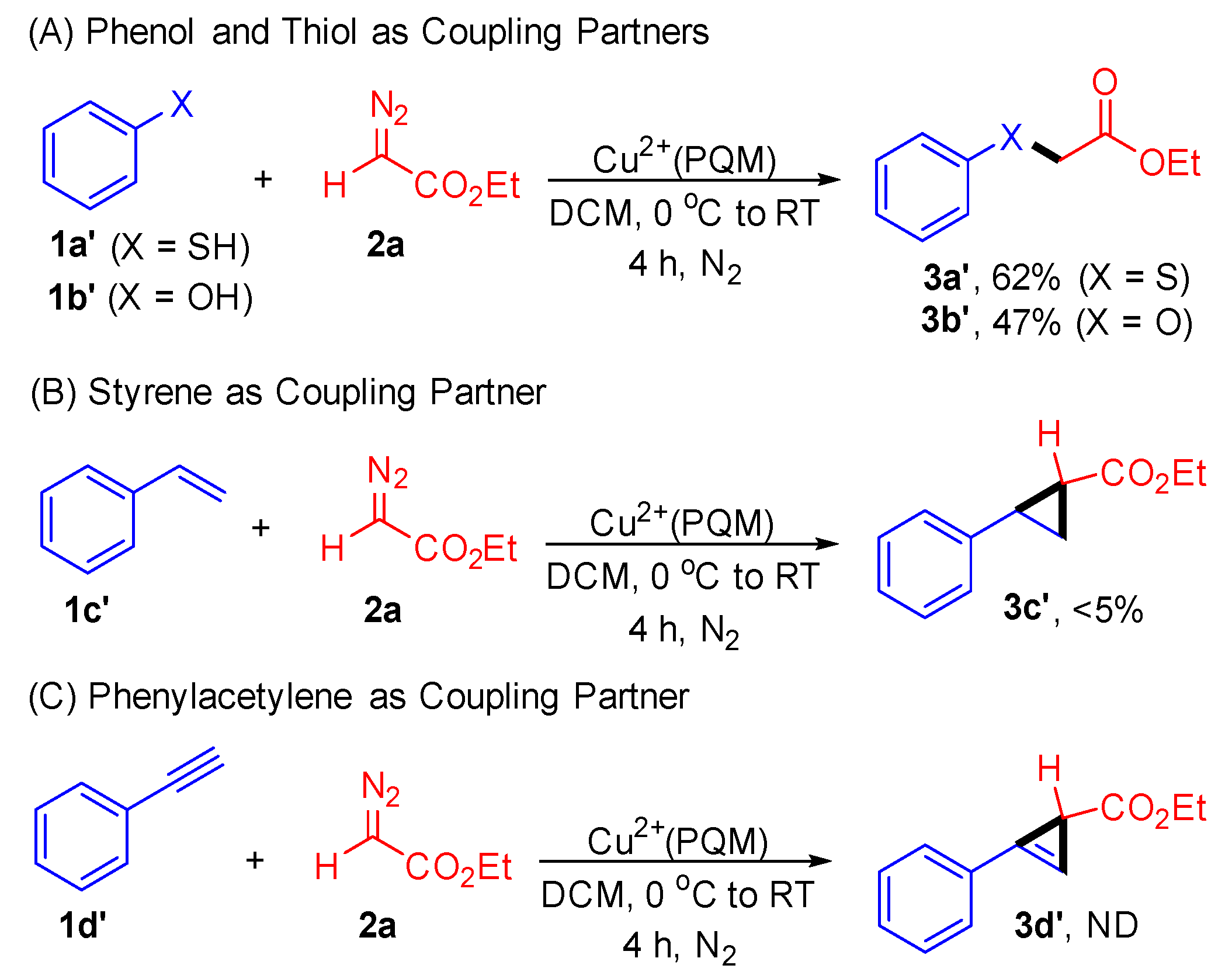
2. Results and Discussion
3. Materials and Methods
3.1. Synthesis of 1-(Pyridin-2-yl)-N-(Quinolin-8-yl)Methanimine (PQM) Ligand and Resulting Copper(II) Complexes
3.2. General Procedure for C–N Bond Construction via Cu2+(PQM)-Catalyzed Redox-Neutral Type Cross-Coupling (3a–3o)
3.3. Other Experimental Methods
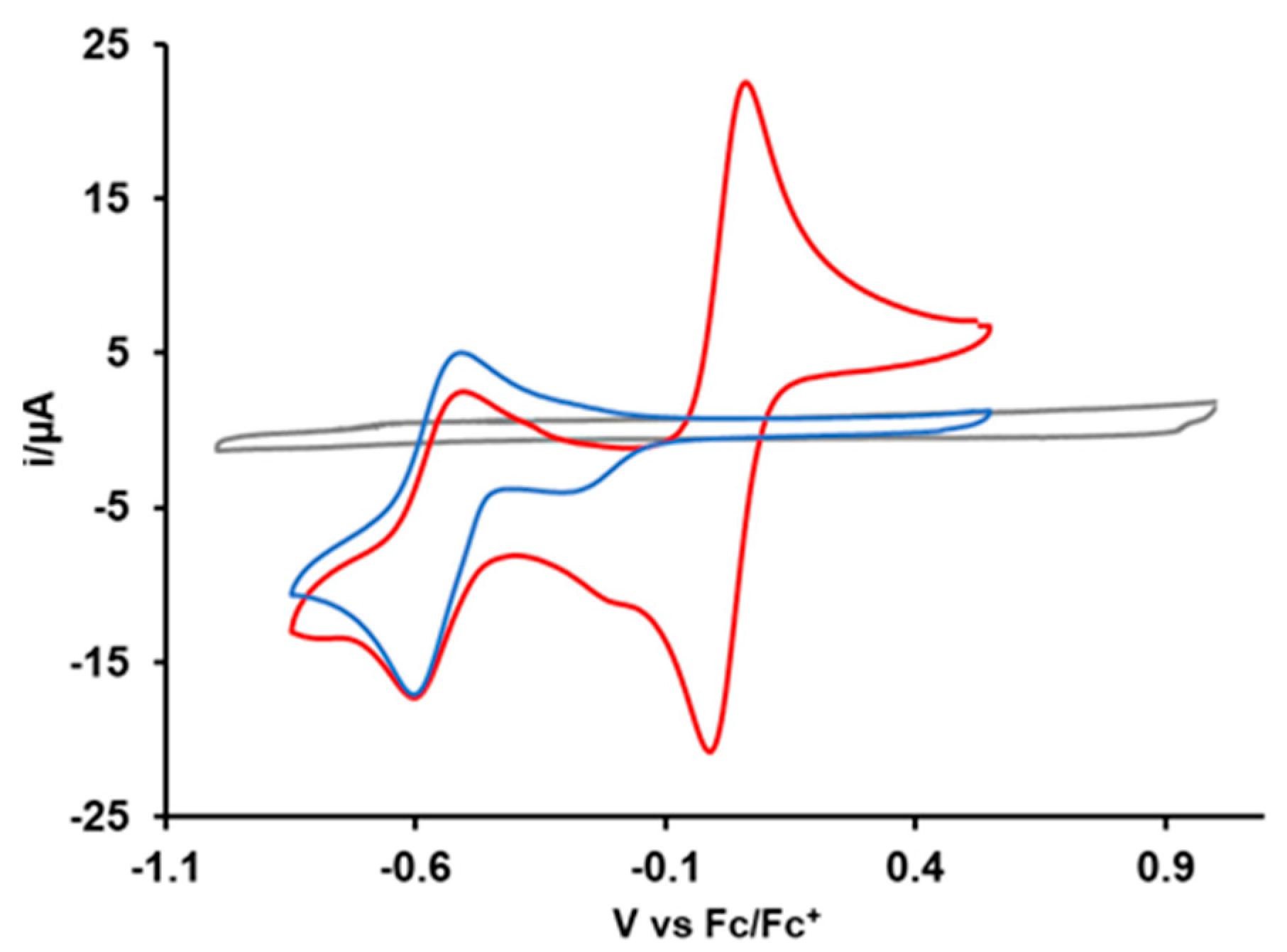
3.3.1. Cyclic Voltammetry
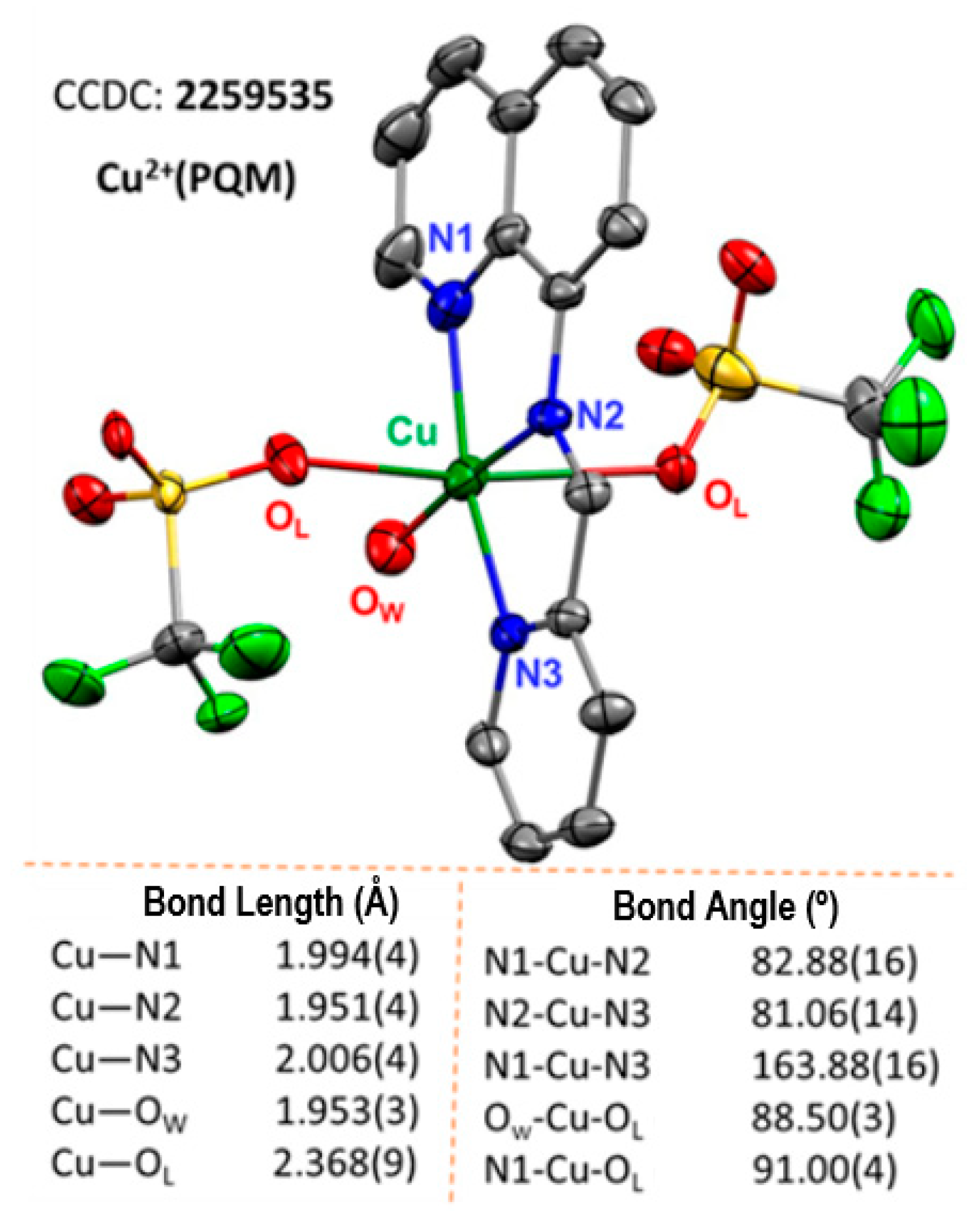
3.3.2. Single-Crystal X-ray Crystallography
3.3.3. UV–Vis Spectroscopy and Continuous Wave Electron Paramagnetic Resonance (CW EPR) Spectroscopy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Itoh, S. Developing Mononuclear Copper–Active-Oxygen Complexes Relevant to Reactive Intermediates of Biological Oxidation Reactions. Acc. Chem. Res. 2015, 48, 2066–2074. [Google Scholar] [CrossRef]
- Suzuki, M. Ligand Effects on Dioxygen Activation by Copper and Nickel Complexes: Reactivity and Intermediates. Acc. Chem. Res. 2007, 40, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Pelosi, G.; Bisceglie, F.; Bignami, F.; Ronzi, P.; Schiavone, P.; Re, M.C.; Casoli, C.; Pilotti, E. Antiretroviral Activity of Thiosemicarbazone Metal Complexes. J. Med. Chem. 2010, 53, 8765–8769. [Google Scholar] [CrossRef]
- Gou, Y.; Chen, M.; Li, S.; Deng, J.; Li, J.; Fang, G.; Yang, F.; Huang, G. Dithiocarbazate-Copper Complexes for Bioimaging and Treatment of Pancreatic Cancer. J. Med. Chem. 2021, 64, 5485–5499. [Google Scholar] [CrossRef] [PubMed]
- Bormio Nunes, J.H.; Hager, S.; Mathuber, M.; Pósa, V.; Roller, A.; Enyedy, É.A.; Stefanelli, A.; Berger, W.; Keppler, B.K.; Heffeter, P.; et al. Cancer Cell Resistance Against the Clinically Investigated Thiosemicarbazone COTI-2 Is Based on Formation of Intracellular Copper Complex Glutathione Adducts and ABCC1-Mediated Efflux. J. Med. Chem. 2020, 63, 13719–13732. [Google Scholar] [CrossRef] [PubMed]
- Tardito, S.; Barilli, A.; Bassanetti, I.; Tegoni, M.; Bussolati, O.; Franchi-Gazzola, R.; Mucchino, C.; Marchiò, L. Copper-Dependent Cytotoxicity of 8-Hydroxyquinoline Derivatives Correlates with Their Hydrophobicity and Does Not Require Caspase Activation. J. Med. Chem. 2012, 55, 10448–10459. [Google Scholar] [CrossRef]
- Santini, C.; Pellei, M.; Gandin, V.; Porchia, M.; Tisato, F.; Marzano, C. Advances in Copper Complexes as Anticancer Agents. Chem. Rev. 2014, 114, 815–862. [Google Scholar] [CrossRef]
