
Synthesis of New Pyrazolo[3,4-d]pyrimidine Derivatives: NMR Spectroscopic Characterization, X-Ray, Hirshfeld Surface Analysis, DFT, Molecular Docking, and Antiproliferative Activity Investigations

 , , , , , , , and
, , , , , , , and
Abstract
1. Introduction
2. Results and Discussion
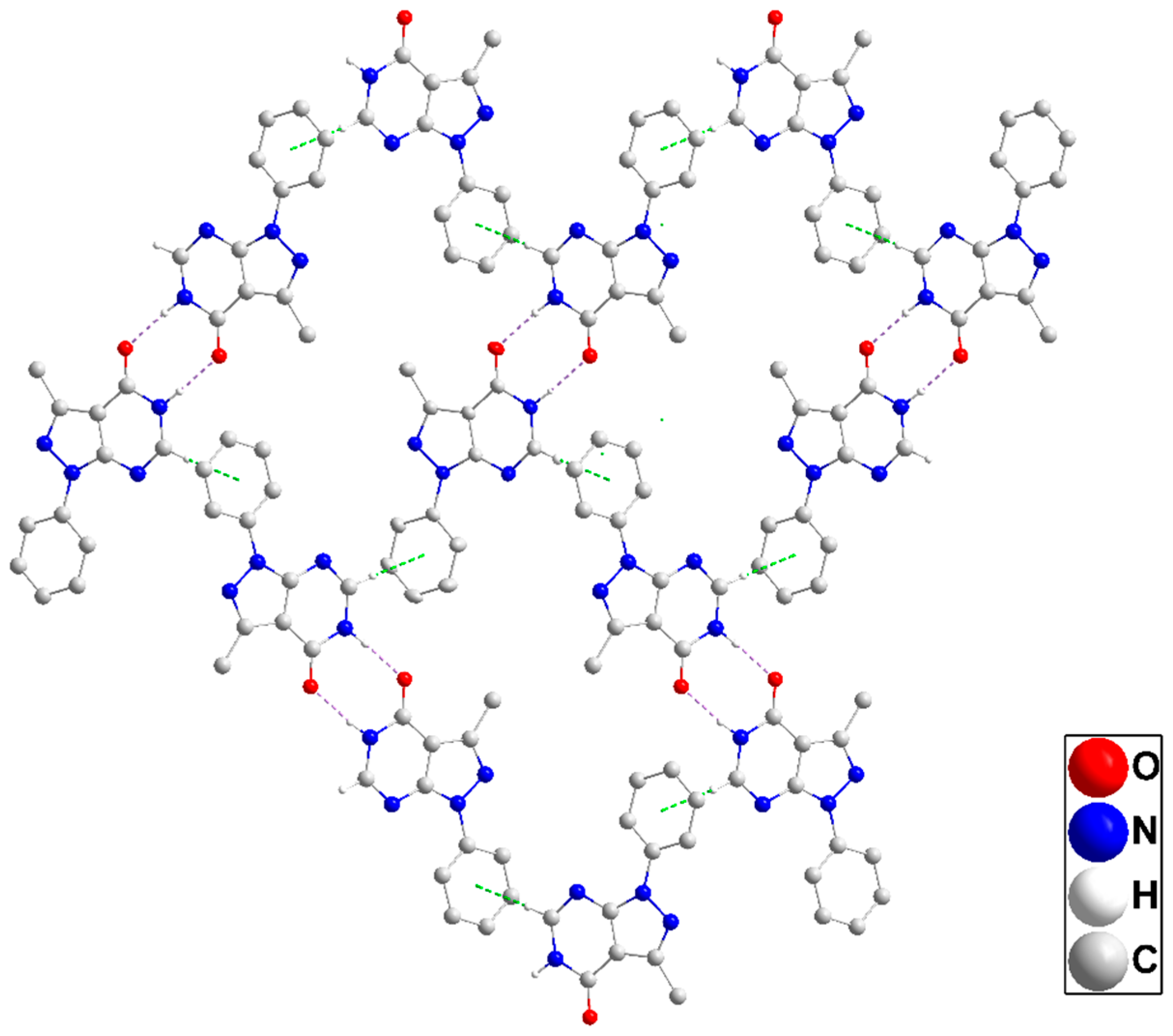
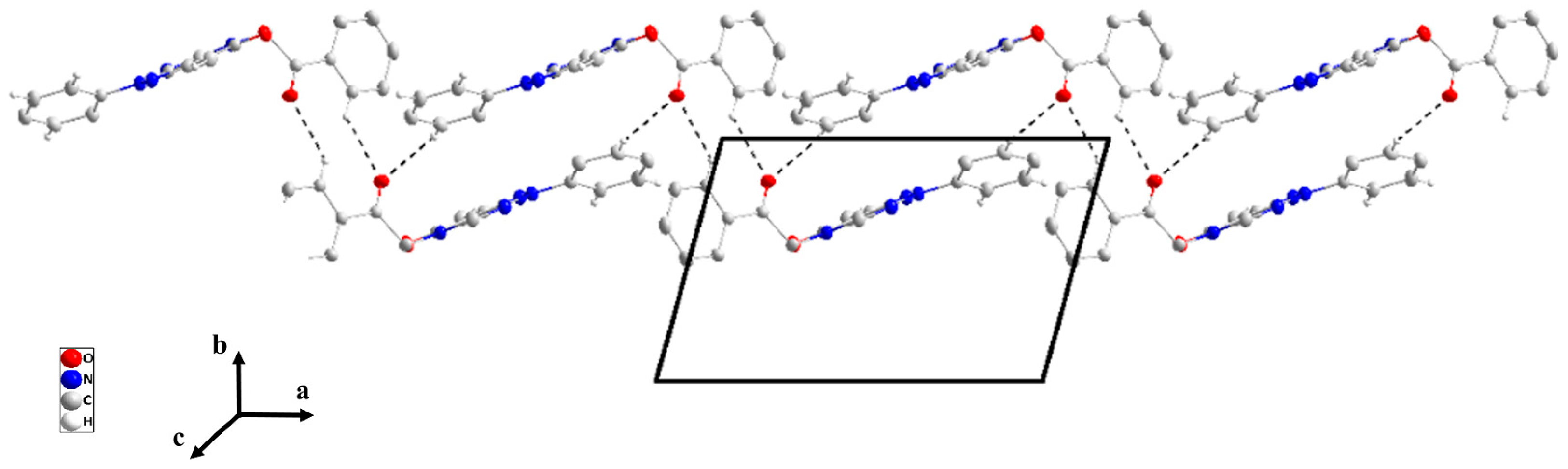
2.1. X-Ray Structure Descriptions
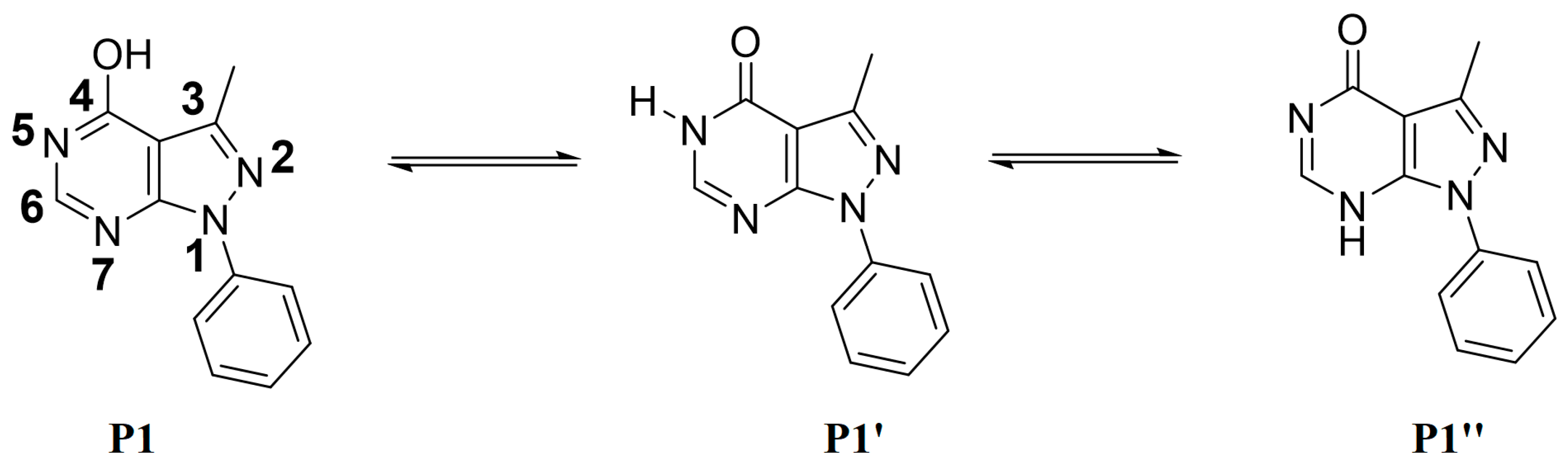
2.1.1. Crystal Structure of 3-methyl-1-phenyl-1,5-dihydro-4H-pyrazolo[3,4-d]pyrimidin-4-one (P1)
2.1.2. Crystal Structure of 3,5-dimethyl-1-phenyl-1,5-dihydro-4H-pyrazolo[3,4-d]pyrimidin-4-one (P2)
2.1.3. Crystal Structure of 3-methyl-1-phenyl-5-(prop-2-yn-1-yl)-1,5-dihydro-4H-pyrazolo[3,4-d]pyrimidinone (P3)
2.1.4. Crystal Structure of 3-methyl-5-(2-oxo-2-phenylethyl)-1-phenyl-1,5-dihydro-4H-pyrazolo[3,4-d]pyrimidin-4-one (P4)
2.1.5. General Structural Comments
2.2. Optimized Geometries
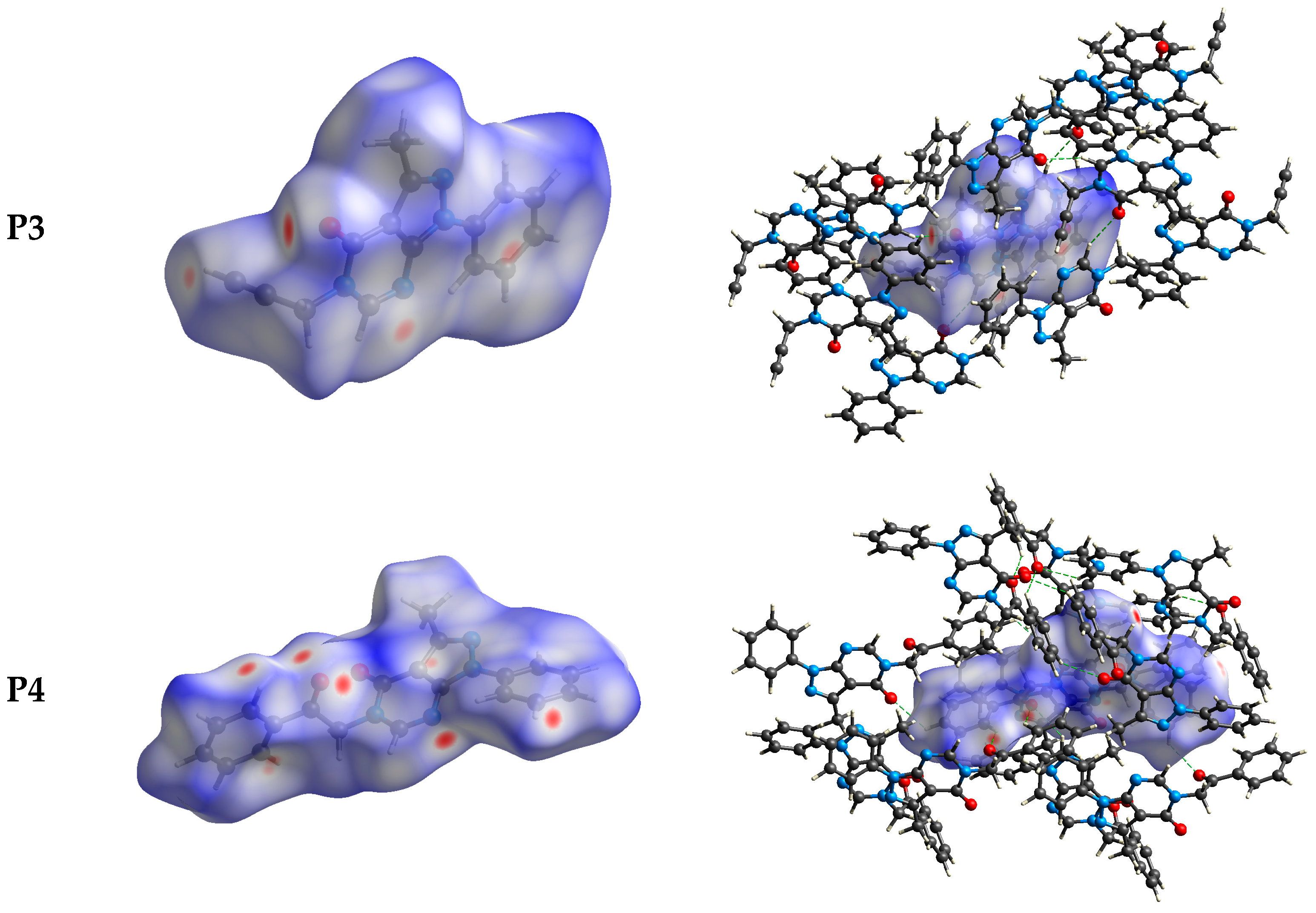
2.3. Hirshfeld Surface Analysis
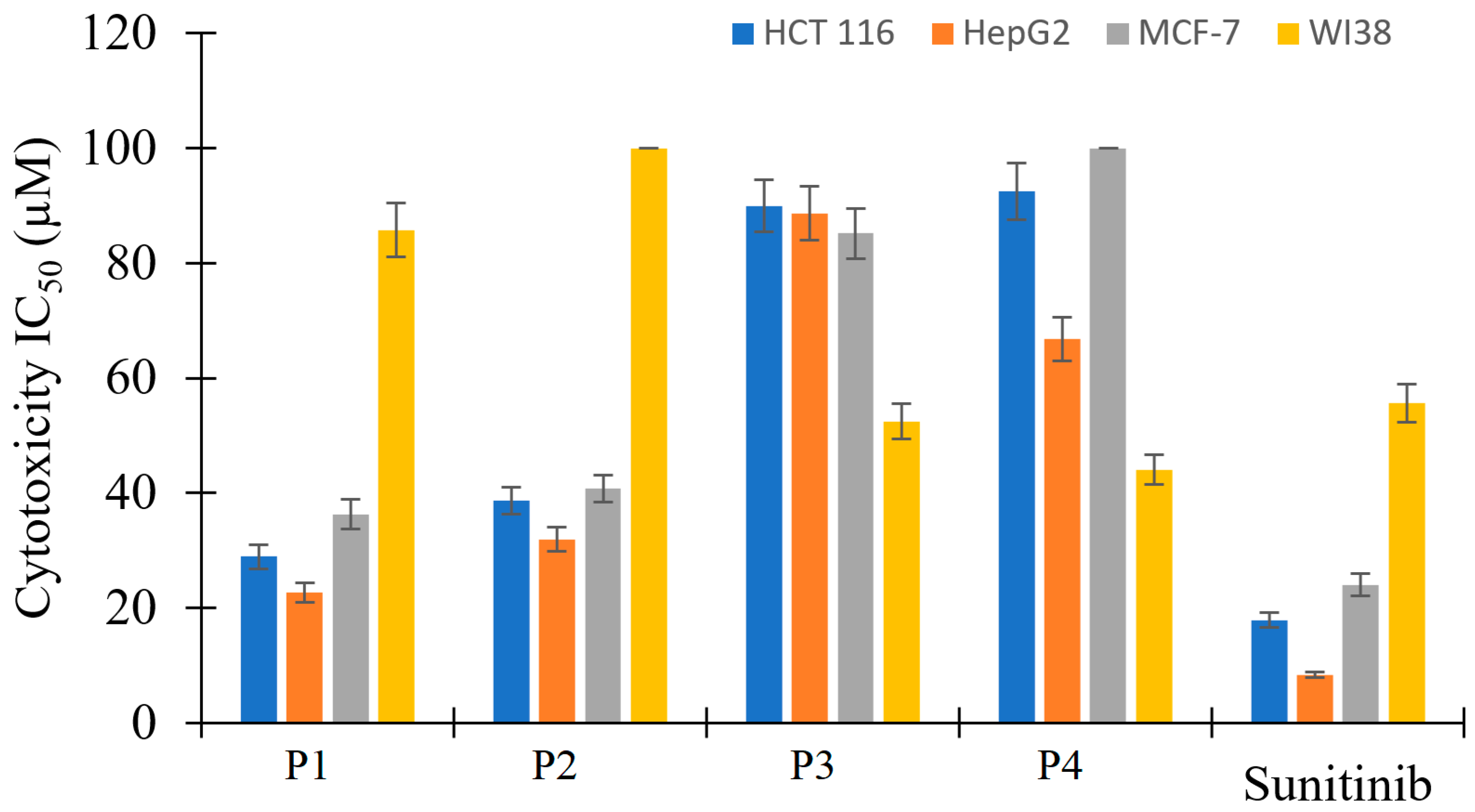
2.4. Antiproliferative Activity
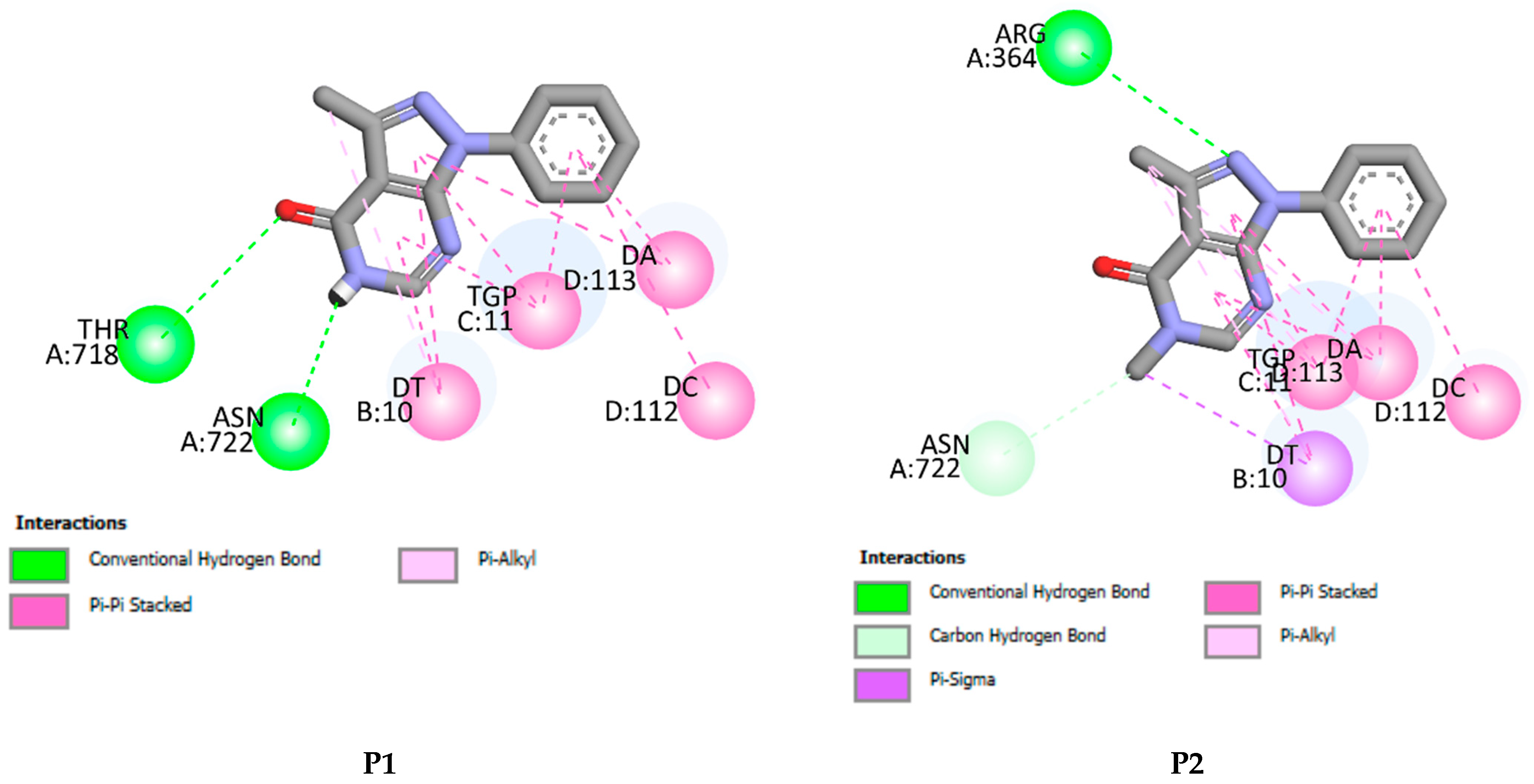
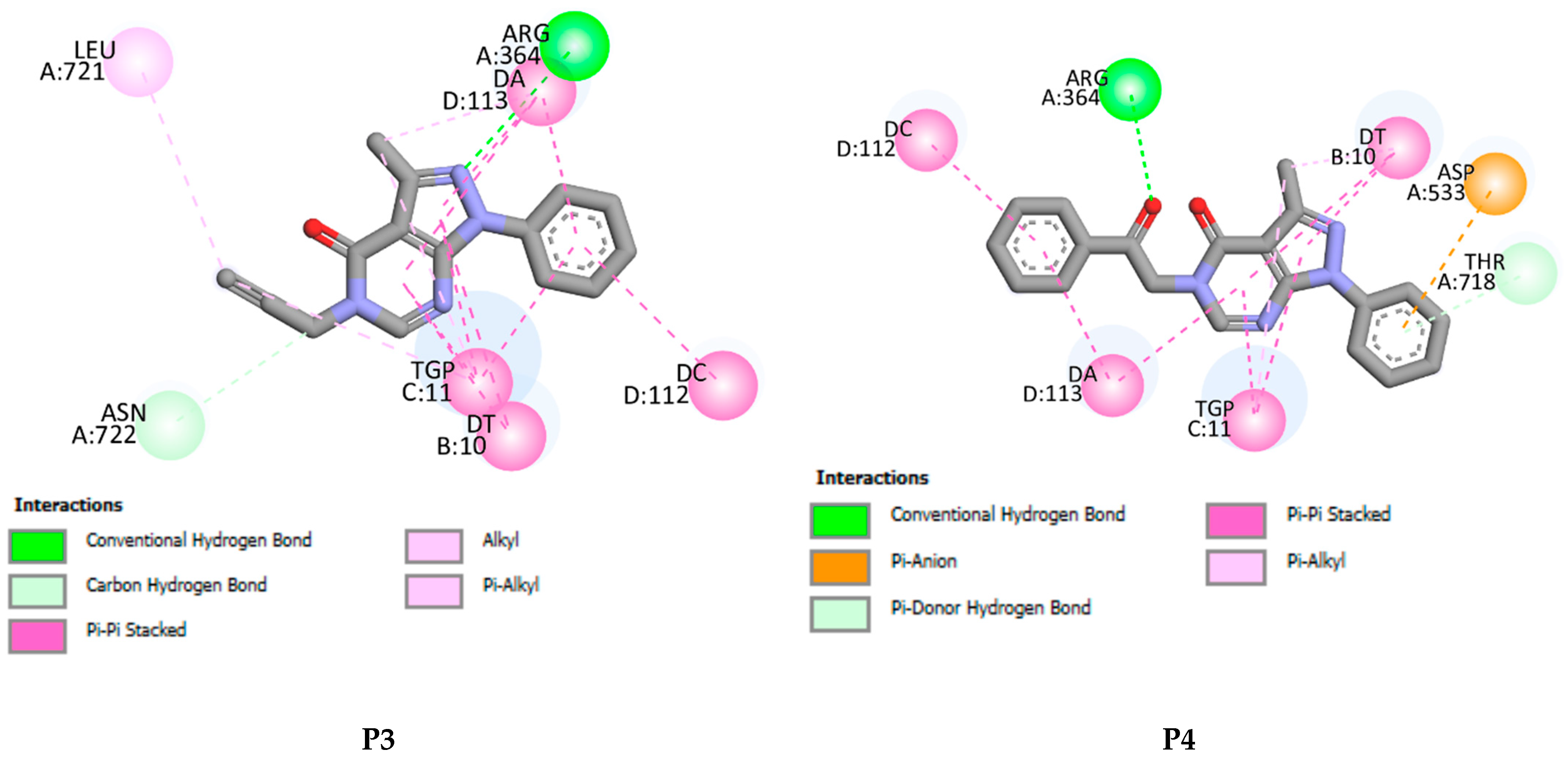
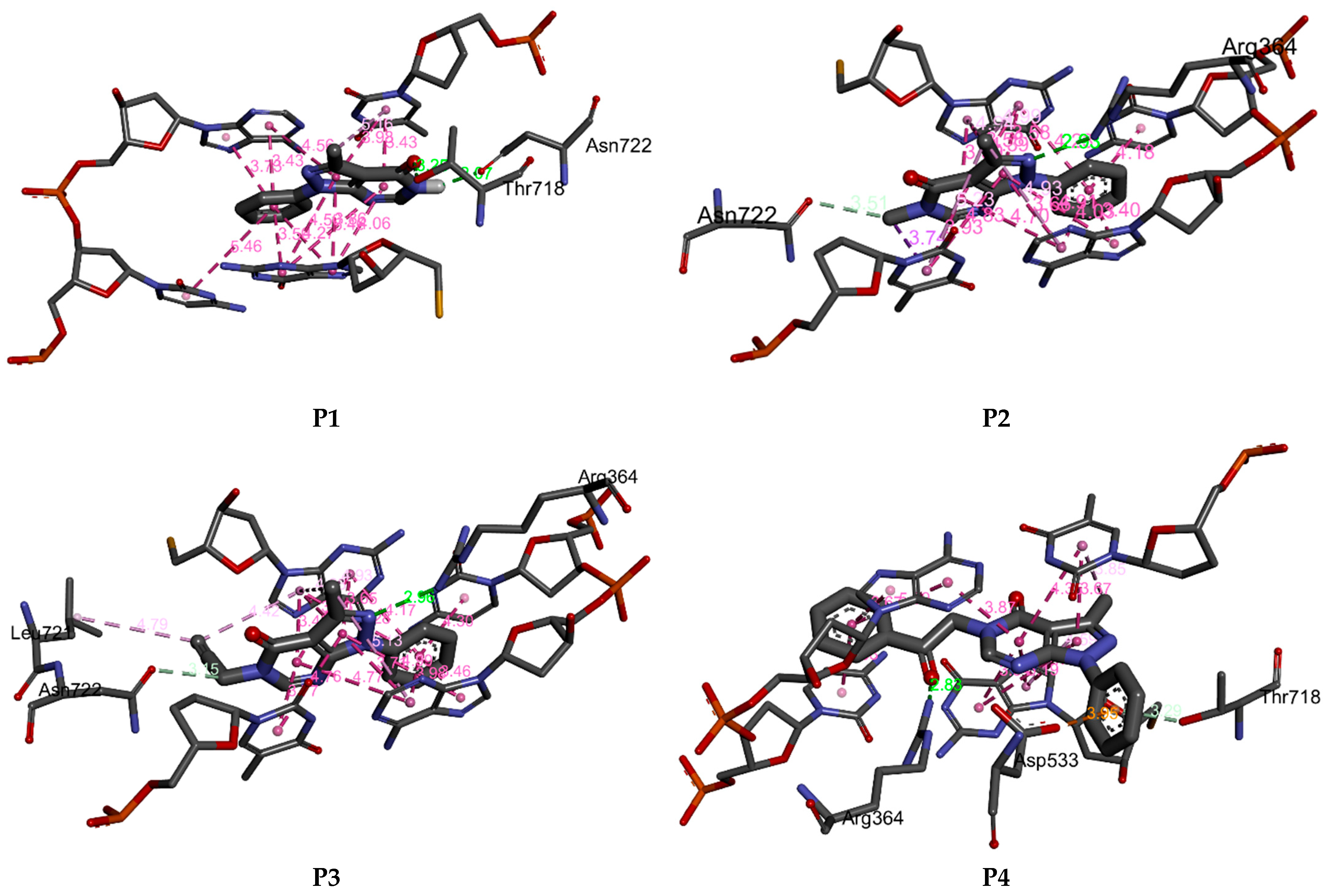
2.5. Molecular Docking Results
3. Materials and Methods
3.1. Reagents and Instruments
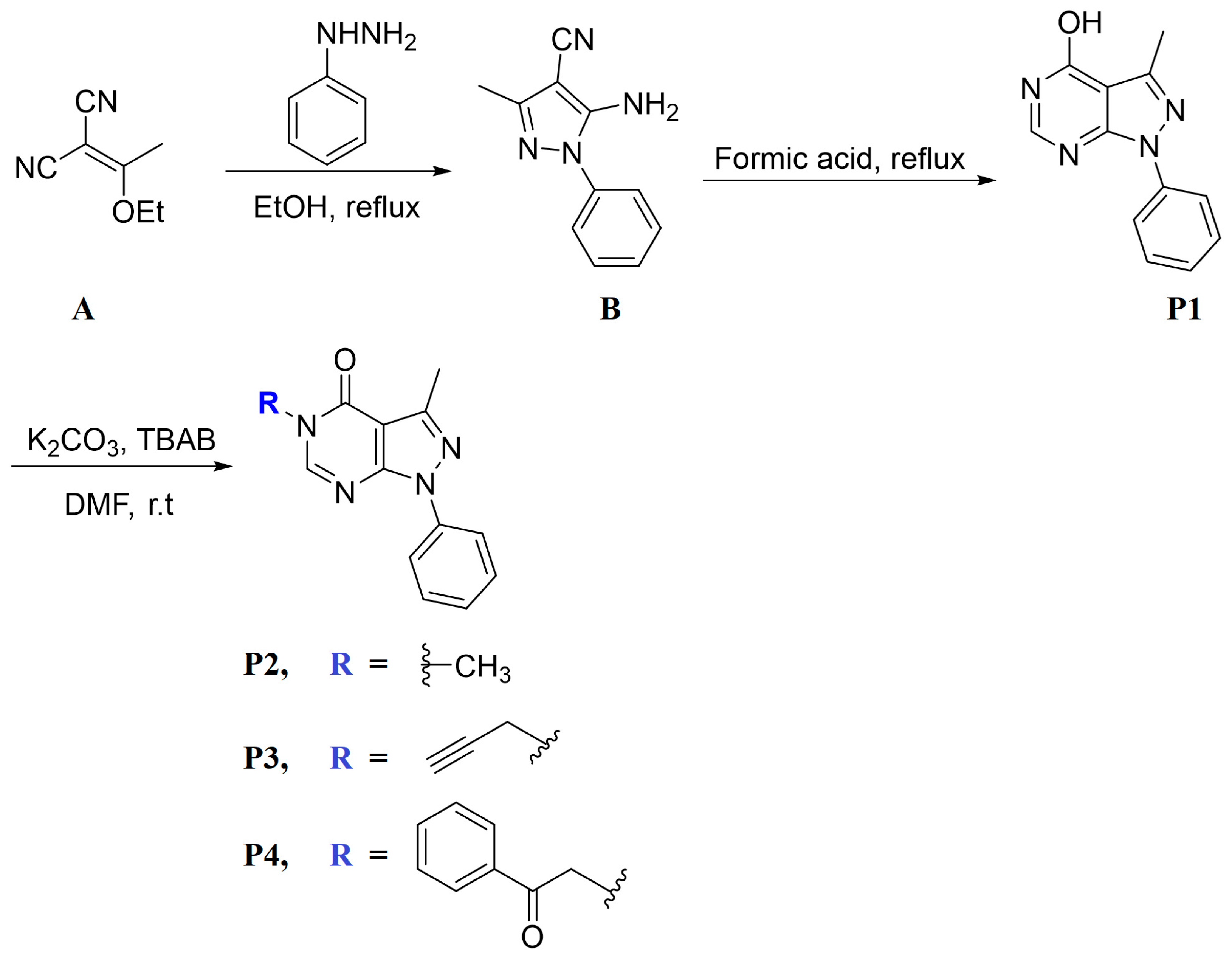
3.2. Synthesis
3.2.1. Synthesis of P1
3.2.2. Synthesis of P2–P4
3.3. Single Crystal X-Ray Diffraction
3.4. Hirshfeld Surface Study
3.5. DFT Calculations
3.6. Molecular Docking Study
3.7. Cytotoxicity Assay
4. Summary
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kreeger, P.K.; Lauffenburger, D.A. Cancer systems biology: A network modeling perspective. Carcinogenesis 2010, 31, 2–8. [Google Scholar] [CrossRef]
