
Domino Reactions Enable Zn-Mediated Direct Synthesis of Spiro-Fused 2-Oxindole-α-Methylene-γ-Butyrolactones/Lactams from Isatin Derivatives and 2-(Bromomethyl)acrylates

Abstract

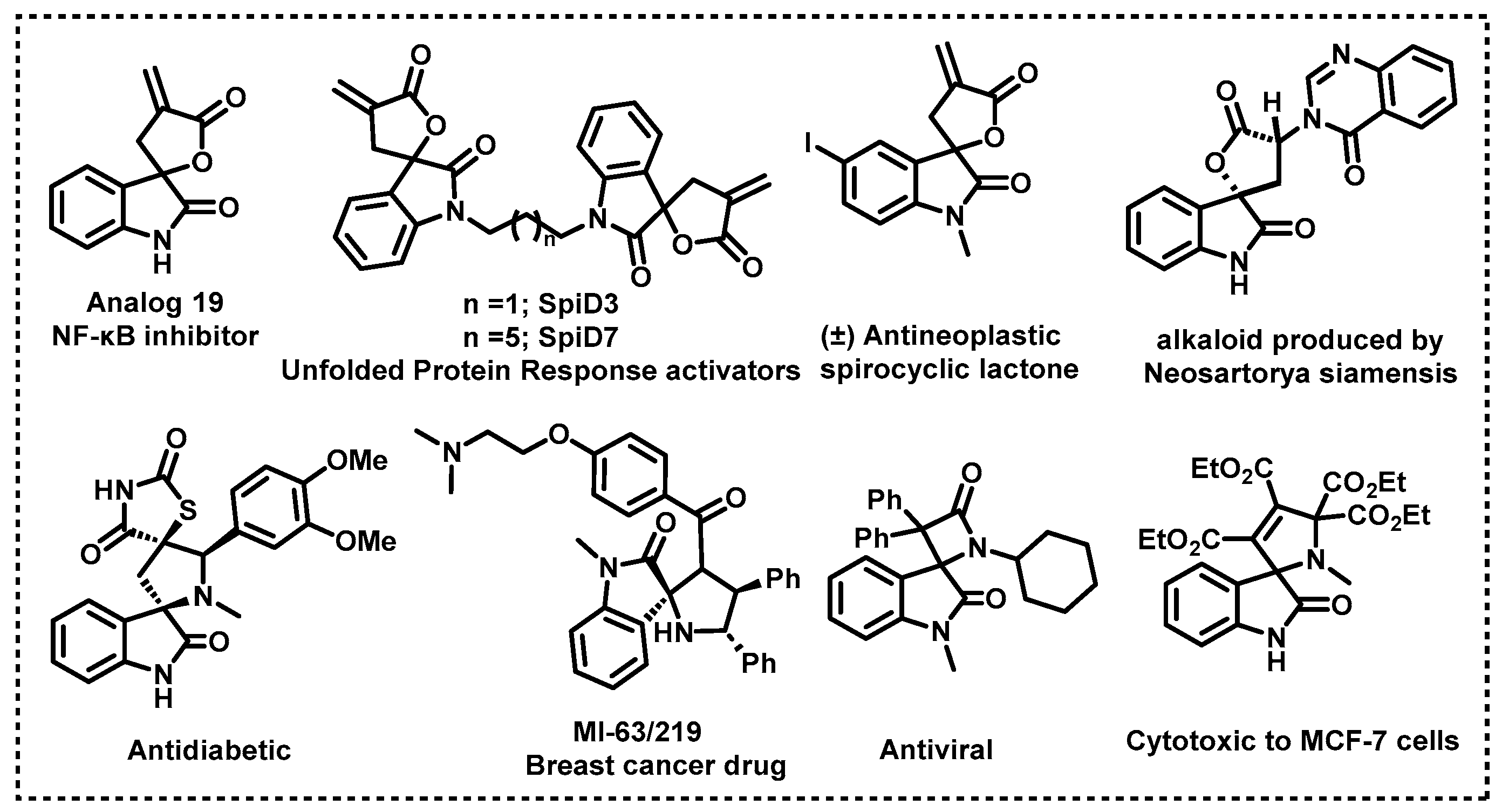
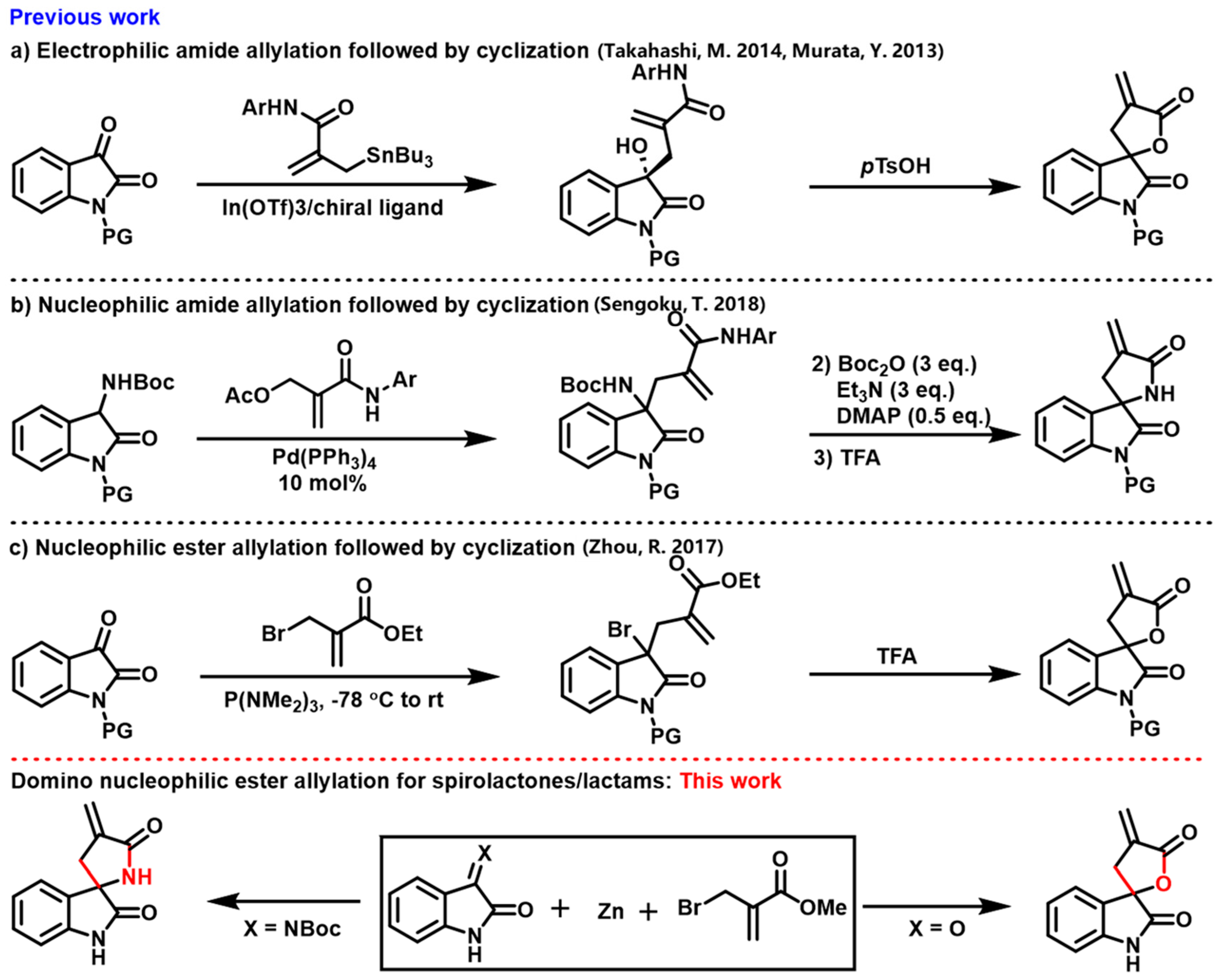
1. Introduction
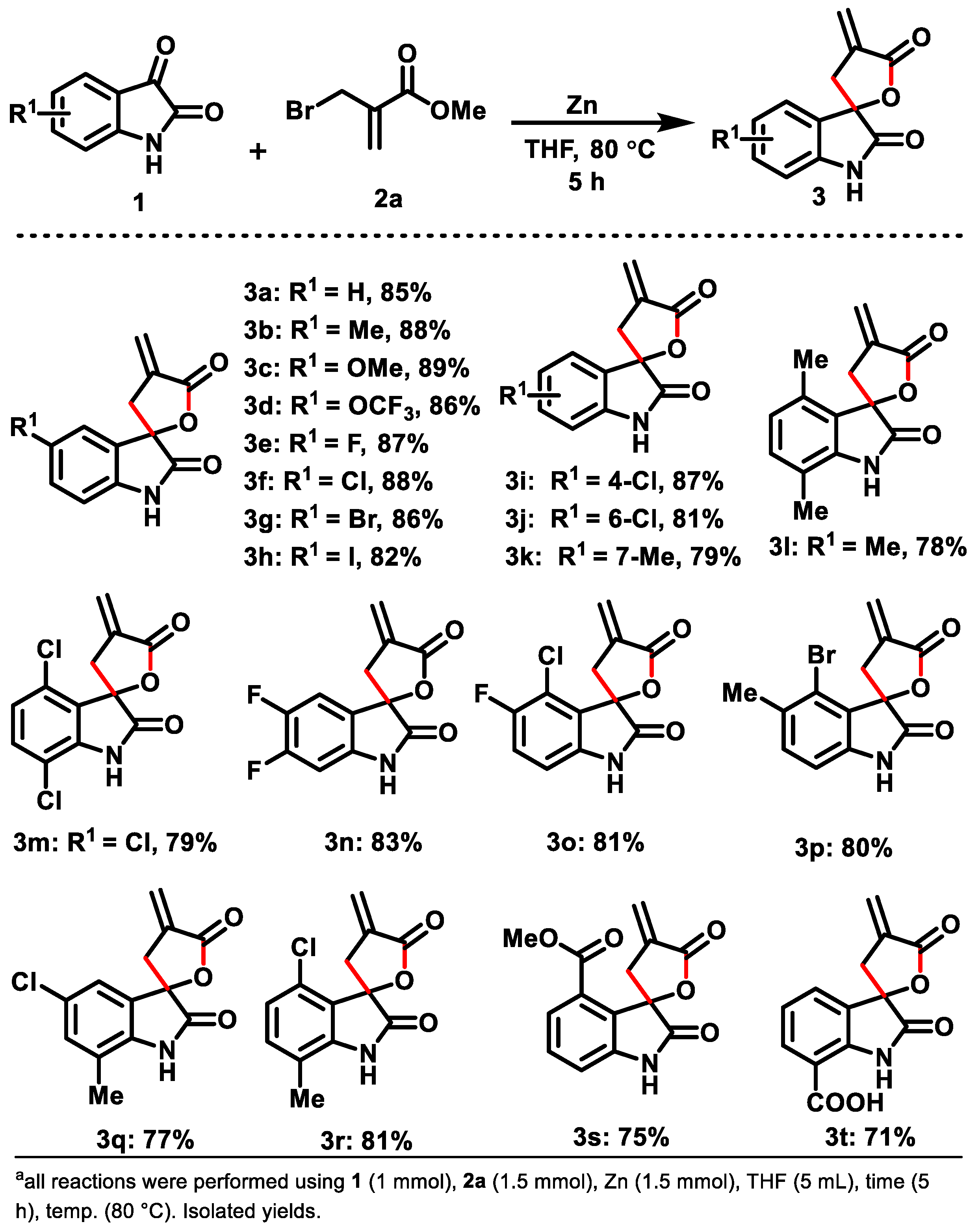
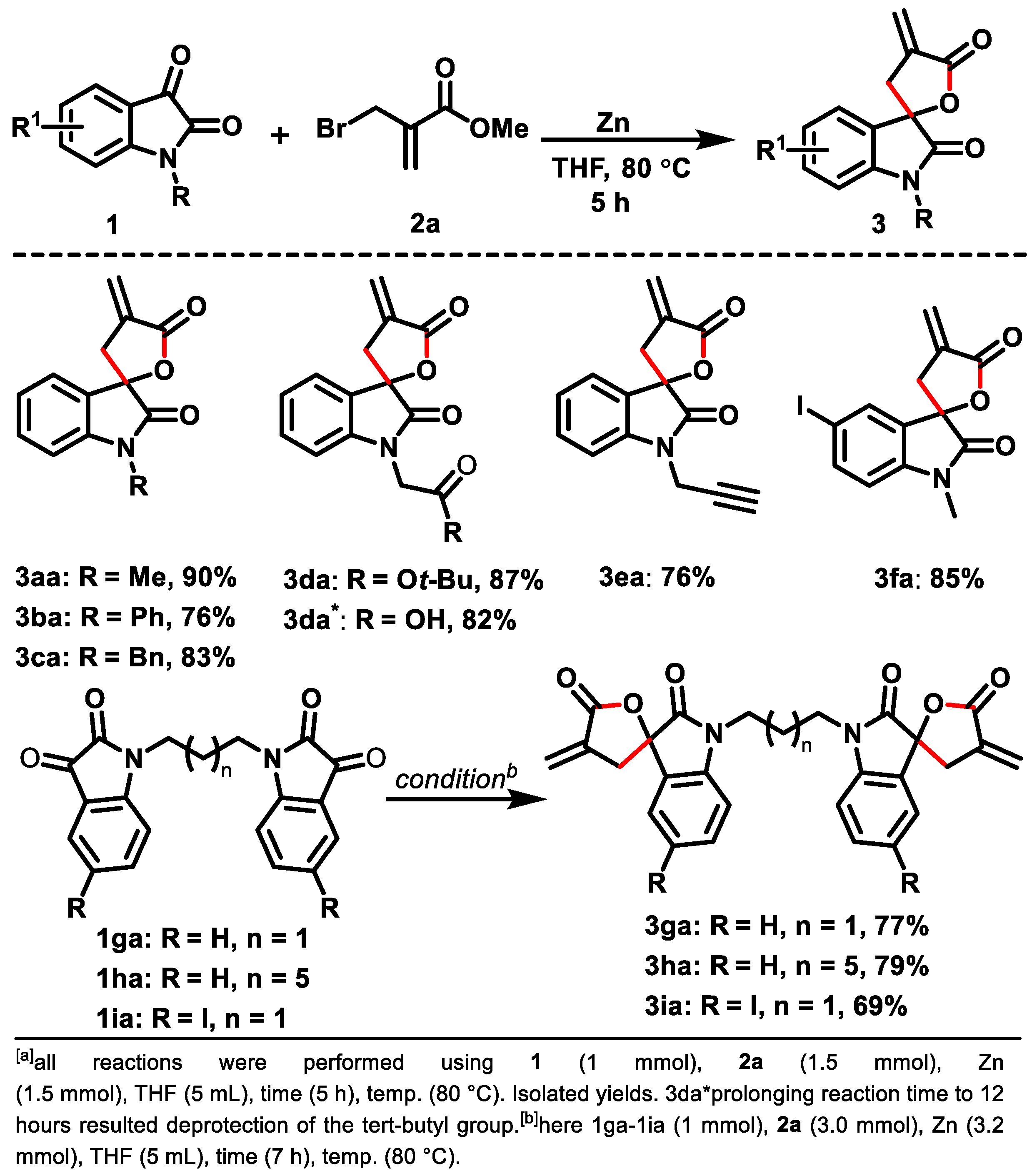
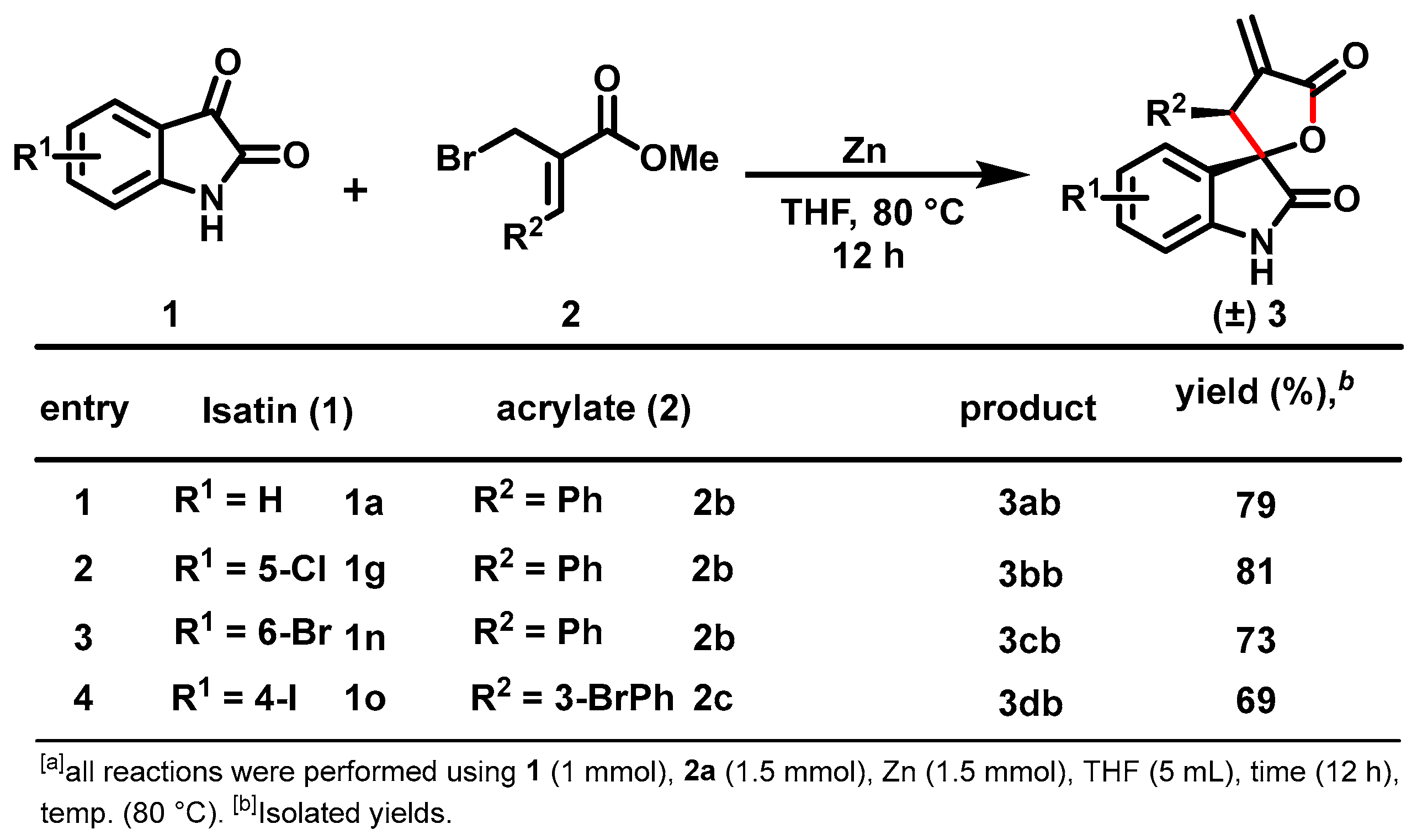
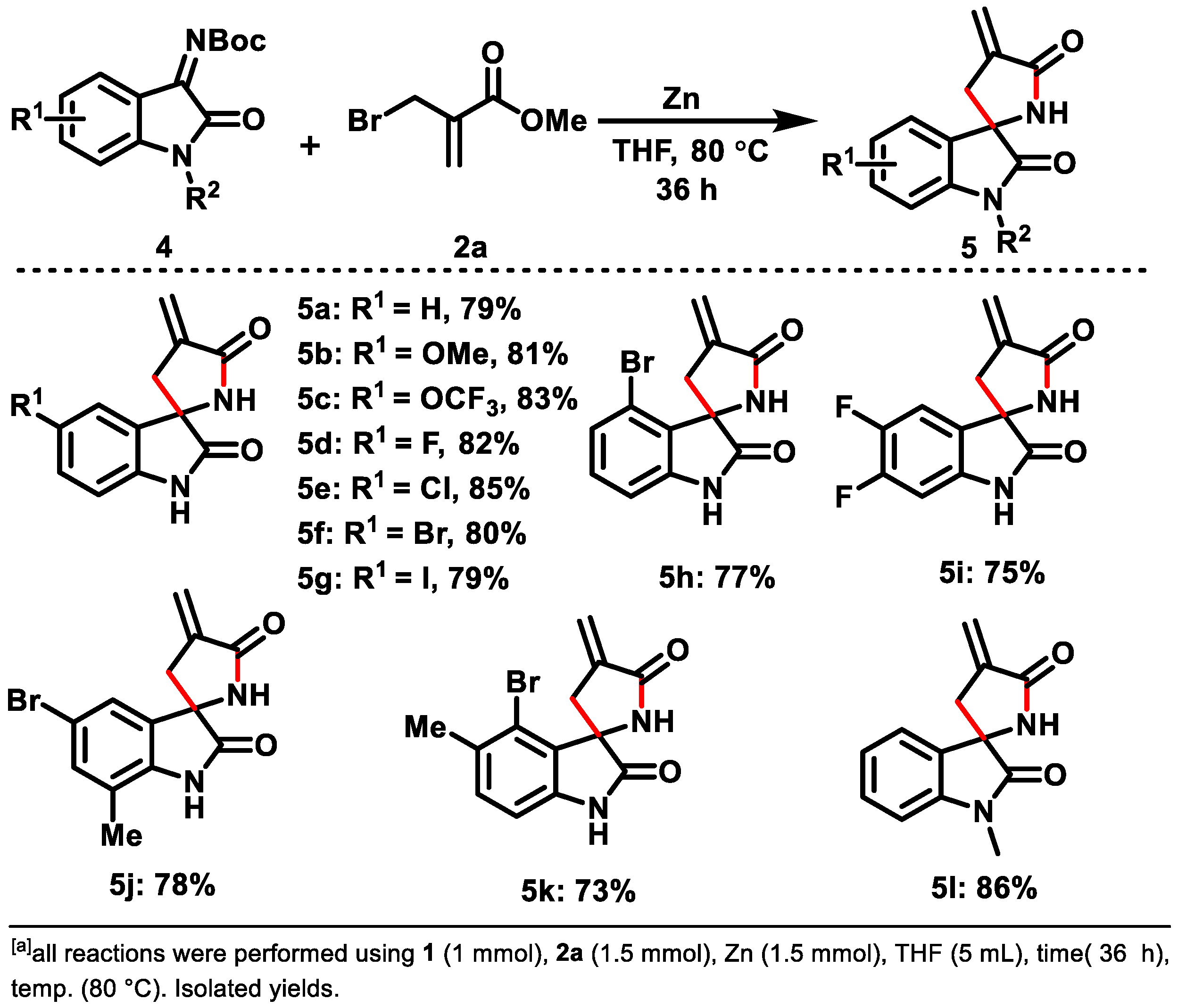
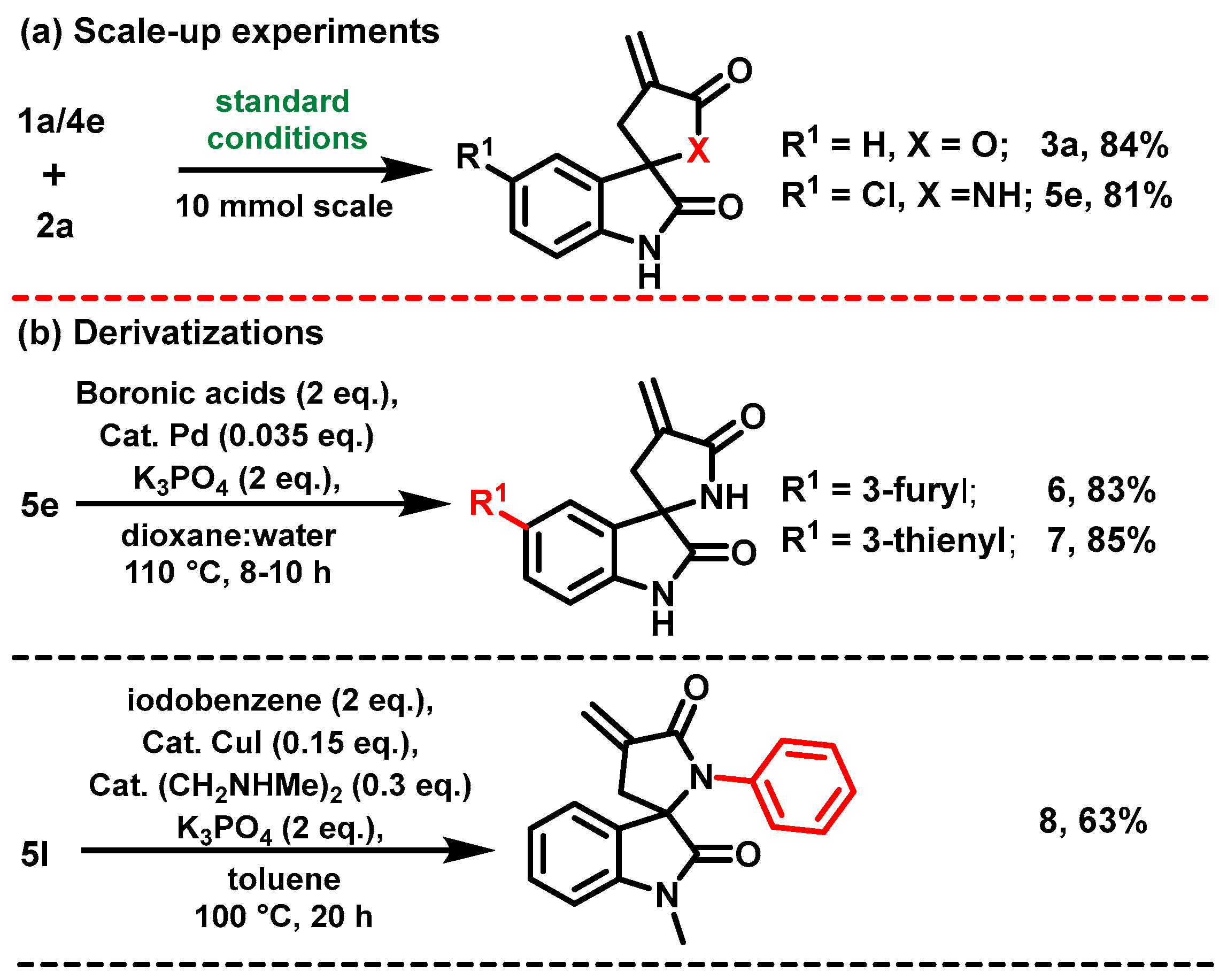
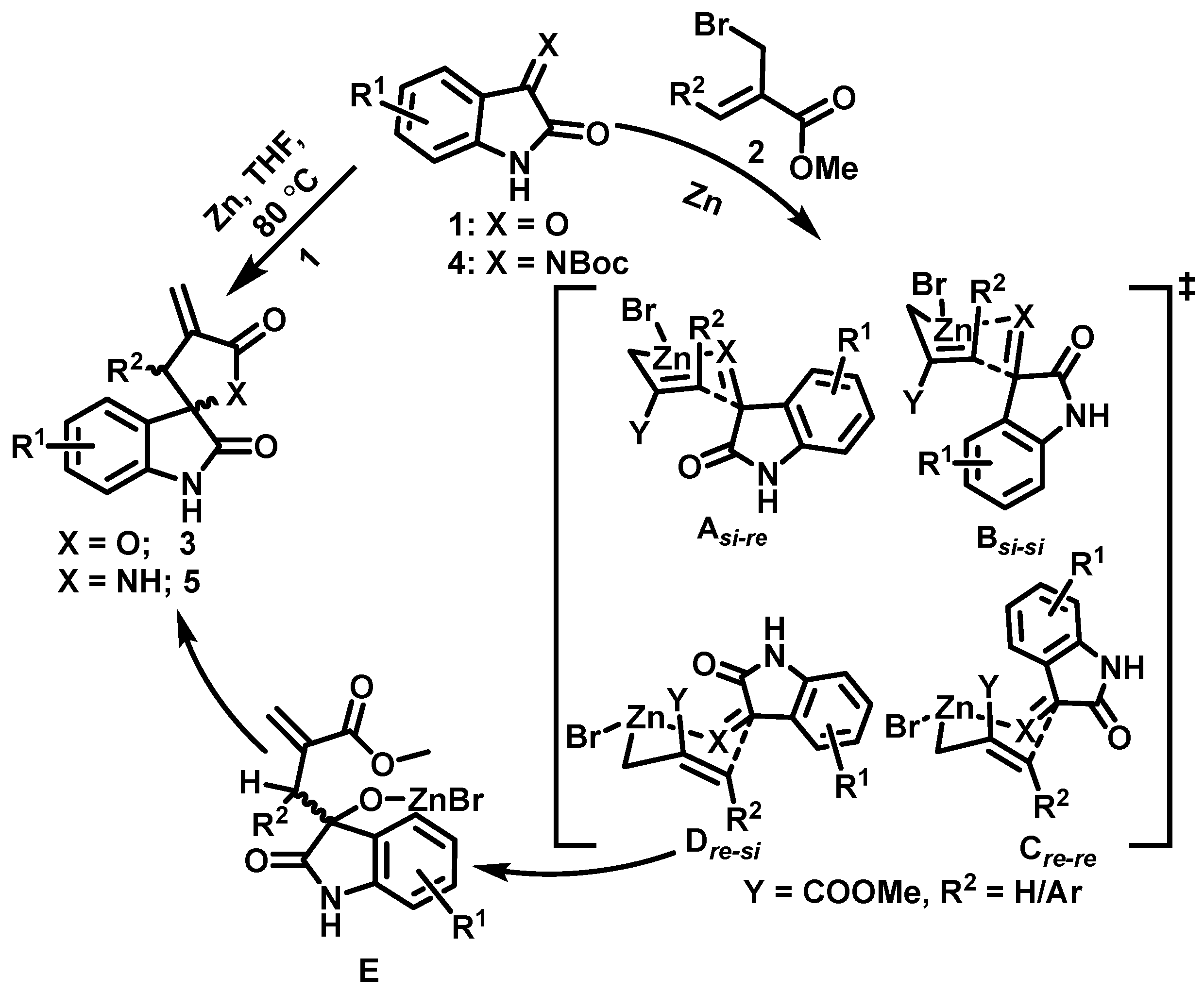
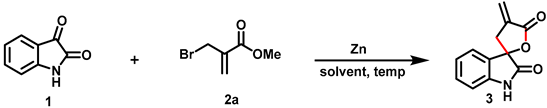
2. Results and Discussion
3. Materials and Methods
3.1. General Method for the Synthesis of Spiro-Fused 2-Oxindole/α-Methylene-γ-Butyrolactone
3.2. General Method for the Synthesis of Spiro-Fused 2-Oxindole/α-Methylene-γ-Butyrolactam
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tietze, L.F. Domino Reactions in Organic Synthesis. Chem. Rev. 1996, 96, 115–136. [Google Scholar] [CrossRef]
- Delayre, B.; Wang, Q.; Zhu, J. Natural Product Synthesis Enabled by Domino Processes Incorporating a 1,2-Rearrangement Step. ACS Cent. Sci. 2021, 7, 559–569. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Zhu, X.; Liu, X.; He, F.; Yang, G.; Xu, C.; Yang, X. Recent Advances in the Synthesis of 3,n-Fused Tricyclic Indole Skeletons via Palladium-Catalyzed Domino Reactions. Molecules 2023, 28, 1647. [Google Scholar] [CrossRef] [PubMed]
- Marti, C.; Carreira, E.M. Construction of Spiro[pyrrolidine-3,3′-oxindoles]—Recent Applications to the Synthesis of Oxindole Alkaloids. Eur. J. Org. Chem. 2003, 2003, 2209–2219. [Google Scholar] [CrossRef]
