
Micro-Structure Engineering in Pd-InOx Catalysts and Mechanism Studies for CO2 Hydrogenation to Methanol


 , ,
, ,  ,
,
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. Structure Characterization of Pd-InOx
2.2. The Structure–Function Relationship
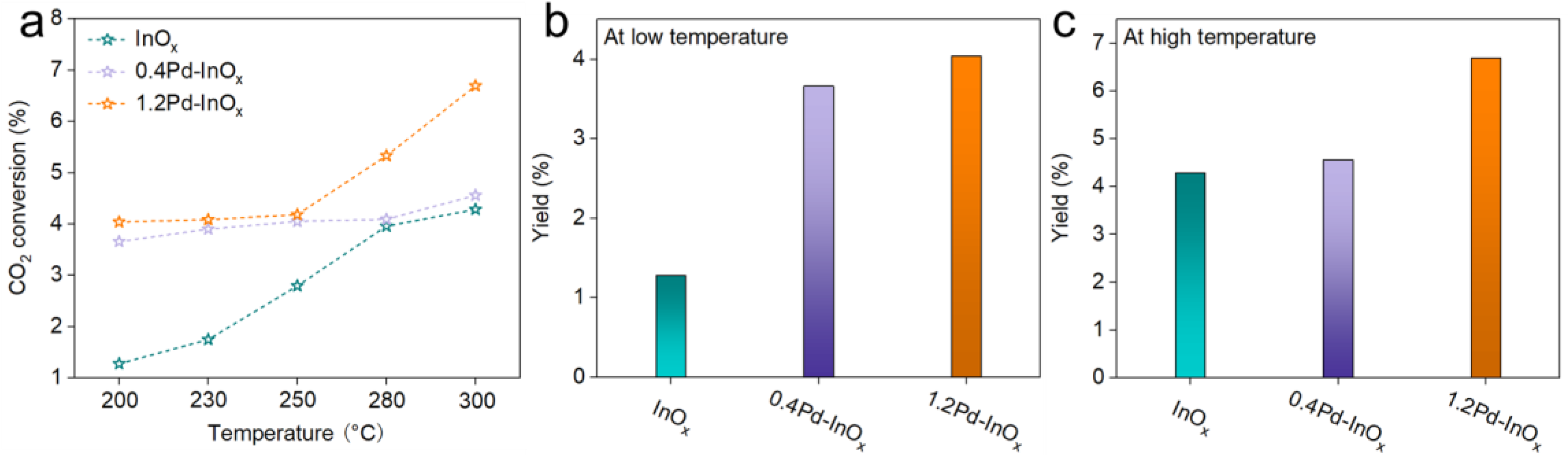
2.3. Catalytic Performance for CO2 Hydrogenation
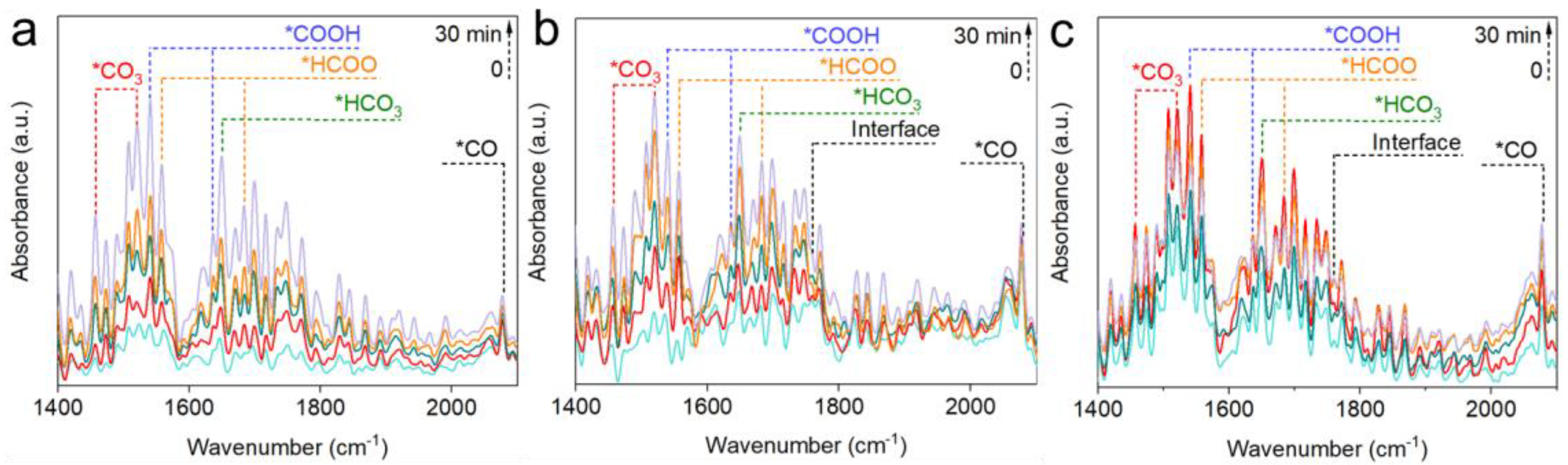
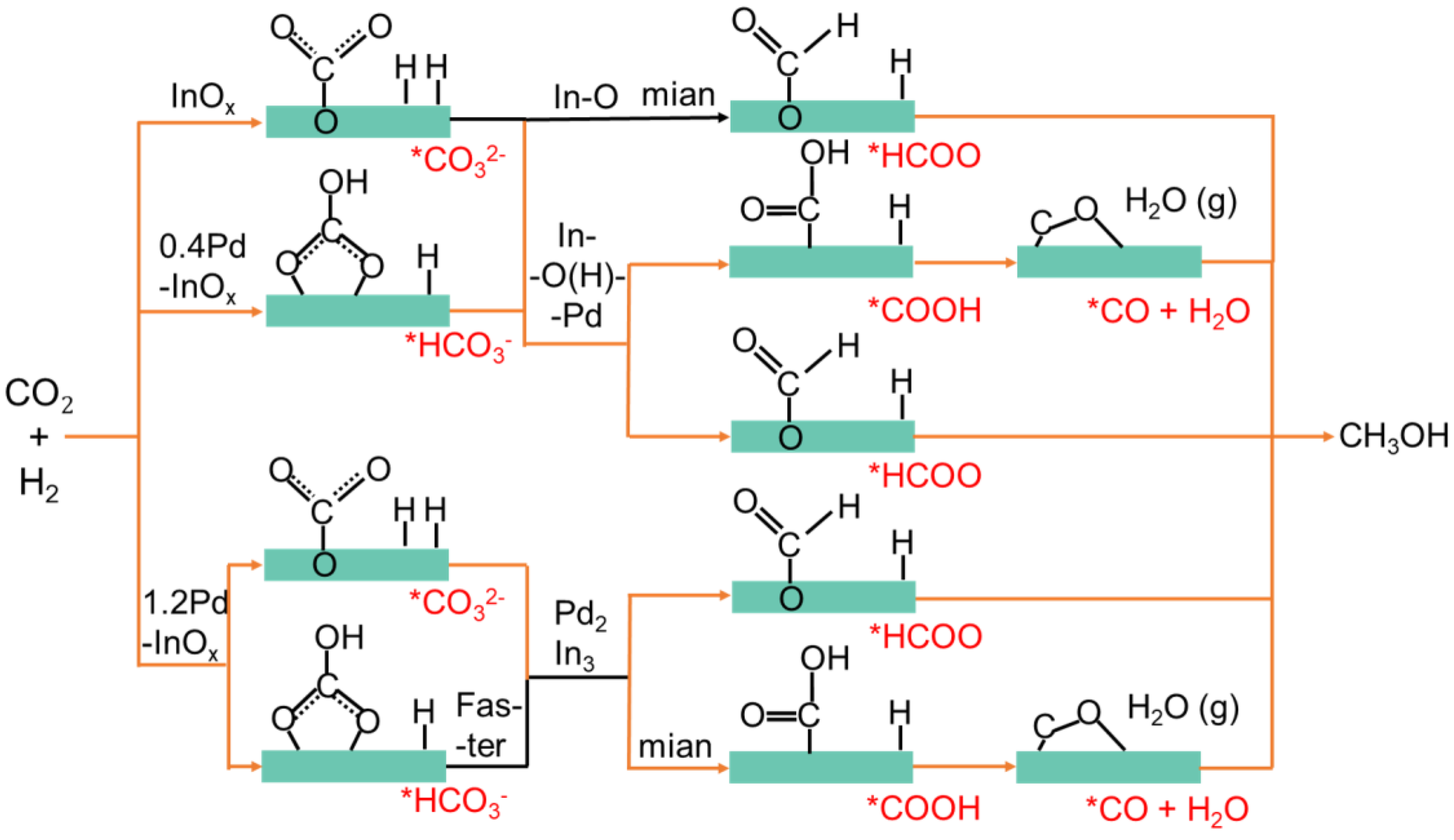
2.4. Reaction Mechanisms
3. Experimental Section
3.1. Preparation of InOx Catalysts
3.2. Preparation of Pd-InOx Catalysts
3.3. Catalyst Characterizations
3.4. Catalytic Performance Evaluation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, W.; Yang, Y.; Li, Y.; Li, F.; Luo, M. Recent progress on integrated CO2 capture and electrochemical upgrading. Mater. Today Catal. 2023, 2, 100006. [Google Scholar] [CrossRef]
- Zhao, H.; Yu, R.; Ma, S.; Xu, K.; Chen, Y.; Jiang, K.; Fang, Y.; Zhu, C.; Liu, X.; Tang, Y.; et al. The role of Cu1–O3 species in single-atom Cu/ZrO2 catalyst for CO2 hydrogenation. Nat. Catal. 2022, 5, 818–831. [Google Scholar] [CrossRef]
- Tebar-Soler, C.; Martin-Diaconescu, V.; Simonelli, L.; Missyul, A.; Perez-Dieste, V.; Villar-Garcia, I.J.; Brubach, J.B.; Roy, P.; Haro, M.L.; Calvino, J.J.; et al. Low-oxidation-state Ru sites stabilized in carbon-doped RuO2 with low-temperature CO2 activation to yield methane. Nat. Mater. 2023, 22, 762–768. [Google Scholar] [CrossRef] [PubMed]
- Claver, C.; Yeamin, M.B.; Reguero, M.; Masdeu-Bultó, A.M. Recent advances in the use of catalysts based on natural products for the conversion of CO2 into cyclic carbonates. Green Chem. 2020, 22, 7665–7706. [Google Scholar] [CrossRef]
- Yang, H.; Zhang, C.; Gao, P.; Wang, H.; Li, X.; Zhong, L.; Wei, W.; Sun, Y. A review of the catalytic hydrogenation of carbon dioxide into value-added hydrocarbons. Catal. Sci. Technol. 2017, 7, 4580–4598. [Google Scholar] [CrossRef]
- Nikolaeva, D.; Luis, P. Top-Down Polyelectrolytes for Membrane-Based Post-Combustion CO2 Capture. Molecules 2020, 25, 323. [Google Scholar] [CrossRef] [PubMed]