- Karlin, K.D.; Gultneh, Y. Bioinorganic Chemical Modeling of Dioxygen-Activating Copper Proteins. J. Chem. Educ. 1985, 62, 983. [Google Scholar] [CrossRef]
- Lewis, E.A.; Tolman, W.B. Reactivity of Dioxygen-Copper Systems. Chem. Rev. 2004, 104, 1047–1076. [Google Scholar] [CrossRef]
- Hu, Q.-Q.; Su, X.-J.; Zhang, M.-T. Electrocatalytic Water Oxidation by an Unsymmetrical Di-Copper Complex. Inorg. Chem. 2018, 57, 10481–10484. [Google Scholar] [CrossRef]
- Shimizu, D.; Kurose, A.; Nishikata, T. Remote Nucleophilic Substitution at a C(sp3)–H Bond of α-Bromocarboxamides via 1,4-Hydrogen Atom Transfer To Access N-Acyl-N,O-Acetal Compounds. Org. Lett. 2022, 24, 7873–7877. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Górski, B.; Leonori, D. Merging Halogen-Atom Transfer (XAT) and Copper Catalysis for the Modular Suzuki–Miyaura-Type Cross-Coupling of Alkyl Iodides and Organoborons. J. Am. Chem. Soc. 2022, 144, 1986–1992. [Google Scholar] [CrossRef] [PubMed]
- Cope, J.D.; Valle, H.U.; Hall, R.S.; Riley, K.M.; Goel, E.; Biswas, S.; Hendrich, M.P.; Wipf, D.O.; Stokes, S.L.; Emerson, J.P. Tuning the Copper(II)/Copper(I) Redox Potential for More Robust Copper-Catalyzed C–N Bond Forming Reactions. Eur. J. Inorg. Chem. 2020, 2020, 1278–1285. [Google Scholar] [CrossRef] [PubMed]
- Cope, J.D.; Sheridan, P.E.; Galloway, C.J.; Awoyemi, R.F.; Stokes, S.L.; Emerson, J.P. Synthesis and Characterization of a Tetradentate, N-Heterocyclic Carbene Copper(II) Complex and Its Use as a Chan–Evans–Lam Coupling Catalyst. Organometallics 2020, 39, 4457–4464. [Google Scholar] [CrossRef]
- Raju, S.; Sheridan, P.E.; Hauer, A.K.; Garrett, A.E.; McConnell, D.E.; Thornton, J.A.; Stokes, S.L.; Emerson, J.P. Cu-Catalyzed Chan–Evans–Lam Coupling Reactions of 2-Nitroimidazole with Aryl Boronic Acids: An Effort toward New Bioactive Agents against S. Pneumoniae. Chem. Biodivers. 2022, 19, e202200327. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, B.; Teimouri, M.; Akin, J.W.; Raju, S.; Stokes, S.L.; Emerson, J.P. Cu−NHC Complex for Chan-Evans-Lam Cross-Coupling Reactions of N-Heterocyclic Compounds and Arylboronic Acids. Eur. J. Org. Chem. 2023, 26, e202300620. [Google Scholar] [CrossRef]
- Hassan, J.; Sévignon, M.; Gozzi, C.; Schulz, E.; Lemaire, M. Aryl−Aryl Bond Formation One Century after the Discovery of the Ullmann Reaction. Chem. Rev. 2002, 102, 1359–1470. [Google Scholar] [CrossRef] [PubMed]