- Baran, P.S.; Maimone, T.J.; Richter, J.M. Total synthesis of marine natural products without using protecting groups. Nature 2007, 446, 404–408. [Google Scholar] [CrossRef] [PubMed]
- Galliford, C.V.; Scheidt, K.A. Pyrrolidinyl-Spirooxindole Natural Products as Inspirations for the Development of Potential Therapeutic Agents. Angew. Chem. Int. Ed. 2007, 46, 8748–8758. [Google Scholar] [CrossRef] [PubMed]
- Richter, J.M.; Ishihara, Y.; Masuda, T.; Whitefield, B.W.; Llamas, T.; Pohjakallio, A.; Baran, P.S. Enantiospecific Total Synthesis of the Hapalindoles, Fischerindoles, and Welwitindolinones via a Redox Economic Approach. J. Am. Chem. Soc. 2008, 130, 17938–17954. [Google Scholar] [CrossRef]
- Reisman, S.E.; Ready, J.M.; Weiss, M.M.; Hasuoka, A.; Hirata, M.; Tamaki, K.; Ovaska, T.V.; Smith, C.J.; Wood, J.L. Evolution of a Synthetic Strategy: Total Synthesis of (±)-Welwitindolinone A Isonitrile. J. Am. Chem. Soc. 2008, 130, 2087–2100. [Google Scholar] [CrossRef]
- Singh, G.S.; Desta, Z.Y. Isatins as privileged molecules in design and synthesis of spiro-fused cyclic frameworks. Chem. Rev. 2012, 112, 6104–6155. [Google Scholar] [CrossRef]
- Hong, S.; Jung, M.; Park, Y.; Ha, M.W.; Park, C.; Lee, M.; Park, H.-G. Efficient Enantioselective Total Synthesis of (−)-Horsfiline. Chem. A Eur. J. 2013, 19, 9599–9605. [Google Scholar] [CrossRef]
- Zhou, X.; Xiao, T.; Iwama, Y.; Qin, Y. Biomimetic Total Synthesis of (+)-Gelsemine. Angew. Chem. Int. Ed. 2012, 51, 4909–4912. [Google Scholar] [CrossRef] [PubMed]