- Martin, O.; Martin, A.J.; Mondelli, C.; Mitchell, S.; Segawa, T.F.; Hauert, R.; Drouilly, C.; Curulla-Ferre, D.; Perez-Ramirez, J. Indium Oxide as a Superior Catalyst for Methanol Synthesis by CO2 Hydrogenation. Angew. Chem. Int. Ed. 2016, 55, 6261–6265. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Shen, C.; Sun, K.; Mei, D.; Liu, C.-j. Enhanced Surface Charge Localization Over Nitrogen-Doped In2O3 for CO2 Hydrogenation to Methanol with Improved Stability. ACS Catal. 2023, 13, 6154–6168. [Google Scholar] [CrossRef]
- Li, Y.; Fei, N.; Li, W.; Cao, Y.; Ge, X.; Dai, S.; Yan, K.; Yuwen, Q.; Zhou, X.; Yuan, W.; et al. H2 activation on metal oxides promoted by highly dispersed Pd. Catal. Commun. 2023, 177, 106645. [Google Scholar] [CrossRef]
- Rui, N.; Wang, Z.; Sun, K.; Ye, J.; Ge, Q.; Liu, C.-J. CO2 hydrogenation to methanol over Pd/In2O3: Effects of Pd and oxygen vacancy. Appl. Catal. B Environ. 2017, 218, 488–497. [Google Scholar] [CrossRef]
- Gao, D.; Zhou, H.; Wang, J.; Miao, S.; Yang, F.; Wang, G.; Wang, J.; Bao, X. Size-dependent electrocatalytic reduction of CO2 over Pd nanoparticles. J. Am. Chem. Soc. 2015, 137, 4288–4291. [Google Scholar] [CrossRef] [PubMed]
- Bahruji, H.; Bowker, M.; Hutchings, G.; Dimitratos, N.; Wells, P.; Gibson, E.; Jones, W.; Brookes, C.; Morgan, D.; Lalev, G. Pd/ZnO catalysts for direct CO2 hydrogenation to methanol. J. Catal. 2016, 343, 133–146. [Google Scholar] [CrossRef]
- Jiang, X.; Koizumi, N.; Guo, X.; Song, C. Bimetallic Pd–Cu catalysts for selective CO2 hydrogenation to methanol. Appl. Catal. B Environ. 2015, 170–171, 173–185. [Google Scholar] [CrossRef]
- Snider, J.L.; Streibel, V.; Hubert, M.A.; Choksi, T.S.; Valle, E.; Upham, D.C.; Schumann, J.; Duyar, M.S.; Gallo, A.; Abild-Pedersen, F.; et al. Revealing the Synergy between Oxide and Alloy Phases on the Performance of Bimetallic In–Pd Catalysts for CO2 Hydrogenation to Methanol. ACS Catal. 2019, 9, 3399–3412. [Google Scholar] [CrossRef]