- Evano, G.; Blanchard, N.; Toumi, M. Copper-Mediated Coupling Reactions and Their Applications in Natural Products and Designed Biomolecules Synthesis. Chem. Rev. 2008, 108, 3054–3131. [Google Scholar] [CrossRef]
- Allen, S.E.; Walvoord, R.R.; Padilla-Salinas, R.; Kozlowski, M.C. Aerobic Copper-Catalyzed Organic Reactions. Chem. Rev. 2013, 113, 6234–6458. [Google Scholar] [CrossRef]
- Guo, X.-X.; Gu, D.-W.; Wu, Z.; Zhang, W. Copper-Catalyzed C–H Functionalization Reactions: Efficient Synthesis of Heterocycles. Chem. Rev. 2015, 115, 1622–1651. [Google Scholar] [CrossRef]
- Maestre, L.; Ozkal, E.; Ayats, C.; Beltrán, Á.; Díaz-Requejo, M.M.; Pérez, P.J.; Pericàs, M.A. A Fully Recyclable Heterogenized Cu Catalyst for the General Carbene Transfer Reaction in Batch and Flow. Chem. Sci. 2015, 6, 1510–1515. [Google Scholar] [CrossRef]
- Kroitor, A.P.; Cailler, L.P.; Martynov, A.G.; Gorbunova, Y.G.; Tsivadze, A.Y.; Sorokin, A.B. Unexpected Formation of a μ-Carbido Diruthenium(Iv) Complex during the Metalation of Phthalocyanine with Ru3(CO)12 and Its Catalytic Activity in Carbene Transfer Reactions. Dalt. Trans. 2017, 46, 15651–15655. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-Q.; Li, J.-H.; Dong, Z.-B. A Review on the Latest Progress of Chan-Lam Coupling Reaction. Adv. Synth. Catal. 2020, 362, 3311–3331. [Google Scholar] [CrossRef]
- Seifinoferest, B.; Tanbakouchian, A.; Larijani, B.; Mahdavi, M. Ullmann-Goldberg and Buchwald-Hartwig C−N Cross Couplings: Synthetic Methods to Pharmaceutically Potential N-Heterocycles. Asian J. Org. Chem. 2021, 10, 1319–1344. [Google Scholar] [CrossRef]
- Hari, D.P.; Waser, J. Copper-Catalyzed Oxy-Alkynylation of Diazo Compounds with Hypervalent Iodine Reagents. J. Am. Chem. Soc. 2016, 138, 2190–2193. [Google Scholar] [CrossRef] [PubMed]
- Durka, J.; Turkowska, J.; Gryko, D. Lightening Diazo Compounds? ACS Sustain. Chem. Eng. 2021, 9, 8895–8918. [Google Scholar] [CrossRef]
- Dong, S.; Liu, X.; Feng, X. Asymmetric Catalytic Rearrangements with α-Diazocarbonyl Compounds. Acc. Chem. Res. 2022, 55, 415–428. [Google Scholar] [CrossRef]
- Ford, A.; Miel, H.; Ring, A.; Slattery, C.N.; Maguire, A.R.; McKervey, M.A. Modern Organic Synthesis with α-Diazocarbonyl Compounds. Chem. Rev. 2015, 115, 9981–10080. [Google Scholar] [CrossRef]
- Lee, E.C.; Fu, G.C. Copper-Catalyzed Asymmetric N−H Insertion Reactions: Couplings of Diazo Compounds with Carbamates to Generate α-Amino Acids. J. Am. Chem. Soc. 2007, 129, 12066–12067. [Google Scholar] [CrossRef]