- Trost, B.M.; Bringley, D.A.; Zhang, T.; Cramer, N. Rapid Access to Spirocyclic Oxindole Alkaloids: Application of the Asymmetric Palladium-Catalyzed [3 + 2] Trimethylenemethane Cycloaddition. J. Am. Chem. Soc. 2013, 135, 16720–16735. [Google Scholar] [CrossRef] [PubMed]
- Bian, Z.; Marvin, C.C.; Pettersson, M.; Martin, S.F. Enantioselective total syntheses of citrinadins A and B. Stereochemical revision of their assigned structures. J. Am. Chem. Soc. 2014, 136, 14184–14192. [Google Scholar] [CrossRef] [PubMed]
- Rana, S.; Kour, S.; Kizhake, S.; King, H.M.; Mallareddy, J.R.; Case, A.J.; Huxford, T.; Natarajan, A. Dimers of isatin derived α-methylene-γ-butyrolactone as potent anti-cancer agents. Bioorg. Med. Chem. Lett. 2022, 65, 128713–1218719. [Google Scholar] [CrossRef] [PubMed]
- Kour, S.; Rana, S.; Kubica, S.P.; Kizhake, S.; Ahmad, M.; Muñoz-Trujillo, C.; Klinkebiel, D.; Singh, S.; Mallareddy, J.R.; Chandra, S.; et al. Spirocyclic dimer SpiD7 activates the unfolded protein response to selectively inhibit growth and induce apoptosis of cancer cells. J. Biol. Chem. 2022, 298, 101890. [Google Scholar] [CrossRef] [PubMed]
- Kour, S.; Rana, S.; Kizhake, S.; Lagundžin, D.; Klinkebiel, D.; Mallareddy, J.R.; Huxford, T.; Woods, N.T.; Natarajan, A. Stapling proteins in the RELA complex inhibits TNFα-induced nuclear translocation of RELA. RSC Chem. Biol. 2022, 3, 32–36. [Google Scholar] [CrossRef] [PubMed]