- Zhang, M.; Li, F.; Dou, M.; Yu, Y.; Chen, Y. The synergetic effect of Pd, In and Zr on the mechanism of Pd/In2O3–ZrO2 for CO2 hydrogenation to methanol. React. Chem. Eng. 2022, 7, 2433–2444. [Google Scholar] [CrossRef]
- Bi, F.; Feng, X.; Zhou, Z.; Zhang, Y.; Wei, J.; Yuan, L.; Liu, B.; Huang, Y.; Zhang, X. Mn-based catalysts derived from the non-thermal treatment of Mn-MIL-100 to enhance its water-resistance for toluene oxidation: Mechanism study. Chem. Eng. J. 2024, 485, 149776. [Google Scholar] [CrossRef]
- Wang, X.; Yang, X.; Pei, G.; Yang, J.; Liu, J.; Zhao, F.; Jin, F.; Jiang, W.; Ben, H.; Zhang, L. Strong metal–support interaction boosts the electrocatalytic hydrogen evolution capability of Ru nanoparticles supported on titanium nitride. Carbon Energy 2023, 6, e391. [Google Scholar] [CrossRef]
- García-Trenco, A.; Regoutz, A.; White, E.R.; Payne, D.J.; Shaffer, M.S.P.; Williams, C.K. PdIn intermetallic nanoparticles for the Hydrogenation of CO2 to Methanol. Appl. Catal. B Environ. 2018, 220, 9–18. [Google Scholar] [CrossRef]
- Wang, Z.; Men, G.; Zhang, R.; Gu, F.; Han, D. Pd loading induced excellent NO2 gas sensing of 3DOM In2O3 at room temperature. Sens. Actuators B Chem. 2018, 263, 218–228. [Google Scholar] [CrossRef]
- Sun, J.; Zhang, J.; Fu, H.; Wan, H.; Wan, Y.; Qu, X.; Xu, Z.; Yin, D.; Zheng, S. Enhanced catalytic hydrogenation reduction of bromate on Pd catalyst supported on CeO2 modified SBA-15 prepared by strong electrostatic adsorption. Appl. Catal. B Environ. 2018, 229, 32–40. [Google Scholar] [CrossRef]
- Wu, Y.; Lu, X.; Cui, P.; Jia, W.; Zhou, J.; Wang, Y.; Zahid, H.; Wu, Y.; Rafique, M.U.; Yin, X.; et al. Enhancing alkyne semi-hydrogenation through engineering metal–support interactions of Pd on oxides. Nano Res. 2023, 17, 3707–3713. [Google Scholar] [CrossRef]
- Tian, G.; Wu, Y.; Wu, S.; Huang, S.; Gao, J. Solid-State Synthesis of Pd/In2O3 Catalysts for CO2 Hydrogenation to Methanol. Catal. Lett. 2022, 153, 903–910. [Google Scholar] [CrossRef]