- Gaber, A.A.; Sobhy, M.; Turky, A.; Abdulwahab, H.G.; Al-Karmalawy, A.A.; Elhendawy, M.A.; Radwan, M.M.; Elkaeed, E.B.; Ibrahim, I.M.; Elzahabi, H.S. Discovery of new 1 H-pyrazolo [3,4-d] pyrimidine derivatives as anticancer agents targeting EGFRWT and EGFRT790M. J. Enzym. Inhib. Med. Chem. 2022, 37, 2283–2303. [Google Scholar] [CrossRef]
- Bondock, S.; Rabie, R.; Etman, H.A.; Fadda, A.A. Synthesis and antimicrobial activity of some new heterocycles incorporating antipyrine moiety. Eur. J. Med. Chem. 2008, 43, 2122–2129. [Google Scholar] [CrossRef]
- Trivedi, A.R.; Dholariya, B.H.; Vakhariya, C.P.; Dodiya, D.K.; Ram, H.K.; Kataria, V.B.; Siddiqui, A.B.; Shah, V.H. Synthesis and anti-tubercular evaluation of some novel pyrazolo [3,4-d] pyrimidine derivatives. Med. Chem. Res. 2012, 21, 1887–1891. [Google Scholar] [CrossRef]
- Tintori, C.; Fallacara, A.L.; Radi, M.; Zamperini, C.; Dreassi, E.; Crespan, E.; Maga, G.; Schenone, S.; Musumeci, F.; Brullo, C. Combining X-ray crystallography and molecular modeling toward the optimization of pyrazolo [3,4-d] pyrimidines as potent c-Src inhibitors active in vivo against neuroblastoma. J. Med. Chem. 2015, 58, 347–361. [Google Scholar] [CrossRef]
- Yeom, H.; Achary, R.; Choi, Y.; Park, C.H.; Lee, J.Y.; Lee, H.K.; Kim, P.; Cho, S.Y. Pyrazolo [3,4-d] pyrimidine derivatives as irreversible Bruton’s tyrosine kinase inhibitors. Bull. Korean Chem. Soc. 2022, 43, 577–584. [Google Scholar] [CrossRef]
- Podolski-Renić, A.; Dinić, J.; Stanković, T.; Tsakovska, I.; Pajeva, I.; Tuccinardi, T.; Botta, L.; Schenone, S.; Pešić, M. New therapeutic strategy for overcoming multidrug resistance in cancer cells with pyrazolo [3,4-d] pyrimidine tyrosine kinase inhibitors. Cancers 2021, 13, 5308. [Google Scholar] [CrossRef]