- Rana, S.; Kour, S.; Sonawane, Y.A.; Robb, C.M.; Contreras, J.I.; Kizhake, S.; Zahid, M.; Karpf, A.R.; Natarajan, A. Symbiotic prodrugs (SymProDs) dual targeting of NFkappaB and CDK. Chem. Biol. Drug Des. 2020, 96, 773–784. [Google Scholar] [CrossRef]
- Rana, S.; Blowers, E.C.; Tebbe, C.; Contreras, J.I.; Radhakrishnan, P.; Kizhake, S.; Zhou, T.; Rajule, R.N.; Arnst, J.L.; Munkarah, A.R.; et al. Isatin Derived Spirocyclic Analogues with α-Methylene-γ-butyrolactone as Anticancer Agents: A Structure-Activity Relationship Study. J. Med. Chem. 2016, 59, 5121–5127. [Google Scholar] [CrossRef]
- Rana, S.; Natarajan, A. Face selective reduction of the exocyclic double bond in isatin derived spirocyclic lactones. Org. Biomol. Chem. 2013, 11, 244–247. [Google Scholar] [CrossRef]
- Murata, K.; Kaneko, S.; Kitazume, T. Preparation and biological evaluation of γ-fluoromethyl-α-methylene-γ-butyrolactone and γ-butyrolactam. Bioorg. Med. Chem. Lett. 1993, 3, 2685–2688. [Google Scholar] [CrossRef]
- Qiao, L.; Wang, S.; George, C.; Lewin, N.E.; Blumberg, P.M.; Kozikowski, A.P. Structure-Based Design of a New Class of Protein Kinase C Modulators. J. Am. Chem. Soc. 1998, 120, 6629–6630. [Google Scholar] [CrossRef]
- Janecki, T.; Błaszczyk, E.; Studzian, K.; Janecka, A.; Krajewska, U.; Rózalski, M. Novel synthesis, cytotoxic evaluation, and structure-activity relationship studies of a series of alpha-alkylidene-gamma-lactones and lactams. J. Med. Chem. 2005, 48, 3516–3521. [Google Scholar] [CrossRef] [PubMed]
- Kitson, R.R.; Millemaggi, A.; Taylor, R.J. The renaissance of alpha-methylene-gamma-butyrolactones: New synthetic approaches. Angew. Chem. Int. Ed. 2009, 48, 9426–9451. [Google Scholar] [CrossRef]