- Neumann, M.; Teschner, D.; Knop-Gericke, A.; Reschetilowski, W.; Armbrüster, M. Controlled synthesis and catalytic properties of supported In–Pd intermetallic compounds. J. Catal. 2016, 340, 49–59. [Google Scholar] [CrossRef]
- Ojelade, O.A.; Zaman, S.F. A Review on Pd Based Catalysts for CO2 Hydrogenation to Methanol: In-Depth Activity and DRIFTS Mechanistic Study. Catal. Surv. Asia 2019, 24, 11–37. [Google Scholar] [CrossRef]
- Shen, C.; Sun, K.; Zhang, Z.; Rui, N.; Jia, X.; Mei, D.; Liu, C.-J. Highly Active Ir/In2O3 Catalysts for Selective Hydrogenation of CO2 to Methanol: Experimental and Theoretical Studies. ACS Catal. 2021, 11, 4036–4046. [Google Scholar] [CrossRef]
- Yang, Y.; Guo, M.; Zhao, F. Cr2O3 Promoted In2O3 Catalysts for CO2 Hydrogenation to Methanol. Chemphyschem 2024, 25, e202300530. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Dai, J.; Li, W.; Tan, K.B.; Huang, Z.; Zhan, G.; Huang, J.; Li, Q. Pd Supported on MIL-68(In)-Derived In2O3 Nanotubes as Superior Catalysts to Boost CO2 Hydrogenation to Methanol. ACS Catal. 2020, 10, 13275–13289. [Google Scholar] [CrossRef]
- Furukawa, S.; Endo, M.; Komatsu, T. Bifunctional Catalytic System Effective for Oxidative Dehydrogenation of 1-Butene and n-Butane Using Pd-Based Intermetallic Compounds. ACS Catal. 2014, 4, 3533–3542. [Google Scholar] [CrossRef]
- Zhang, H.; Mao, D.; Zhang, J.; Wu, D. Regulating the crystal structure of layered double hydroxide-derived Co-In catalysts for highly selective CO2 hydrogenation to methanol. Chem. Eng. J. 2023, 452, 139144. [Google Scholar] [CrossRef]
- Chen, J.; Zha, Y.; Liu, B.; Li, Y.; Xu, Y.; Liu, X. Rationally Designed Water Enriched Nano Reactor for Stable CO2 Hydrogenation with Near 100% Ethanol Selectivity over Diatomic Palladium Active Sites. ACS Catal. 2023, 13, 7110–7121. [Google Scholar] [CrossRef]
- Yang, X.; Duan, H.; Wang, R.; Zhao, F.; Jin, F.; Jiang, W.; Han, G.; Guan, Q.; Ben, H. Tailoring Zeolite L-Supported-Cu Catalysts for CO2 Hydrogenation: Insights into the Mechanism of CH3OH and CO Formation. Inorg. Chem. 2023, 62, 13419–13427. [Google Scholar] [CrossRef] [PubMed]