- Xiang, Y.; Wang, C.; Ding, Q.; Peng, Y. Diazo Compounds: Versatile Synthons for the Synthesis of Nitrogen Heterocycles via Transition Metal-Catalyzed Cascade C–H Activation/Carbene Insertion/Annulation Reactions. Adv. Synth. Catal. 2019, 361, 919–944. [Google Scholar] [CrossRef]
- Khanal, H.D.; Thombal, R.S.; Maezono, S.M.B.; Lee, Y.R. Designs and Strategies for the Halo-Functionalization of Diazo Compounds. Adv. Synth. Catal. 2018, 360, 3185–3212. [Google Scholar] [CrossRef]
- Melby, T.; Hughes, R.A.; Hansen, T. H. Diastereoselective Synthesis of 1-Aryl-2-Amino-Cyclopropane Carboxylates. Synlett 2007, 2007, 2277–2279. [Google Scholar] [CrossRef]
- Cailler, L.P.; Kroitor, A.P.; Martynov, A.G.; Gorbunova, Y.G.; Sorokin, A.B. Selective Carbene Transfer to Amines and Olefins Catalyzed by Ruthenium Phthalocyanine Complexes with Donor Substituents. Dalt. Trans. 2021, 50, 2023–2031. [Google Scholar] [CrossRef] [PubMed]
- Kantam, M.L.; Laha, S.; Yadav, J.; Jha, S. Nanocrystalline Copper(II) Oxide Catalyzed Aza-Michael Reaction and Insertion of α-Diazo Compounds into N–H Bonds of Amines. Tetrahedron Lett. 2009, 50, 4467–4469. [Google Scholar] [CrossRef]
- Kantam, M.L.; Neelima, B.; Reddy, C.V. Cu(Acac)2 Immobilized in Ionic Liquids: A Reusable Catalytic System for the Insertion of α-Diazo Compounds into NH Bonds of Amines. J. Mol. Catal. A Chem. 2006, 256, 269–272. [Google Scholar] [CrossRef]
- Han, F.; Choi, P.H.; Ye, C.-X.; Grell, Y.; Xie, X.; Ivlev, S.I.; Chen, S.; Meggers, E. Cyclometalated Chiral-at-Ruthenium Catalyst for Enantioselective Ring-Closing C(sp3)–H Carbene Insertion to Access Chiral Flavanones. ACS Catal. 2022, 12, 10304–10312. [Google Scholar] [CrossRef]
- Tanbouza, N.; Keipour, H.; Ollevier, T. FeII-Catalysed Insertion Reaction of α-Diazocarbonyls into X–H Bonds (X = Si, S, N, and O) in Dimethyl Carbonate as a Suitable Solvent Alternative. RSC Adv. 2019, 9, 31241–31246. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, Y.-T.; Zhou, T.; Chen, M.-Q.; Li, Y.-P.; Huang, M.-Y.; Xu, Z.-C.; Zhu, S.-F.; Zhou, Q.-L. Highly Enantioselective O–H Bond Insertion Reaction of α-Alkyl- and α-Alkenyl-α-Diazoacetates with Water. J. Am. Chem. Soc. 2020, 142, 10557–10566. [Google Scholar] [CrossRef]
- Liu, B.; Zhu, S.-F.; Zhang, W.; Chen, C.; Zhou, Q.-L. Highly Enantioselective Insertion of Carbenoids into N−H Bonds Catalyzed by Copper Complexes of Chiral Spiro Bisoxazolines. J. Am. Chem. Soc. 2007, 129, 5834–5835. [Google Scholar] [CrossRef] [PubMed]