- Lepoittevin, J.P.; Berl, V.; Giménez-Arnau, E. Alpha-methylene-gamma-butyrolactones: Versatile skin bioactive natural products. Chem. Rec. 2009, 9, 258–270. [Google Scholar] [CrossRef]
- Delong, W.; Lanying, W.; Yongling, W.; Shuang, S.; Juntao, F.; Xing, Z. Natural α-methylenelactam analogues: Design, synthesis and evaluation of α-alkenyl-γ and δ-lactams as potential antifungal agents against Colletotrichum orbiculare. Eur. J. Med. Chem. 2017, 130, 286–307. [Google Scholar] [CrossRef]
- Jun-Tao, F.; De-Long, W.; Yong-Ling, W.; He, Y.; Xing, Z. New antifungal scaffold derived from a natural pharmacophore: Synthesis of α-methylene-γ-butyrolactone derivatives and their antifungal activity against Colletotrichum lagenarium. Bioorg. Med. Chem. Lett. 2013, 23, 4393–4397. [Google Scholar] [CrossRef]
- Jackson, P.A.; Schares, H.A.M.; Jones, K.F.M.; Widen, J.C.; Dempe, D.P.; Grillet, F.; Cuellar, M.E.; Walters, M.A.; Harki, D.A.; Brummond, K.M. Synthesis of Guaianolide Analogues with a Tunable α-Methylene−γ-lactam Electrophile and Correlating Bioactivity with Thiol Reactivity. J. Med. Chem. 2020, 63, 14951–14978. [Google Scholar] [CrossRef]
- Siedle, B.; García-Piñeres, A.J.; Murillo, R.; Schulte-Mönting, J.; Castro, V.; Rüngeler, P.; Klaas, C.A.; Da Costa, F.B.; Kisiel, W.; Merfort, I. Quantitative Structure−Activity Relationship of Sesquiterpene Lactones as Inhibitors of the Transcription Factor NF-κB. J. Med. Chem. 2004, 47, 6042–6054. [Google Scholar] [CrossRef]