- Di Maria, S.; Picarazzi, F.; Mori, M.; Cianciusi, A.; Carbone, A.; Crespan, E.; Perini, C.; Sabetta, S.; Deplano, S.; Poggialini, F. Novel pyrazolo [3,4-d] pyrimidines as dual Src/Bcr-Abl kinase inhibitors: Synthesis and biological evaluation for chronic myeloid leukemia treatment. Bioorganic Chem. 2022, 128, 106071. [Google Scholar] [CrossRef]
- Pavlović, K.T.; Kocić, G.; Šmelcerović, A. Myt1 kinase inhibitors-Insight into structural features, offering potential frameworks. Chem.-Biol. Interact. 2024, 391, 110901. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Abdellattif, M.H.; Ali, A.; AbuAli, O.; Shahbaaz, M.; Ahsan, M.J.; Hussien, M.A. Computational approaches for the design of novel anticancer compounds based on Pyrazolo [3,4-d] pyrimidine derivatives as TRAP1 inhibitor. Molecules 2021, 26, 5932. [Google Scholar] [CrossRef] [PubMed]
- Hanke, J.H.; Gardner, J.P.; Dow, R.L.; Changelian, P.S.; Brissette, W.H.; Weringer, E.J.; Pollok, B.A.; Connelly, P.A. Discovery of a Novel, Potent, and Src Family-selective Tyrosine Kinase Inhibitor: STUDY OF Lck-AND FynT-DEPENDENT T CELL ACTIVATION (∗). J. Biol. Chem. 1996, 271, 695–701. [Google Scholar] [CrossRef] [PubMed]
- Robins, R.K. Potential purine antagonists. I. Synthesis of some 4, 6-substituted pyrazolo [3,4-d] pyrimidines1. J. Am. Chem. Soc. 1956, 78, 784–790. [Google Scholar] [CrossRef]
- Smyth, L.A.; Matthews, T.P.; Horton, P.N.; Hursthouse, M.B.; Collins, I. Divergent cyclisations of 2-(5-amino-4-carbamoyl-1H-pyrazol-3-yl) acetic acids with formyl and acetyl electrophiles. Tetrahedron 2007, 63, 9627–9634. [Google Scholar] [CrossRef]