- Nam, D.; Tinoco, A.; Shen, Z.; Adukure, R.D.; Sreenilayam, G.; Khare, S.D.; Fasan, R. Enantioselective Synthesis of α-Trifluoromethyl Amines via Biocatalytic N–H Bond Insertion with Acceptor-Acceptor Carbene Donors. J. Am. Chem. Soc. 2022, 144, 2590–2602. [Google Scholar] [CrossRef]
- Ramakrishna, K.; Murali, M.; Sivasankar, C. Chemoselective Carbene Insertion into the N–H Bond over O–H Bond Using a Well-Defined Single Site (P–P)Cu(I) Catalyst. Org. Lett. 2015, 17, 3814–3817. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Zhang, Y.; Wang, J. Recent Developments in Copper-Catalyzed Reactions of Diazo Compounds. Chem. Commun. 2012, 48, 10162–10173. [Google Scholar] [CrossRef] [PubMed]
- Álvarez, M.; Besora, M.; Molina, F.; Maseras, F.; Belderrain, T.R.; Pérez, P.J. Two Copper-Carbenes from One Diazo Compound. J. Am. Chem. Soc. 2021, 143, 4837–4843. [Google Scholar] [CrossRef] [PubMed]
- Namitharan, K.; Zhu, T.; Cheng, J.; Zheng, P.; Li, X.; Yang, S.; Song, B.-A.; Chi, Y.R. Metal and Carbene Organocatalytic Relay Activation of Alkynes for Stereoselective Reactions. Nat. Commun. 2014, 5, 3982. [Google Scholar] [CrossRef] [PubMed]
- Efthimiadou, E.K.; Katsaros, N.; Karaliota, A.; Psomas, G. Synthesis, Characterization, Antibacterial Activity, and Interaction with DNA of the Vanadyl-Enrofloxacin Complex. Bioorg. Med. Chem. Lett. 2007, 17, 1238–1242. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, S.; Palaniandavar, M. Interaction of rac-[Cu(Diimine)3]2+ and rac-[Zn(Diimine)3]2+ Complexes with CT DNA: Effect of Fluxional Cu(II) Geometry on DNA Binding, Ligand–Promoted Exciton Coupling and Prominent DNA Cleavage. Dalt. Trans. 2008, 3866–3878. [Google Scholar] [CrossRef]
- Murphy, B.; Aljabri, M.; Ahmed, A.M.; Murphy, G.; Hathaway, B.J.; Light, M.E.; Geilbrich, T.; Hursthouse, M.B. Structural Systematics of the [Cu(Chelate)3][Y]2 Series. An Interesting Crystallographic Structural Insight Involving Vibronic Coupling and the Jahn–Teller Effect (JTE). The Syntheses and Low Temperature Crystal Structures of Tris(2,2′bipyridyl)Copper(II) Tetraphenylborate and Tris(2,2′bipyridyl)Zinc(II) Tetraphenylborate. Dalt. Trans. 2006, 357–367. [Google Scholar] [CrossRef]
- Lu, J.; Sun, Q.; Li, J.-L.; Jiang, L.; Gu, W.; Liu, X.; Tian, J.-L.; Yan, S.-P. Two Water-Soluble Copper(II) Complexes: Synthesis, Characterization, DNA Cleavage, Protein Binding Activities and in Vitro Anticancer Activity Studies. J. Inorg. Biochem. 2014, 137, 46–56. [Google Scholar] [CrossRef]