- Dempe, D.P.; Ji, C.-L.; Liu, P.; Brummond, K.M. Thiol Reactivity of N-Aryl α-Methylene-γ-lactams: Influence of the Guaianolide Structure. J. Org. Chem. 2022, 87, 11204–11217. [Google Scholar] [CrossRef] [PubMed]
- González-Saiz, B.; Carreira-Barral, I.; Pertejo, P.; Gómez-Ayuso, J.; Quesada, R.; García-Valverde, M. One-Pot Diastereoselective Synthesis of Pyrrolopiperazine-2,6-diones by a Ugi/Nucleophilic Substitution/N-Acylation Sequence. J. Org. Chem. 2022, 87, 9391–9398. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Murata, Y.; Yagishita, F.; Sakamoto, M.; Sengoku, T.; Yoda, H. Catalytic Enantioselective Amide Allylation of Isatins and Its Application in the Synthesis of 2-Oxindole Derivatives Spiro-Fused to the α-Methylene-γ-Butyrolactone Functionality. Chem.-Eur. J. 2014, 20, 11091–11100. [Google Scholar] [CrossRef]
- Murata, Y.; Takahashi, M.; Yagishita, F.; Sakamoto, M.; Sengoku, T.; Yoda, H. Construction of Spiro-Fused 2-Oxindole/α-Methylene- γ-Butyrolactone Systems with Extremely High Enantioselectivity via Indium-Catalyzed Amide Allylation of N-Methyl Isatin. Org. Lett. 2013, 15, 6182–6185. [Google Scholar] [CrossRef]
- Sengoku, T.; Hayashi, D.; Takahashi, M.; Yoda, H. Electrophilic Amide Allylation of 3-Heterosubstituted Oxindoles: A Route to Spirocyclic 2-Oxindoles Containing the α-Methylene-γ-butyrolactam Structure. Eur. J. Org. Chem. 2018, 2018, 1813–1820. [Google Scholar] [CrossRef]