- Abd El Hamid, M.K.; Mihovilovic, M.D.; El-Nassan, H.B. Synthesis of novel pyrazolo [3,4-d] pyrimidine derivatives as potential anti-breast cancer agents. Eur. J. Med. Chem. 2012, 57, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Li, J.; Chai, H.; Zhang, K.; Yang, D.; Zhang, Q.; Shi, D. A convenient four-component one-pot strategy toward the synthesis of pyrazolo [3,4-d] pyrimidines. Beilstein J. Org. Chem. 2015, 11, 2125–2131. [Google Scholar] [CrossRef]
- Gaonkar, S.; Savanur, M.A.; Nadaf, A.A.; Najare, M.S.; Mantur, S.; Garbhagudi, M.; Mulla, S.I.; Khazi, I.A.M. Novel pyrazolo [3,4-d] pyrimidine derivatives inhibit human cancer cell proliferation and induce apoptosis by ROS generation. Arch. Der Pharm. 2020, 353, 1900296. [Google Scholar] [CrossRef]
- Salem, I.M.; Mostafa, S.M.; Salama, I.; El-Sabbagh, O.I.; Hegazy, W.A.; Ibrahim, T.S. Design, synthesis and antitumor evaluation of novel pyrazolo [3,4-d] pyrimidines incorporating different amino acid conjugates as potential DHFR inhibitors. J. Enzym. Inhib. Med. Chem. 2023, 38, 203–215. [Google Scholar] [CrossRef]
- Bassoude, I.; Tber, Z.; Essassi, E.M.; Guillaumet, G.; Berteina-Raboin, S. A one-pot process for the microwave-assisted synthesis of 7-substituted pyrazolo [1, 5-a] pyrimidine. RSC Adv. 2016, 6, 3301–3306. [Google Scholar] [CrossRef]
- El Hafi, M.; Kansiz, S.; Lahmidi, S.; Boulhaoua, M.; Ramli, Y.; Dege, N.; Essassi, E.M.; Mague, J.T. Crystal structure and Hirshfeld surface analysis of 3-(4-methoxyphenyl)-1-methyl-4-phenyl-1H-pyrazolo [3,4-d] pyrimidine. Acta Crystallogr. Sect. E Crystallogr. Commun. 2019, 75, 638–641. [Google Scholar] [CrossRef]
- Somakala, K.; Tariq, S.; Amir, M. Synthesis, evaluation and docking of novel pyrazolo pyrimidines as potent p38α MAP kinase inhibitors with improved anti-inflammatory, ulcerogenic and TNF-α inhibitory properties. Bioorganic Chem. 2019, 87, 550–559. [Google Scholar] [CrossRef]