- Valle, H.U.; Riley, K.M.; Russell, D.E.; Wolgemuth, D.K.; Voges-Haupt, C.F.; Rogers, T.A.; Redd, S.L.; Stokes, S.L.; Emerson, J.P. Synthesis, Characterization, and Structure of a [Cu(Phen)2(OTf)]OTf Complex: An Efficient Nitrogen Transfer Pre-Catalyst. ChemistrySelect 2018, 3, 5143–5146. [Google Scholar] [CrossRef]
- Doyle, M.P. Catalytic Methods for Metal Carbene Transformations. Chem. Rev. 1986, 86, 919–939. [Google Scholar] [CrossRef]
- Lions, F.; Martin, K.V. Tridentate Chelate Compounds. I. J. Am. Chem. Soc. 1957, 79, 2733–2738. [Google Scholar] [CrossRef]
- Kinunda, G.; Jaganyi, D. Understanding the Electronic and π-Conjugation Roles of Quinoline on Ligand Substitution Reactions of Platinum(II) Complexes. Transit. Met. Chem. 2014, 39, 451–459. [Google Scholar] [CrossRef]
- Sorouraddin, M.-H.; Rashidi, M.-R.; Shabani, B.; Ghorbani-Kalhor, E. Spectrofluorimetric Determination of Cu2+ Using New Fluorogenic Reagent. Chin. J. Chem. 2005, 23, 160–165. [Google Scholar] [CrossRef]
- Palmer, G. Electron Paramagnetic Resonance of Metalloproteins. In Physical Methods in Bioinorganic Chemistry Spectroscopy and Magnetism; Que, L.J., Ed.; University Science Books: Sausalito, CA, USA, 2000; pp. 121–186. [Google Scholar]
- Hales, B.J. Electron Paramagnetic Resonance (EPR) Spectroscopy. In Applications of Physical Methods to Inorganic and Bioinorganic Chemistry; Scott, R.A., Lukehart, C.M., Eds.; John Wiley & Sons Ltd.: Chichester, UK, 2007; pp. 39–78. [Google Scholar]
- Petasis, D.T.; Hendrich, M.P. Chapter Eight—Quantitative Interpretation of Multifrequency Multimode EPR Spectra of Metal Containing Proteins, Enzymes, and Biomimetic Complexes. In Methods in Enzymology; Qin, P.Z., Warncke, K., Eds.; Academic Press: Cambridge, MA, USA, 2015; Volume 563, pp. 171–208. ISBN 0076-6879. [Google Scholar]
- Quach, T.D.; Batey, R.A. Ligand- and Base-Free Copper(II)-Catalyzed C−N Bond Formation:Cross-Coupling Reactions of Organoboron Compounds with Aliphatic Amines and Anilines. Org. Lett. 2003, 5, 4397–4400. [Google Scholar] [CrossRef]
- Sreenilayam, G.; Fasan, R. Myoglobin-Catalyzed Intermolecular Carbene N-H Insertion with Arylamine Substrates. Chem Comm. 2015, 51, 1532–1534. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, L.; Li, J.; Zheng, J.; Huang, J.; Qi, C.; Wu, W.; Jiang, H. Access to α-Amino Acid Esters through Palladium-Catalyzed Oxidative Amination of Vinyl Ethers with Hydrogen Peroxide as the Oxidant and Oxygen Source. Angew. Chem. Int. Ed. 2017, 56, 15926–15930. [Google Scholar] [CrossRef]