- Rameshan, C.; Lorenz, H.; Mayr, L.; Penner, S.; Zemlyanov, D.; Arrigo, R.; Haevecker, M.; Blume, R.; Knop-Gericke, A.; Schlogl, R.; et al. CO2-selective methanol steam reforming on In-doped Pd studied by in situ X-ray photoelectron spectroscopy. J. Catal. 2012, 295, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Chávez-Rocha, R.; Mercado-Sánchez, I.; Vargas-Rodriguez, I.; Hernández-Lima, J.; Bazán-Jimínez, A.; Robles, J.; García-Revilla, M.A. Modeling Adsorption of CO2 in Rutile Metallic Oxide Surfaces: Implications in CO2 Catalysis. Molecules 2023, 28, 1776. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhao, G.; Bing, H.; Zhong, J.; Yuan, H.; Xie, J.; Chen, Y. High-performance Ni–Ce1-xZrxO2 nanoparticles for biogas reforming: Enhanced CO2 activation and stability. Sustain. Energy Fuels 2021, 5, 6449–6459. [Google Scholar] [CrossRef]
- Li, K.; Chang, X.; Pei, C.; Li, X.; Chen, S.; Zhang, X.; Assabumrungrat, S.; Zhao, Z.-J.; Zeng, L.; Gong, J. Ordered mesoporous Ni/La2O3 catalysts with interfacial synergism towards CO2 activation in dry reforming of methane. Appl. Catal. B Environ. 2019, 259, 118092. [Google Scholar] [CrossRef]
- Malik, A.S.; Zaman, S.F.; Al-Zahrani, A.A.; Daous, M.A.; Driss, H.; Petrov, L.A. Development of highly selective PdZn/CeO2 and Ca-doped PdZn/CeO2 catalysts for methanol synthesis from CO2 hydrogenation. Appl. Catal. A Gen. 2018, 560, 42–53. [Google Scholar] [CrossRef]
- Ma, J.; Xu, L.; Xu, L.; Wang, H.; Xu, S.; Li, H.; Xie, S.; Li, H. Highly Dispersed Pd on Co–B Amorphous Alloy: Facile Synthesis via Galvanic Replacement Reaction and Synergetic Effect between Pd and Co. ACS Catal. 2013, 3, 985–992. [Google Scholar] [CrossRef]
- Kattel, S.; Yan, B.; Yang, Y.; Chen, J.G.; Liu, P. Optimizing Binding Energies of Key Intermediates for CO2 Hydrogenation to Methanol over Oxide-Supported Copper. J. Am. Chem. Soc. 2016, 138, 12440–12450. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Qu, Z.; Song, L.; Fu, Q. CO2 hydrogenation to methanol over Cu/CeO2 and Cu/ZrO2 catalysts: Tuning methanol selectivity via metal-support interaction. J. Energy Chem. 2020, 40, 22–30. [Google Scholar] [CrossRef]