- Zhou, R.; Liu, R.; Zhang, K.; Han, L.; Zhang, H.; Gao, W.; Li, R. Metal-free formal carbon-halogen bond insertion: Facile syntheses of 3-halo 3,3′-disubstituted oxindoles and spirooxindole-γ-butyrolactones. Chem. Commun. 2017, 53, 6860–6863. [Google Scholar] [CrossRef]
- Jayakumar, S.; Muthusamy, S.; Prakash, M.; Kesavan, V. Enantioselective Synthesis of Spirooxindole α-exo-Methylene-γ-butyrolactones from 3-OBoc-Oxindoles. Eur. J. Org. Chem. 2014, 2014, 1893–1898. [Google Scholar] [CrossRef]
- Dilman, A.D.; Levin, V.V. Advances in the chemistry of organozinc reagents. Tetrahedron Lett. 2016, 57, 3986–3992. [Google Scholar] [CrossRef]
- Heindel, N.D.; Minatelli, J.A. Synthesis and antibacterial and anticancer evaluations of alpha-methylene-gamma-butyrolactones. J. Pharm. Sci. 1981, 70, 84–86. [Google Scholar] [CrossRef]
- Lee, A.S.-Y.; Chang, Y.-T.; Wang, S.-H.; Chu, S.-F. Synthesis of 3,4-disubstituted α-methylene-γ-lactones via sonochemical Barbier-type reaction. Tetrahedron Lett. 2002, 43, 8489–8492. [Google Scholar] [CrossRef]
- Csuk, R.; Schröder, C.; Hutter, S.; Mohr, K. Enantioselective Dreiding-Schmidt reactions: Asymmetric synthesis and analysis of α-methylene-γ-butyrolactones. Tetrahedron Asymmetry 1997, 8, 1411–1429. [Google Scholar] [CrossRef]
- Yang, Z.; Huang, D.; Wen, L.; Wang, J.; Wang, K.; Hu, Y. Tin Powder-Promoted “One-Pot” Synthesis of α-Methylene-γ-butyrolactones. Chin. J. Org. Chem. 2018, 38, 1725–1732. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Entry | 2a (equiv.) | Zn (equiv.) | Solvent | Temperature (°C) | 3 Yield (%) b |
| 1 | 1 | 1 | THF | 60 | 63 |
| 2 | 1 | 1 | THF | 80 | 71 |