- Aviv, I.; Gross, Z. Iron(III) Corroles and Porphyrins as Superior Catalysts for the Reactions of Diazoacetates with Nitrogen- or Sulfur-Containing Nucleophilic Substrates: Synthetic Uses and Mechanistic Insights. Chem. A Eur. J. 2008, 14, 3995–4005. [Google Scholar] [CrossRef]
- Rohlmann, R.; Stopka, T.; Richter, H.; García Mancheño, O. Iron-Catalyzed Oxidative Tandem Reactions with TEMPO Oxoammonium Salts: Synthesis of Dihydroquinazolines and Quinolines. J. Org. Chem. 2013, 78, 6050–6064. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M.W.; Vargas, D.A.; Lehnert, N. Engineering of RuMb: Toward a Green Catalyst for Carbene Insertion Reactions. Inorg. Chem. 2017, 56, 5623–5635. [Google Scholar] [CrossRef]
- Abu-Elfotoh, A.-M. NH Insertion Reactions Catalyzed by Reusable Water-Soluble Ruthenium(II)-Hm-Phenyloxazoline Complex. Tetrahedron Lett. 2017, 58, 4750–4754. [Google Scholar] [CrossRef]
- Ueda, T.; Konishi, H.; Manabe, K. Trichlorophenyl Formate: Highly Reactive and Easily Accessible Crystalline CO Surrogate for Palladium-Catalyzed Carbonylation of Aryl/Alkenyl Halides and Triflates. Org. Lett. 2012, 14, 5370–5373. [Google Scholar] [CrossRef] [PubMed]
- Hattori, G.; Sakata, K.; Matsuzawa, H.; Tanabe, Y.; Miyake, Y.; Nishibayashi, Y. Copper-Catalyzed Enantioselective Propargylic Amination of Propargylic Esters with Amines: Copper−Allenylidene Complexes as Key Intermediates. J. Am. Chem. Soc. 2010, 132, 10592–10608. [Google Scholar] [CrossRef] [PubMed]
- Ugarriza, I.; Uria, U.; Carrillo, L.; Vicario, J.L.; Reyes, E. Base-Promoted C→N Acyl Rearrangement:An Unconventional Approach to α-Amino Acid Derivatives. Chem. A Eur. J. 2014, 20, 11650–11654. [Google Scholar] [CrossRef] [PubMed]
- Steck, V.; Sreenilayam, G.; Gillingham, D.; Fei, N.; Moody, C.; Zhu, S.; Zhou, Q.; Yates, P.; Moyer, M.; Feldman, P.; et al. Selective Functionalization of Aliphatic Amines via Myoglobin-Catalyzed Carbene N–H Insertion. Synlett 2020, 31, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Baumann, L.K.; Mbuvi, H.M.; Du, G.; Woo, L.K. Iron Porphyrin Catalyzed N−H Insertion Reactions with Ethyl Diazoacetate. Organometallics 2007, 26, 3995–4002. [Google Scholar] [CrossRef]
- Miura, T.; Morimoto, M.; Murakami, M. Copper-Catalyzed Amination of Silyl Ketene Acetals with N-Chloroamines. Org. Lett. 2012, 14, 5214–5217. [Google Scholar] [CrossRef]