- Daifeng, L.; Zhen, Z.; Yinye, C.; Lingxing, Z.; Xiaochuan, C.; Xuhui, Y.; Baoquan, H.; Yongjin, L.; Qingrong, Q.; Qinghua, C. The Co-In2O3 interaction concerning the effect of amorphous Co metal on CO2 hydrogenation to methanol. J. CO2 Util. 2022, 65, 102209. [Google Scholar] [CrossRef]
- Jiao, J.; Yuan, Q.; Tan, M.; Han, X.; Gao, M.; Zhang, C.; Yang, X.; Shi, Z.; Ma, Y.; Xiao, H.; et al. Constructing asymmetric double-atomic sites for synergistic catalysis of electrochemical CO2 reduction. Nat. Commun. 2023, 14, 6164. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, G.; Zhu, J.; Zhang, X.; Ding, F.; Zhang, A.; Guo, X.; Song, C. CO2 Hydrogenation to Methanol over In2O3-Based Catalysts: From Mechanism to Catalyst Development. ACS Catal. 2021, 11, 1406–1423. [Google Scholar] [CrossRef]
- Liu, Z.; Liu, H.; Li, M.; Meng, Y.; Wang, X.; Yan, T.; Fan, Q.; Lou, S.N.; Cui, W.; Zhang, S. Pd-doped In2O3 for CO2 Electroreduction to Ethanol through CO Binding Regulation. ChemCatChem 2024, 16, e202301700. [Google Scholar] [CrossRef]
- Ning, H.H.; Li, Y.D.; Zhang, C.J. Recent Progress in the Integration of CO2 Capture and Utilization. Molecules 2023, 28, 4500. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.Y.; Liu, C.J.; Ge, Q. DFT Study of CO2 Adsorption and Hydrogenation on the In2O3 Surface. J. Phys. Chem. C 2012, 116, 7817–7825. [Google Scholar] [CrossRef]
- Xu, J.; Su, X.; Liu, X.; Pan, X.; Pei, G.; Huang, Y.; Wang, X.; Zhang, T.; Geng, H. Methanol synthesis from CO2 and H2 over Pd/ZnO/Al2O3: Catalyst structure dependence of methanol selectivity. Appl. Catal. A Gen. 2016, 514, 51–59. [Google Scholar] [CrossRef]
- Khobragade, R.; Roškarič, M.; Žerjav, G.; Košiček, M.; Zavašnik, J.; Van de Velde, N.; Jerman, I.; Tušar, N.N.; Pintar, A. Exploring the effect of morphology and surface properties of nanoshaped Pd/CeO2 catalysts on CO2 hydrogenation to methanol. Appl. Catal. A Gen. 2021, 627, 118394. [Google Scholar] [CrossRef]