- Lazrak, F.; Lahmidi, S.; Anouar, E.H.; Alanazi, M.M.; Alanazi, A.S.; Essassi, E.M.; Mague, J.T. Synthesis, X-ray Crystal Structure, Anticancer, Hirshfeld Surface Analysis, DFT, TD-DFT, ADMET, and Molecular Docking of 3-Phenyl-1,2,4-triazolo[3,4-h]-13,4-thiaza-11-crown-4. Molecules 2023, 28, 3166. [Google Scholar] [CrossRef] [PubMed]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer: A program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J. Appl. Crystallogr. 2021, 54, 1006–1011. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.; McKinnon, J.; Wolff, S.; Grimwood, D.; Spackman, P.; Jayatilaka, D.; Spackman, M. CrystalExplorer17; University of Western Australia: Crawley, WA, Australia, 2017. [Google Scholar]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Petersson, G.; Nakatsuji, H. Gaussian 16; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Staker, B.L.; Feese, M.D.; Cushman, M.; Pommier, Y.; Zembower, D.; Stewart, L.; Burgin, A.B. Structures of three classes of anticancer agents bound to the human topoisomerase I−DNA covalent complex. J. Med. Chem. 2005, 48, 2336–2345. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| D—H···A | D—H | H···A | D···A | D—H···A |
|---|---|---|---|---|
| N1—H1···O1 i | 0.917 (9) | 1.877 (9) | 2.7912 (13) | 174.9 (14) |
| C2—H2···Cg3 ii | 0.95 | 2.86 | 3.7328 (14) | 154 |
| C7—H7···N2 | 0.95 | 2.30 | 2.9746 (17) | 127 |
| D—H···A | D—H | H···A | D···A | D—H···A |
|---|---|---|---|---|
| C5—H5···N2 i | 0.95 | 2.57 | 3.4517 (17) | 154 |
| C6—H6A···Cg3 ii | 0.98 | 2.92 | 3.5166 (19) | 121 |
| C9—H9···Cg3 iii | 0.95 | 2.99 | 3.7890 (19) | 142 |
| D—H···A | D—H | H···A | D···A | D—H···A |
|---|---|---|---|---|
| C2—H2···O1 i | 0.95 | 2.42 | 3.3115 (15) | 156 |
| C8—H8···N3 ii | 0.95 | 2.48 | 3.3915 (18) | 160 |
| C15—H15···N1 | 0.95 | 2.28 | 2.9506 (17) | 127 |