| 3 | 1.5 | 2 | THF | 80 | 84 |
| 4 | 1.5 | 1.6 | THF | 80 | 85 |
| 5 | 2.0 | 2.5 | THF | 80 | 81 |
| 6 | 1.5 | 1.6 | DMF | 80 | 63 |
| 7 | 1.5 | 1.6 | Dioxane | 80 | 59 |
| 8 | 1.5 | 1.6 | DCE | 80 | 65 |
| 9 | 1.5 | 1.6 | MeOH | 80 | 73 |
| 10 | 1.5 | 1.6 | THF | 70 | 76 |
| 11 | 1.5 | 1.6 | THF | 100 | 81 |
| 12 c | 1.5 | 1.6 | THF | 80 | 82 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mukthapuram, P.R.; Natarajan, A. Domino Reactions Enable Zn-Mediated Direct Synthesis of Spiro-Fused 2-Oxindole-α-Methylene-γ-Butyrolactones/Lactams from Isatin Derivatives and 2-(Bromomethyl)acrylates. Molecules 2024, 29, 3612. https://doi.org/10.3390/molecules29153612
Mukthapuram PR, Natarajan A. Domino Reactions Enable Zn-Mediated Direct Synthesis of Spiro-Fused 2-Oxindole-α-Methylene-γ-Butyrolactones/Lactams from Isatin Derivatives and 2-(Bromomethyl)acrylates. Molecules. 2024; 29(15):3612. https://doi.org/10.3390/molecules29153612
Chicago/Turabian StyleMukthapuram, Prathap Reddy, and Amarnath Natarajan. 2024. "Domino Reactions Enable Zn-Mediated Direct Synthesis of Spiro-Fused 2-Oxindole-α-Methylene-γ-Butyrolactones/Lactams from Isatin Derivatives and 2-(Bromomethyl)acrylates" Molecules 29, no. 15: 3612. https://doi.org/10.3390/molecules29153612
APA StyleMukthapuram, P. R., & Natarajan, A. (2024). Domino Reactions Enable Zn-Mediated Direct Synthesis of Spiro-Fused 2-Oxindole-α-Methylene-γ-Butyrolactones/Lactams from Isatin Derivatives and 2-(Bromomethyl)acrylates. Molecules, 29(15), 3612. https://doi.org/10.3390/molecules29153612