- Lawes, N.; Aggett, K.J.; Smith, L.R.; Slater, T.J.A.; Dearg, M.; Morgan, D.J.; Dummer, N.F.; Taylor, S.H.; Hutchings, G.J.; Bowker, M. Zn Loading Effects on the Selectivity of PdZn Catalysts for CO2 Hydrogenation to Methanol. Catal. Lett. 2023, 154, 1603–1610. [Google Scholar] [CrossRef]
- Pan, H.; Ma, B.; Zhou, L.; Hu, Y.; Shakouri, M.; Guo, Y.; Liu, X.; Wang, Y. Highly Efficient CuPd0.1/γ-Al2O3 Catalyst with Isolated Pd Species for CO2 Hydrogenation to Methanol. ACS Sustain. Chem. Eng. 2023, 11, 7489–7499. [Google Scholar] [CrossRef]
- Song, J.; Liu, S.; Yang, C.; Wang, G.; Tian, H.; Zhao, Z.-J.; Mu, R.; Gong, J. The role of Al doping in Pd/ZnO catalyst for CO2 hydrogenation to methanol. Appl. Catal. B Environ. 2020, 263, 118367. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, F.; Liang, G.; Yang, X.; Lei, Y.; Jin, F.; Xu, L.; Zhang, C.; Jiang, W.; Ben, H.; Li, X. Micro-Structure Engineering in Pd-InOx Catalysts and Mechanism Studies for CO2 Hydrogenation to Methanol. Molecules 2024, 29, 3715. https://doi.org/10.3390/molecules29163715
Zhao F, Liang G, Yang X, Lei Y, Jin F, Xu L, Zhang C, Jiang W, Ben H, Li X. Micro-Structure Engineering in Pd-InOx Catalysts and Mechanism Studies for CO2 Hydrogenation to Methanol. Molecules. 2024; 29(16):3715. https://doi.org/10.3390/molecules29163715
Chicago/Turabian StyleZhao, Fengwang, Gemeng Liang, Xiaoli Yang, Yang Lei, Fayi Jin, Leilei Xu, Chuanhui Zhang, Wei Jiang, Haoxi Ben, and Xingyun Li. 2024. "Micro-Structure Engineering in Pd-InOx Catalysts and Mechanism Studies for CO2 Hydrogenation to Methanol" Molecules 29, no. 16: 3715. https://doi.org/10.3390/molecules29163715
APA StyleZhao, F., Liang, G., Yang, X., Lei, Y., Jin, F., Xu, L., Zhang, C., Jiang, W., Ben, H., & Li, X. (2024). Micro-Structure Engineering in Pd-InOx Catalysts and Mechanism Studies for CO2 Hydrogenation to Methanol. Molecules, 29(16), 3715. https://doi.org/10.3390/molecules29163715