| D—H···A | D—H | H···A | D···A | D—H···A |
|---|---|---|---|---|
| C6—H6C···N2 i | 0.96 | 2.56 | 3.5153 (12) | 178 |
| C9—H9···Cg4 i | 0.93 | 2.68 | 3.5863 (12) | 166 |
| C11—H11···O2 ii | 0.93 | 2.55 | 3.2895 (13) | 137 |
| C20—H20···O2 iii | 0.93 | 2.55 | 3.2895 (13) | 137 |
| P1 | P2 | P3 | P4 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| X-Ray | Cal | ΔX a | X-Ray | Cal | ΔX a | X-Ray | Cal | ΔX a | X-Ray | Cal | ΔX a | |
| Bond length, B(Å) | ||||||||||||
| C1-O1 | 1.239 | 1.224 | 0.01 | 1.220 | 1.225 | 0.00 | 1.225 | 1.223 | 0.00 | 1.220 | 1.225 | 0.00 |
| C1-N3 | 1.392 | 1.418 | 0.03 | 1.407 | 1.427 | 0.02 | 1.417 | 1.431 | 0.01 | 1.424 | 1.430 | 0.01 |
| N9-N10 | 1.387 | 1.376 | 0.01 | 1.378 | 1.377 | 0.00 | 1.381 | 1.376 | 0.00 | 1.385 | 1.376 | 0.01 |
| N10-C12 | 1.425 | 1.425 | 0.00 | 1.415 | 1.424 | 0.01 | 1.424 | 1.424 | 0.00 | 1.420 | 1.424 | 0.00 |
| N3-C18 | - | - | - | 1.465 | 1.468 | 0.00 | 1.479 | 1.477 | 0.00 | 1.458 | 1.457 | 0.00 |
| C18-C19 | - | - | - | - | - | - | 1.466 | 1.462 | 0.00 | 1.528 | 1.538 | 0.01 |
| C19-O20 | - | - | - | - | - | - | - | - | - | 1.215 | 1.216 | 0.00 |
| Bond angles, A(°) | ||||||||||||
| N3-C1-C7 | 111.90 | 110.46 | 1.45 | 111.81 | 111.65 | 0.16 | 111.25 | 111.24 | 0.01 | 111.23 | 111.43 | 0.20 |
| N5-C6-N10 | 126.72 | 126.56 | 0.16 | 126.17 | 127.07 | 0.90 | 125.89 | 126.92 | 1.03 | 126.31 | 126.94 | 0.64 |
| C1-N3-C18 | - | - | - | 117.27 | 117.01 | 0.26 | 117.96 | 117.75 | 0.21 | 117.10 | 117.52 | 0.42 |
| N3-C18-C19 | - | - | - | - | - | 0.00 | 110.60 | 112.92 | 2.32 | 111.20 | 112.17 | 0.98 |
| C18-C19-C20 | - | - | - | - | - | 0.00 | 176.61 | 179.33 | 2.72 | 0.00 | ||
| C18-C19-O20 | - | - | - | - | - | - | - | - | - | 119.79 | 119.99 | 0.20 |