- Deng, Q.-H.; Xu, H.-W.; Yuen, A.W.-H.; Xu, Z.-J.; Che, C.-M. Ruthenium-Catalyzed One-PotCarbenoid N−H Insertion Reactions and Diastereoselective Synthesis of Prolines. Org. Lett. 2008, 10, 1529–1532. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||
|---|---|---|---|
| Entry | Cu+/Cu2+ (mol %) | Solvent | Yields 3a % 2 |
| 1 | CuCl (10) | DCM | 20 |
| 2 | CuBr (10) | DCM | 15 |
| 3 | Cu(OTf) (10) | DCM | 48 |
| 4 | CuCl2 (10) | DCM | ND |
| 5 | CuBr2 (10) | DCM | ND |
| 6 | Cu(OAc)2 (10) | DCM | 38 |
| 7 | Cu2+(PQM) (10) | DCM | 72 |
| 8 | Cu2+(PQM) (10) | DMSO | ND |
| 9 | Cu2+(PQM) (10) | MeOH | 23 |
| 10 | Cu2+(PQM) (10) | H2O | 10 |
| 11 | Cu2+(PQM) (10) | 1,4-Dioxane | 12 |
| 12 | Cu2+(PQM) (10) | Chlorobenzene | 20 |
| 13 | Cu2+(PQM) (10) | CH3CN | 20 |
| 14 | Cu2+(PQM) (10) | DCM:MeOH | ND |
| 15 | Cu2+(PQM) (10) | DCM:H2O | 23 |
| 16 3 | Cu2+(PQM) (10) | DCM | 58 |
| 17 | Cu2+(PQM) (10) | Buffer 7.2 | 12 |
| 18 4 | Cu2+(PQM) (10) | DCM | 44 |
| 19 5 | Cu2+(PQM) (10) | DCM | 38 |
| 20 | Cu2+(PQM) (10) | oDCB | ND |
| 21 | Cu2+(PQM) (10) | DCE | 57 |
| 22 | Cu2+(PQM) (5) | DCM | 68 |
| 23 | Cu2+(PQM) (2) | DCM | 60 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Teimouri, M.; Raju, S.; Acheampong, E.; Schmittou, A.N.; Donnadieu, B.; Wipf, D.O.; Pierce, B.S.; Stokes, S.L.; Emerson, J.P. Aminoquinoline-Based Tridentate (NNN)-Copper Catalyst for C–N Bond-Forming Reactions from Aniline and Diazo Compounds. Molecules 2024, 29, 730. https://doi.org/10.3390/molecules29030730
Teimouri M, Raju S, Acheampong E, Schmittou AN, Donnadieu B, Wipf DO, Pierce BS, Stokes SL, Emerson JP. Aminoquinoline-Based Tridentate (NNN)-Copper Catalyst for C–N Bond-Forming Reactions from Aniline and Diazo Compounds. Molecules. 2024; 29(3):730. https://doi.org/10.3390/molecules29030730
Chicago/Turabian StyleTeimouri, Mohsen, Selvam Raju, Edward Acheampong, Allison N. Schmittou, Bruno Donnadieu, David O. Wipf, Brad S. Pierce, Sean L. Stokes, and Joseph P. Emerson. 2024. "Aminoquinoline-Based Tridentate (NNN)-Copper Catalyst for C–N Bond-Forming Reactions from Aniline and Diazo Compounds" Molecules 29, no. 3: 730. https://doi.org/10.3390/molecules29030730
APA StyleTeimouri, M., Raju, S., Acheampong, E., Schmittou, A. N., Donnadieu, B., Wipf, D. O., Pierce, B. S., Stokes, S. L., & Emerson, J. P. (2024). Aminoquinoline-Based Tridentate (NNN)-Copper Catalyst for C–N Bond-Forming Reactions from Aniline and Diazo Compounds. Molecules, 29(3), 730. https://doi.org/10.3390/molecules29030730