| Torsion angles, D(°) | ||||||||||||
| O2-C1-C7-C8 | −2.35 | 1.19 | 3.54 | 2.63 | 1.26 | 1.37 | −5.60 | 1.21 | 6.81 | −0.20 | −1.40 | 1.21 |
| N5-C6-N10-C12 | 2.58 | −0.12 | 2.71 | 0.09 | −0.54 | 0.63 | −0.15 | −0.64 | 0.49 | 4.43 | 0.62 | 3.81 |
| N9-N10-C12-C17 | 6.02 | 33.01 | 26.99 | 31.34 | 30.62 | 0.72 | 5.51 | 31.55 | 26.04 | −27.06 | −32.12 | 5.05 |
| O2-C1-N3-C18 | - | - | - | −2.24 | 0.02 | 2.26 | 3.72 | 0.96 | 2.75 | −3.93 | −2.66 | 1.27 |
| C1-N3-C18-C19 | - | - | - | - | - | 0.00 | 82.62 | 91.88 | 9.26 | −78.99 | −90.41 | 11.41 |
| N3-C18-C19-O20 | - | - | - | - | - | - | - | - | - | 5.07 | −6.01 | 11.08 |
| C18-C19-C21-C22 | −2.35 | 1.19 | 3.54 | 2.63 | 1.26 | 1.37 | −5.60 | 1.21 | 6.81 | −0.20 | −1.40 | 1.21 |
| P1 | P2 | P3 | P4 | |
|---|---|---|---|---|
| H···H | 46.9 | 44.0 | 43.2 | 40.0 |
| C···H/H···C | 14.6 | 27.5 | 24.7 | 28.8 |
| O···H/H···O | 10.0 | 11.1 | 10.0 | 15.2 |
| N···H/H···N | 10.8 | 11.5 | 6.7 | 8.7 |
| Compound | In Vitro Cytotoxicity IC50 (µM) * | Selectivity Index | |||
|---|---|---|---|---|---|
| HCT 116 | HepG2 | MCF-7 | WI38 | ||
| P1 | 28.93 ± 2.1 | 22.70 ± 1.7 | 36.32 ± 2.6 | 85.73 ± 4.7 | 0.34 |
| P2 | 38.68 ± 2.3 | 31.93 ± 2.1 | 40.75 ± 2.4 | >100 | 0.37 |
| P3 | 89.92 ± 4.5 | 88.62 ± 4.7 | 85.16 ± 4.4 | 52.49 ± 3.1 | 1.67 |
| P4 | 92.47 ± 4.9 | 66.81 ± 3.8 | >100 | 44.03 ± 2.6 | 1.96 |
| Sunitinib | 17.91 ± 1.3 | 8.38 ± 0.5 | 24.06 ± 2.0 | 55.63 ± 3.3 | 0.30 |
| BE (kcal/mol) | HBs | NR 1 | |
|---|---|---|---|
| P1 | −7.01 | 2 | ASN A722, THR A718, DT B10, TGP C11, DA D113, DC D112 |
| P2 | −7.03 | 1 | ARG A364, ASN A722, DT B10, TGP C11, DA D113, DC D112 |
| P3 | −7.51 | 1 | ARG A364, ASN A722, LEU A721, DT B10, TGP C11, DA D113, DC D112 |
| P4 | −8.52 | 1 | ARG A364, TYR A426, LYS A425, DT B10, TGP C11, DA D113, DC D112 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El Hafi, M.; Anouar, E.H.; Lahmidi, S.; Boulhaoua, M.; Loubidi, M.; Alanazi, A.S.; Filali, I.; Hefnawy, M.; El Ghayati, L.; Mague, J.T.; et al. Synthesis of New Pyrazolo[3,4-d]pyrimidine Derivatives: NMR Spectroscopic Characterization, X-Ray, Hirshfeld Surface Analysis, DFT, Molecular Docking, and Antiproliferative Activity Investigations. Molecules 2024, 29, 5020. https://doi.org/10.3390/molecules29215020
El Hafi M, Anouar EH, Lahmidi S, Boulhaoua M, Loubidi M, Alanazi AS, Filali I, Hefnawy M, El Ghayati L, Mague JT, et al. Synthesis of New Pyrazolo[3,4-d]pyrimidine Derivatives: NMR Spectroscopic Characterization, X-Ray, Hirshfeld Surface Analysis, DFT, Molecular Docking, and Antiproliferative Activity Investigations. Molecules. 2024; 29(21):5020. https://doi.org/10.3390/molecules29215020
Chicago/Turabian StyleEl Hafi, Mohamed, El Hassane Anouar, Sanae Lahmidi, Mohammed Boulhaoua, Mohammed Loubidi, Ashwag S. Alanazi, Insaf Filali, Mohamed Hefnawy, Lhoussaine El Ghayati, Joel T. Mague, and et al. 2024. "Synthesis of New Pyrazolo[3,4-d]pyrimidine Derivatives: NMR Spectroscopic Characterization, X-Ray, Hirshfeld Surface Analysis, DFT, Molecular Docking, and Antiproliferative Activity Investigations" Molecules 29, no. 21: 5020. https://doi.org/10.3390/molecules29215020
APA StyleEl Hafi, M., Anouar, E. H., Lahmidi, S., Boulhaoua, M., Loubidi, M., Alanazi, A. S., Filali, I., Hefnawy, M., El Ghayati, L., Mague, J. T., & Essassi, E. M. (2024). Synthesis of New Pyrazolo[3,4-d]pyrimidine Derivatives: NMR Spectroscopic Characterization, X-Ray, Hirshfeld Surface Analysis, DFT, Molecular Docking, and Antiproliferative Activity Investigations. Molecules, 29(21), 5020. https://doi.org/10.3390/molecules29215020